
Characterization of the Molecular Alterations Induced by the Prolonged Exposure of Normal Colon Mucosa and Colon Cancer Cells to Low-Dose Bisphenol A

Abstract
1. Introduction
2. Results
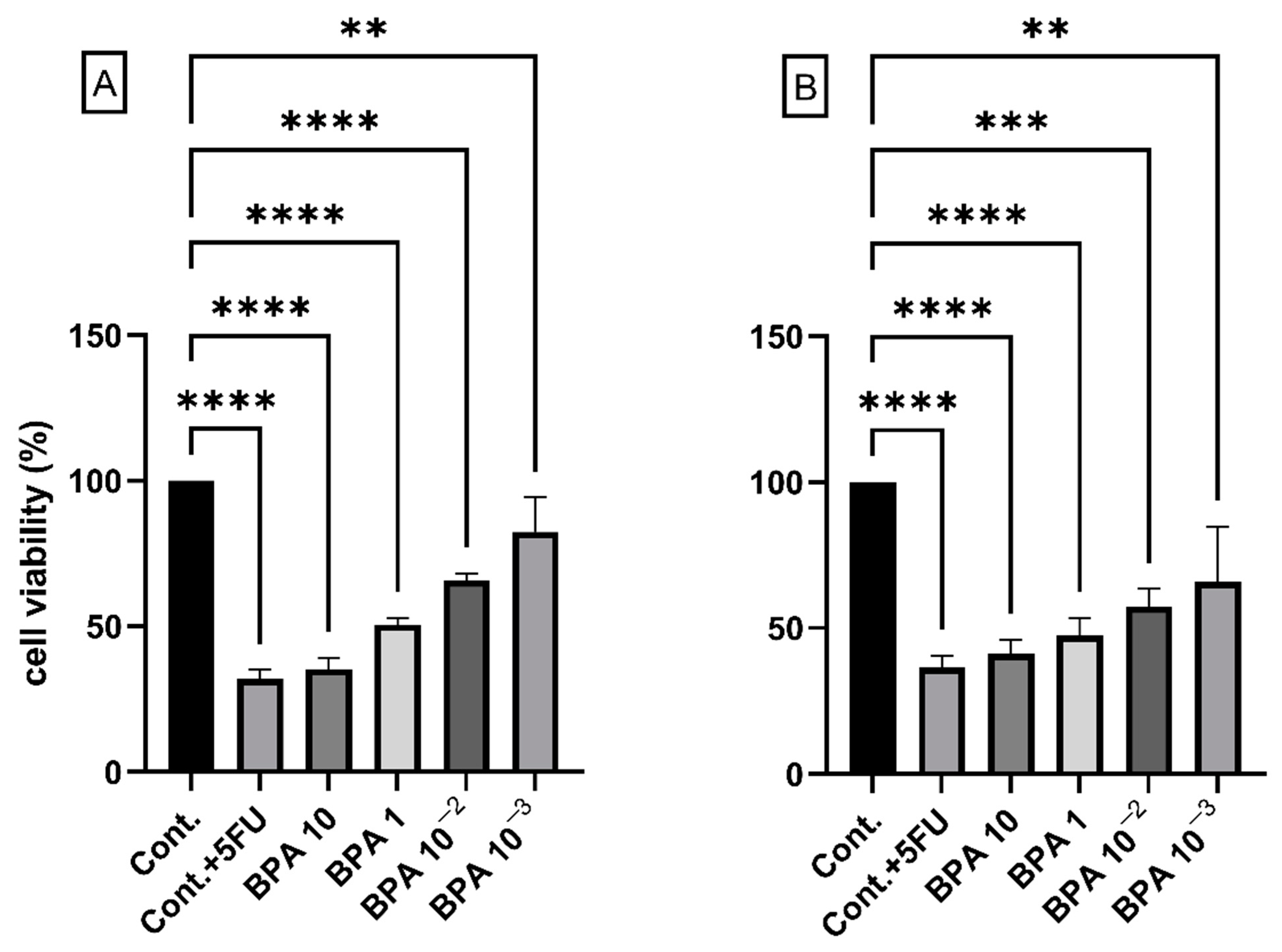
2.1. BPA and Cell Viability
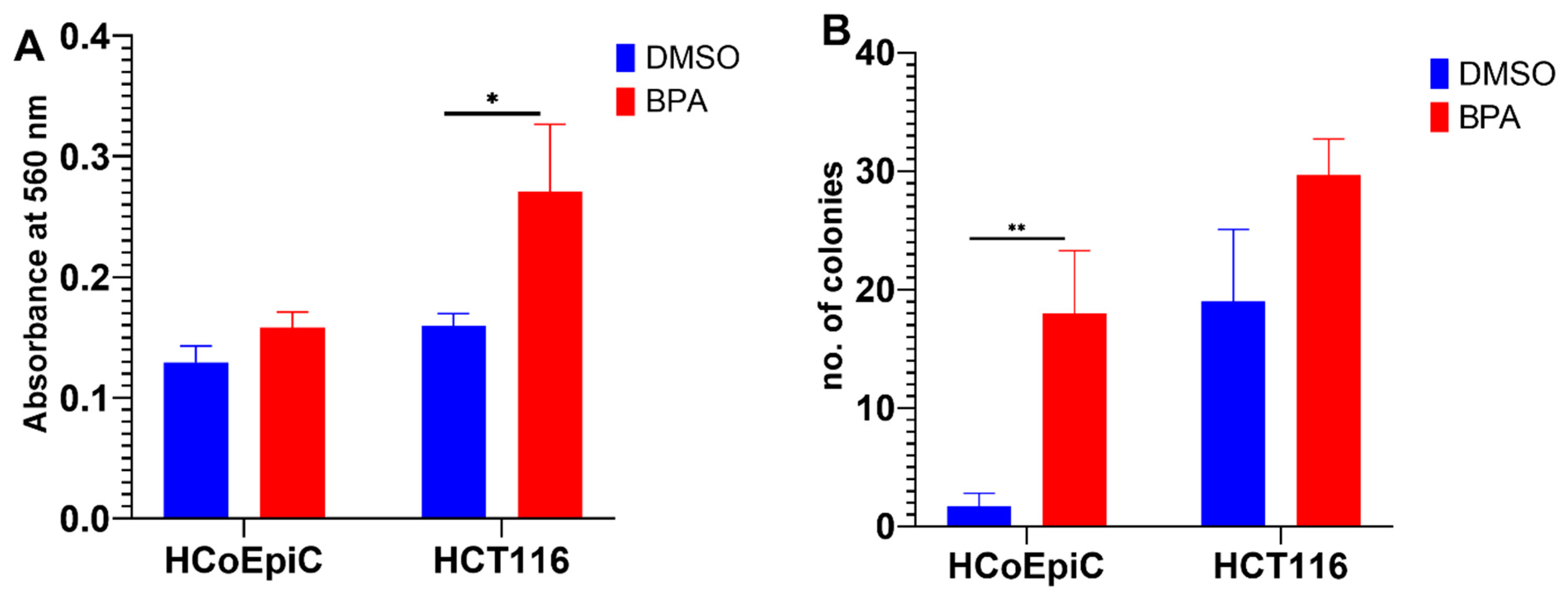
2.2. BPA and Collagen Invasion
2.3. Colony Formation in Soft Agar
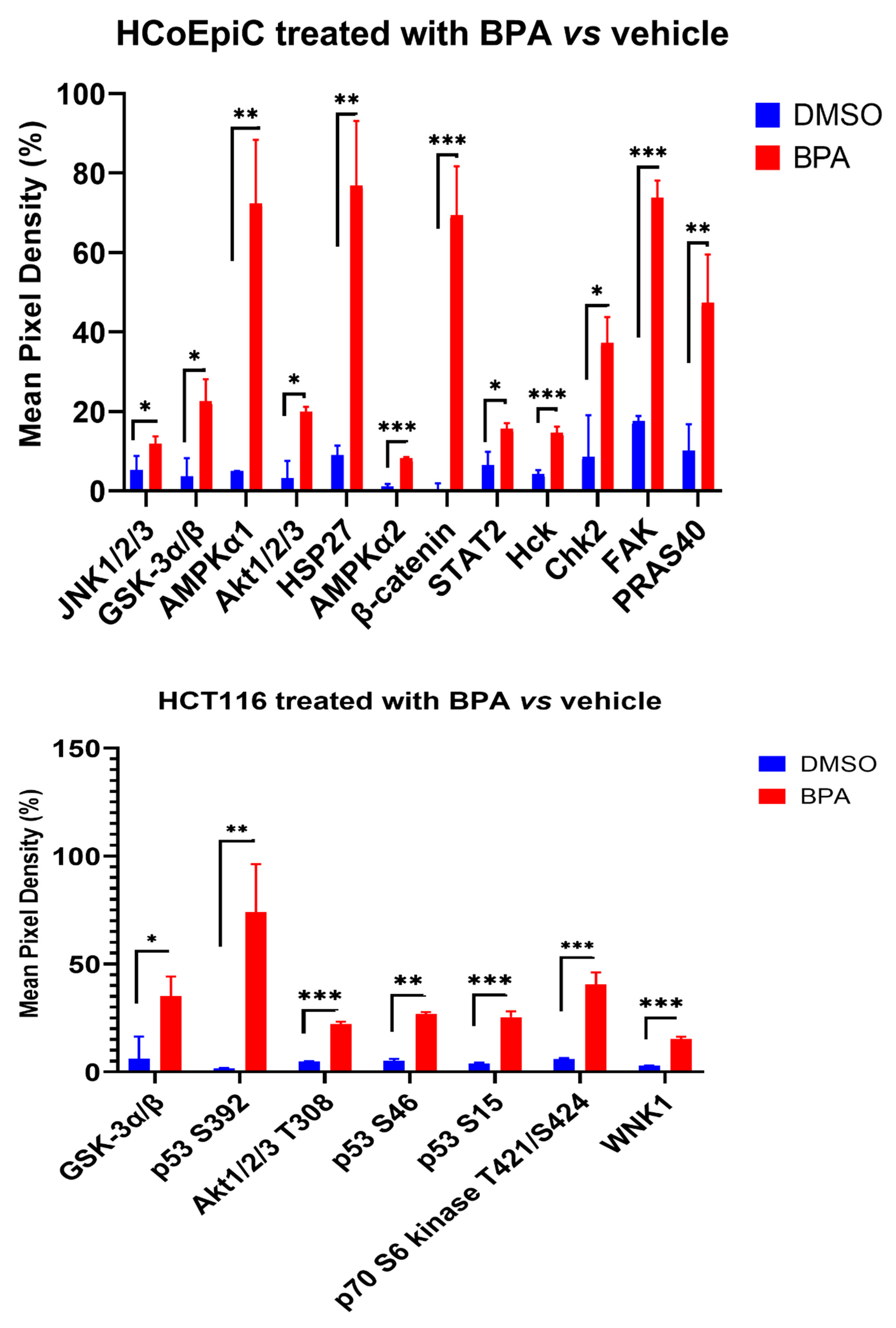
2.4. Proteomic Analysis (Human Phospho Kinase Array)
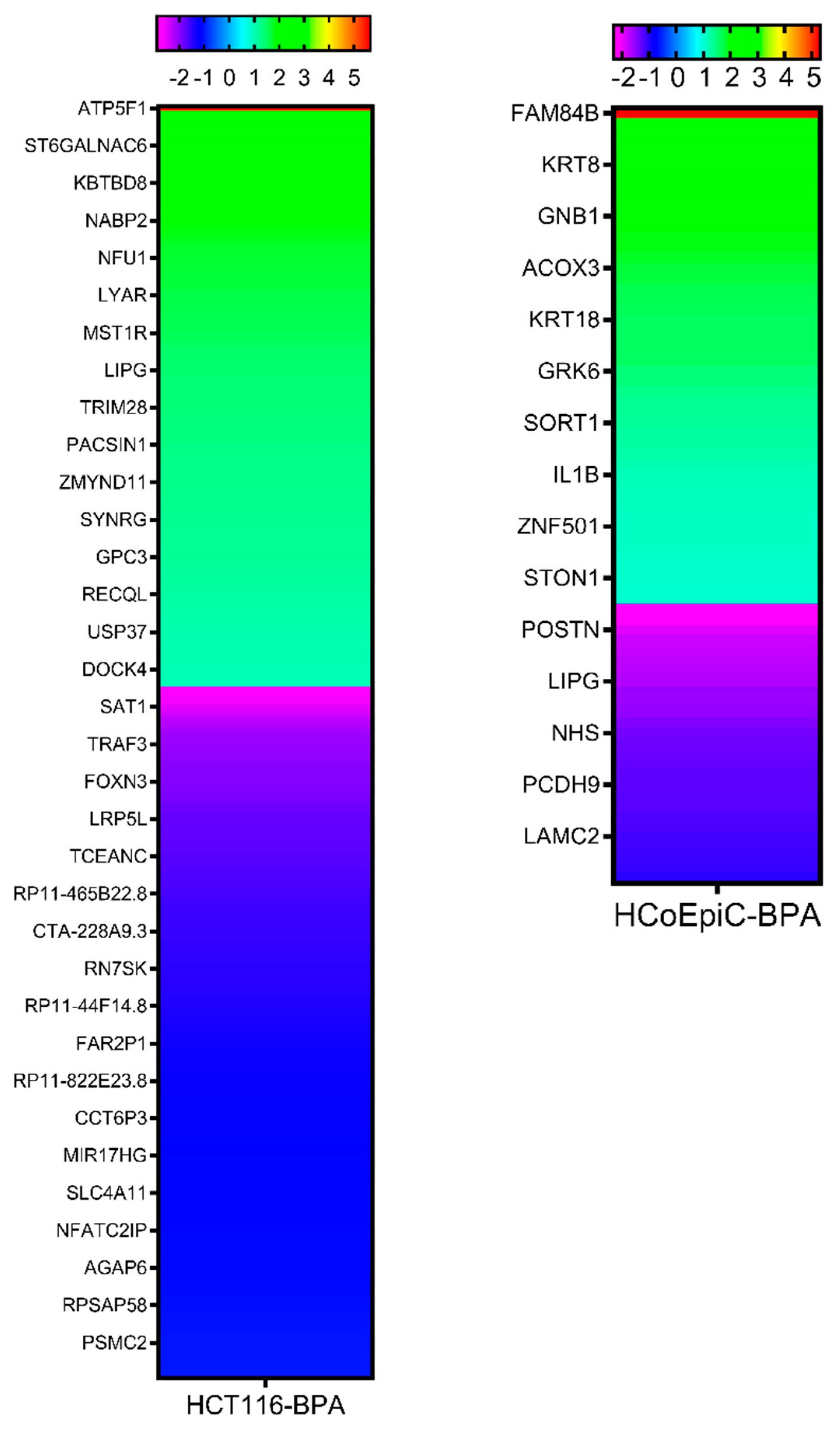
2.5. Next-Generation Sequencing
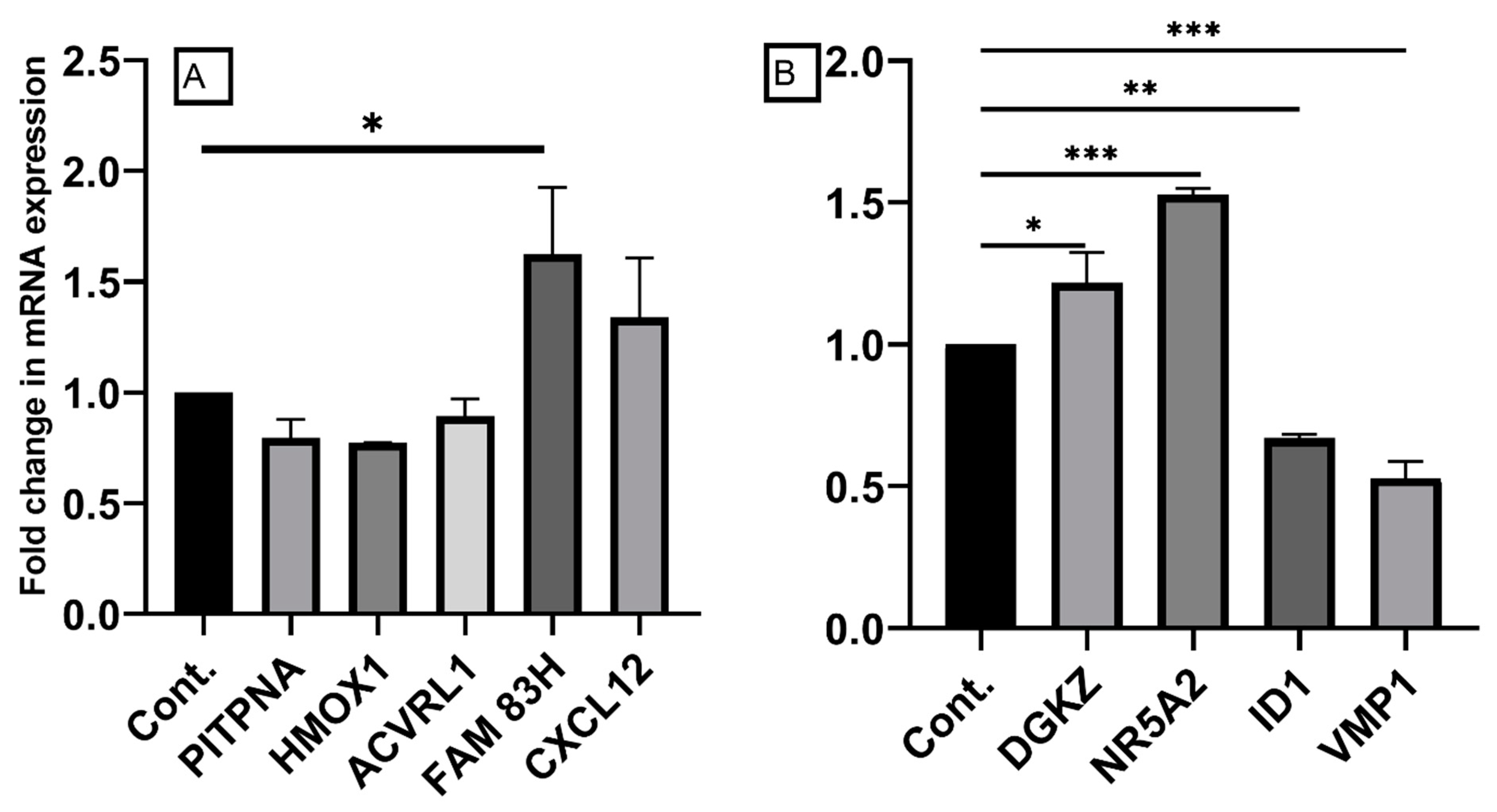
2.6. Confirmation of Differentially Expressed Genes by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Toxic Chemicals and Cytotoxicity Assay
4.3. Invasion Assay
4.4. Colony Formation in Soft Agar
4.5. Next-Generation Sequencing Analysis
4.6. Real Time-Polymerase Chain Reaction (RT-PCR)
4.7. Proteome Profile/Human Phospho-Kinase Array
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdel-Rahman, W.M.; Faris, M.E.; Peltomaki, P. Molecular Determinants of Colon Cancer Susceptibility in the East and West. Curr. Mol. Med. 2017, 17, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, T.T.; Shoman, S.; Eissa, S.; Peltomaki, P.; Abdel-Rahman, W.M. Distinct Genetic and Epigenetic Signatures of Colorectal Cancers According to Ethnic Origin. Cancer Epidemiol. Biomark. Prev. 2012, 21, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, W.M. Genomic Instability and Carcinogenesis: An Update. Curr. Genom. 2008, 9, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Hafezi, S.A.; Abdel-Rahman, W.M. The Endocrine Disruptor Bisphenol A (BPA) Exerts a Wide Range of Effects in Carcinogenesis and Response to Therapy. Curr. Mol. Pharmacol. 2019, 12, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, W.M.; Moustafa, Y.M.; Ahmed, B.O.; Mostafa, R.M. Endocrine Disruptors and Breast Cancer Risk-Time to Consider the Environment. Asian Pac. J. Cancer Prev. 2012, 13, 5937–5946. [Google Scholar] [CrossRef]
- Nair, V.A.; Valo, S.; Peltomaki, P.; Bajbouj, K.; Abdel-Rahman, W.M. Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3735. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Jalal, N.; Surendranath, A.R.; Pathak, J.L.; Yu, S.; Chung, C.Y. Bisphenol A (BPA) the Mighty and the Mutagenic. Toxicol. Rep. 2017, 5, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, H.; Liu, S. Low-Dose Bisphenol A Exposure: A Seemingly Instigating Carcinogenic Effect on Breast Cancer. Adv. Sci. 2016, 4, 1600248. [Google Scholar] [CrossRef]
- Sang, C.; Song, Y.; Jin, T.W.; Zhang, S.; Fu, L.; Zhao, Y.; Zou, X.; Wang, Z.; Gao, H.; Liu, S. Bisphenol A Induces Ovarian Cancer Cell Proliferation and Metastasis through Estrogen Receptor-Alpha Pathways. Environ. Sci. Pollut. Res. Int. 2021, 28, 36060–36068. [Google Scholar] [CrossRef] [PubMed]
- Zahra, A.; Dong, Q.; Hall, M.; Jeyaneethi, J.; Silva, E.; Karteris, E.; Sisu, C. Identification of Potential Bisphenol A (BPA) Exposure Biomarkers in Ovarian Cancer. J. Clin. Med. 2021, 10, 1979. [Google Scholar] [CrossRef] [PubMed]
- Prins, G.S.; Hu, W.Y.; Xie, L.; Shi, G.B.; Hu, D.P.; Birch, L.; Bosland, M.C. Evaluation of Bisphenol A (BPA) Exposures on Prostate Stem Cell Homeostasis and Prostate Cancer Risk in the NCTR-Sprague-Dawley Rat: An NIEHS/FDA CLARITY-BPA Consortium Study. Environ. Health Perspect. 2018, 126, 117001. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, Z.; Ji, W. Bisphenol A Induces Apoptosis, Oxidative Stress and Inflammatory Response in Colon and Liver of Mice in a Mitochondria-Dependent Manner. Biomed. Pharmacother. 2019, 117, 109182. [Google Scholar] [CrossRef]
- Wang, K.; Qiu, L.; Zhu, J.; Sun, Q.; Qu, W.; Yu, Y.; Zhao, Z.; Yu, Y.; Shao, G. Environmental Contaminant BPA Causes Intestinal Damage by Disrupting Cellular Repair and Injury Homeostasis in Vivo and in Vitro. Biomed. Pharmacother. 2021, 137, 111270. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Huang, X.; Lin, X.; Chan, T.F.; Lai, K.P.; Li, R. Analyzing the Synergistic Adverse Effects of BPA and its Substitute, BHPF, on Ulcerative Colitis through Comparative Metabolomics. Chemosphere 2022, 287, 132160. [Google Scholar] [CrossRef] [PubMed]
- Malhab, L.J.B.; Saber-Ayad, M.M.; Al-Hakm, R.; Nair, V.A.; Paliogiannis, P.; Pintus, G.; Abdel-Rahman, W.M. Chronic Inflammation and Cancer: The Role of Endothelial Dysfunction and Vascular Inflammation. Curr. Pharm. Des. 2021, 27, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Bou Malhab, L.J.; Abdel-Rahman, W.M. Obesity and Inflammation: Colorectal Cancer Engines. Curr. Mol. Pharmacol. 2022, 15, 620–646. [Google Scholar] [CrossRef]
- Hong, X.; Wang, G.; Liu, X.; Wu, M.; Zhang, X.; Hua, X.; Jiang, P.; Wang, S.; Tang, S.; Shi, X.; et al. Lipidomic Biomarkers: Potential Mediators of Associations between Urinary Bisphenol A Exposure and Colorectal Cancer. J. Hazard. Mater. 2022, 427, 127863. [Google Scholar] [CrossRef]
- Xia, T.; Guo, J.; Zhang, B.; Song, C.; Zhao, Q.; Cui, B.; Liu, Y. Bisphenol A Promotes the Progression of Colon Cancer through Dual-Targeting of NADPH Oxidase and Mitochondrial Electron-Transport Chain to Produce ROS and Activating HIF-1alpha/VEGF/PI3K/AKT Axis. Front. Endocrinol. 2022, 13, 933051. [Google Scholar] [CrossRef]
- Deng, Y.; He, H.; Wan, H.; Shen, N.; Li, J.; Zhang, S.; Zeng, Q.; Chang, J.; Lu, Q.; Zhong, R.; et al. Bisphenol A Exposure, Interaction with Genetic Variants and Colorectal Cancer Via Mediating Oxidative Stress Biomarkers. Environ. Pollut. 2021, 287, 117630. [Google Scholar] [CrossRef]
- Qu, W.; Zhao, Z.; Chen, S.; Zhang, L.; Wu, D.; Chen, Z. Bisphenol A Suppresses Proliferation and Induces Apoptosis in Colonic Epithelial Cells through Mitochondrial and MAPK/AKT Pathways. Life Sci. 2018, 208, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Schroyer, A.L.; Stimes, N.W.; Abi Saab, W.F.; Chadee, D.N. MLK3 Phosphorylation by ERK1/2 is Required for Oxidative Stress-Induced Invasion of Colorectal Cancer Cells. Oncogene 2018, 37, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.H.; Oh, J.E.; Shim, J.K.; Kwak, Y.L.; Cho, J.S. Effects of Bisphenol A on the Proliferation, Migration, and Tumor Growth of Colon Cancer Cells: In Vitro and in Vivo Evaluation with Mechanistic Insights Related to ERK and 5-HT3. Food Chem. Toxicol. 2021, 158, 112662. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhao, C.; Li, Z.; Zhang, Q.; Jin, X. Bisphenol a Exposure Promotes the Migration of NCM460 Cells Via Estrogen Receptor-Mediated Integrin Beta1/MMP-9 Pathway. Environ. Toxicol. 2016, 31, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Yang, X.L.; Liu, H.; Wei, W.; Zhang, K.S.; Huang, H.B.; Giesy, J.P.; Liu, H.L.; Du, J.; Wang, H.S. Bisphenol A Modulates Colorectal Cancer Protein Profile and Promotes the Metastasis Via Induction of Epithelial to Mesenchymal Transitions. Arch. Toxicol. 2015, 89, 1371–1381. [Google Scholar] [CrossRef]
- Snijders, A.M.; Lee, S.Y.; Hang, B.; Hao, W.; Bissell, M.J.; Mao, J.H. FAM83 Family Oncogenes are Broadly Involved in Human Cancers: An Integrative Multi-Omics Approach. Mol. Oncol. 2017, 11, 167–179. [Google Scholar] [CrossRef]
- Hussein, U.K.; Ha, S.H.; Ahmed, A.G.; Kim, K.M.; Park, S.H.; Kim, C.Y.; Kwon, K.S.; Zhang, Z.; Lee, S.A.; Park, H.S.; et al. FAM83H and SCRIB Stabilize Beta-Catenin and Stimulate Progression of Gastric Carcinoma. Aging 2020, 12, 11812–11834. [Google Scholar] [CrossRef] [PubMed]
- Kuga, T.; Kume, H.; Adachi, J.; Kawasaki, N.; Shimizu, M.; Hoshino, I.; Matsubara, H.; Saito, Y.; Nakayama, Y.; Tomonaga, T. Casein Kinase 1 is Recruited to Nuclear Speckles by FAM83H and SON. Sci. Rep. 2016, 6, 34472. [Google Scholar] [CrossRef]
- Jeong, T.Y.; Lee, H.I.; Park, M.S.; Seo, M.Y.; Jang, K.Y. Individual and Co-Expression Patterns of FAM83H and SCRIB at Diagnosis are Associated with the Survival of Colorectal Carcinoma Patients. Diagnostics 2022, 12, 1579. [Google Scholar] [CrossRef]
- Yu, X.; Wang, D.; Wang, X.; Sun, S.; Zhang, Y.; Wang, S.; Miao, R.; Xu, X.; Qu, X. CXCL12/CXCR4 Promotes Inflammation-Driven Colorectal Cancer Progression through Activation of RhoA Signaling by Sponging miR-133a-3p. J. Exp. Clin. Cancer Res. 2019, 38, 32. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Huang, W.; Chen, J.; Qiao, C.; Liu, D.; Ji, X.; Xie, M.; Zhang, T.; Wang, Y.; Sun, M.; et al. CXCL12-Mediated HOXB5 Overexpression Facilitates Colorectal Cancer Metastasis through Transactivating CXCR4 and ITGB3. Theranostics 2021, 11, 2612–2633. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Liu, G.; Hao, S.; Zhao, T.; Chang, W.; Wang, J.; Ren, H.; Zhao, X. PITPNA-AS1/miR-98-5p to Mediate the Cisplatin Resistance of Gastric Cancer. J. Oncol. 2022, 2022, 7981711. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, S.; Zeng, L.; Ma, J.; Sun, S.; Zeng, F.; Kong, F.; Cheng, X. HMOX-1 Inhibits TGF-Beta-Induced Epithelial-Mesenchymal Transition in the MCF-7 Breast Cancer Cell Line. Int. J. Mol. Med. 2017, 40, 411–417. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, H.; Li, X.; Liu, Q.; Liu, Y.; Hou, Y.; Jin, W. DGKZ Promotes TGFbeta Signaling Pathway and Metastasis in Triple-Negative Breast Cancer by Suppressing Lipid Raft-Dependent Endocytosis of TGFbetaR2. Cell. Death Dis. 2022, 13, 105. [Google Scholar] [CrossRef]
- Cai, K.; Mulatz, K.; Ard, R.; Nguyen, T.; Gee, S.H. Increased Diacylglycerol Kinase Zeta Expression in Human Metastatic Colon Cancer Cells Augments Rho GTPase Activity and Contributes to Enhanced Invasion. BMC Cancer 2014, 14, 208. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhou, Y.; Guo, H.; Ren, D.; Jin, X.; Wu, H. NR5A2 Transcriptional Activation by BRD4 Promotes Pancreatic Cancer Progression by Upregulating GDF15. Cell. Death Discov. 2021, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, S.; Ju, M.; Yan, P.; Sun, W.; Li, Z.; Wu, S.; Lin, R.; Xian, S.; Yang, D.; et al. Identification of Stemness-Related Genes for Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma by Integrated Bioinformatics Analysis. Front. Cell. Dev. Biol. 2021, 9, 642724. [Google Scholar] [CrossRef]
- Lin, W.; Sun, Y.; Qiu, X.; Huang, Q.; Kong, L.; Lu, J.J. VMP1, a Novel Prognostic Biomarker, Contributes to Glioma Development by Regulating Autophagy. J. Neuroinflamm. 2021, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Z.; Ye, X.L.; Xiao, W.Z.; Wei, X.N.; You, Q.H.; Che, X.H.; Cai, Y.J.; Chen, F.; Yuan, H.; Liu, X.J.; et al. Downregulation of VMP1 Confers Aggressive Properties to Colorectal Cancer. Oncol. Rep. 2015, 34, 2557–2566. [Google Scholar] [CrossRef][Green Version]
- Zhao, Z.; Bo, Z.; Gong, W.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int. J. Med. Sci. 2020, 17, 995–1005. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, M.; Lee, Y.M.; Kim, G.H.; Lee, D.; You, J.S.; Kim, S.H.; Choi, M.; Jang, H.; Park, Y.M.; et al. PGC1alpha Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers 2021, 13, 1772. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Nebozhyn, M.V.; Watters, J.W.; Buser, C.A.; Shaw, P.M.; Huang, P.S.; Van’t Veer, L.; Tollenaar, R.A.; Jackson, D.B.; Agrawal, D.; et al. EMT is the Dominant Program in Human Colon Cancer. BMC Med. Genom. 2011, 4, 9. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt Pathway in Oncology Therapy and Beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef]
- Fleming-de-Moraes, C.D.; Rocha, M.R.; Tessmann, J.W.; de Araujo, W.M.; Morgado-Diaz, J.A. Crosstalk between PI3K/Akt and Wnt/Beta-Catenin Pathways Promote Colorectal Cancer Progression Regardless of Mutational Status. Cancer Biol. Ther. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on Epithelial Mesenchymal Transition and Cancer Stem Cells. Oncotarget 2017, 8, 14221–14250. [Google Scholar] [CrossRef]
- Huang, K.; Gao, N.; Bian, D.; Zhai, Q.; Yang, P.; Li, M.; Wang, X. Correlation between FAK and EGF-Induced EMT in Colorectal Cancer Cells. J. Oncol. 2020, 2020, 5428920. [Google Scholar] [CrossRef]
- Hou, C.Y.; Ma, C.Y.; Yuh, C.H. WNK1 Kinase Signaling in Metastasis and Angiogenesis. Cell. Signal. 2022, 96, 110371. [Google Scholar] [CrossRef]
- Qi, X.M.; Wang, F.; Mortensen, M.; Wertz, R.; Chen, G. Targeting an Oncogenic Kinase/Phosphatase Signaling Network for Cancer Therapy. Acta Pharm. Sin. B 2018, 8, 511–517. [Google Scholar] [CrossRef]
- Turdo, A.; D’Accardo, C.; Glaviano, A.; Porcelli, G.; Colarossi, C.; Colarossi, L.; Mare, M.; Faldetta, N.; Modica, C.; Pistone, G.; et al. Targeting Phosphatases and Kinases: How to Checkmate Cancer. Front. Cell. Dev. Biol. 2021, 9, 690306. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.A.; Al-Khayyal, N.A.; Sivaperumal, S.; Abdel-Rahman, W.M. Calponin 3 Promotes Invasion and Drug Resistance of Colon Cancer Cells. World J. Gastrointest. Oncol. 2019, 11, 971–982. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FAM83H | Forward: GAAGTTCCTGCTGGTGGACTGT |
| Reverse: AGACCAGCTCTCCTTGGAACAC | |
| CXCL12 | Forward: CTCAACACTCCAAACTGTGCCC |
| Reverse: CTCCAGGTACTCCTGAATCCAC | |
| ACVRL1 | Forward: GCGACTTCAAGAGCCGCAATGT |
| Reverse: TAATCGCTGCCCTGTGAGTGCA | |
| PITPNA | Forward: GCAGATCGAAGCCAAGTGCTCA |
| Reverse: GGCAGTCCTTCTGGTTTACAAGC | |
| HMOX1 | Forward: CCAGGCAGAGAATGCTGAGTTC |
| Reverse: AAGACTGGGCTCTCCTTGTTGC | |
| DGKZ | Forward: GCTTTCTCTGACTTCCTGATGGG |
| Reverse: GGTTTCAGGTCCTGGATCTTGG | |
| NR5A2 | Forward: GGCTTATGTGCAAAATGGCAGATC |
| Reverse: GCTCACTCCAGCAGTTCTGAAG | |
| VMP1 | Forward: TTGGAACAGGGCTGCACACCTT |
| Reverse: TCAGGATAGGGTGGTTCGGGAA | |
| ID1 | Forward: GTTGGAGCTGAACTCGGAATCC |
| Reverse: ACACAAGATGCGATCGTCCGCA | |
| GAPDH | Forward: GTCTCCTCTGACTTCAACAGCG |
| Reverse: ACCACCCTGTTGCTGTAGCCAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, V.A.; Malhab, L.J.B.; Abdel-Rahman, W.M. Characterization of the Molecular Alterations Induced by the Prolonged Exposure of Normal Colon Mucosa and Colon Cancer Cells to Low-Dose Bisphenol A. Int. J. Mol. Sci. 2022, 23, 11620. https://doi.org/10.3390/ijms231911620
Nair VA, Malhab LJB, Abdel-Rahman WM. Characterization of the Molecular Alterations Induced by the Prolonged Exposure of Normal Colon Mucosa and Colon Cancer Cells to Low-Dose Bisphenol A. International Journal of Molecular Sciences. 2022; 23(19):11620. https://doi.org/10.3390/ijms231911620
Chicago/Turabian StyleNair, Vidhya A, Lara J Bou Malhab, and Wael M. Abdel-Rahman. 2022. "Characterization of the Molecular Alterations Induced by the Prolonged Exposure of Normal Colon Mucosa and Colon Cancer Cells to Low-Dose Bisphenol A" International Journal of Molecular Sciences 23, no. 19: 11620. https://doi.org/10.3390/ijms231911620
APA StyleNair, V. A., Malhab, L. J. B., & Abdel-Rahman, W. M. (2022). Characterization of the Molecular Alterations Induced by the Prolonged Exposure of Normal Colon Mucosa and Colon Cancer Cells to Low-Dose Bisphenol A. International Journal of Molecular Sciences, 23(19), 11620. https://doi.org/10.3390/ijms231911620