
Hemorrhagic Cerebral Insults and Secondary Takotsubo Syndrome: Findings in a Novel In Vitro Model Using Human Blood Samples



Abstract
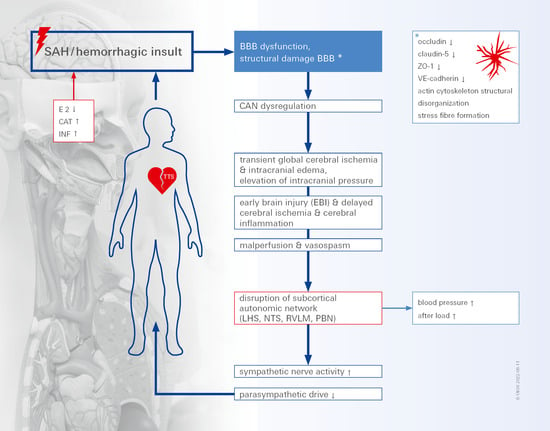
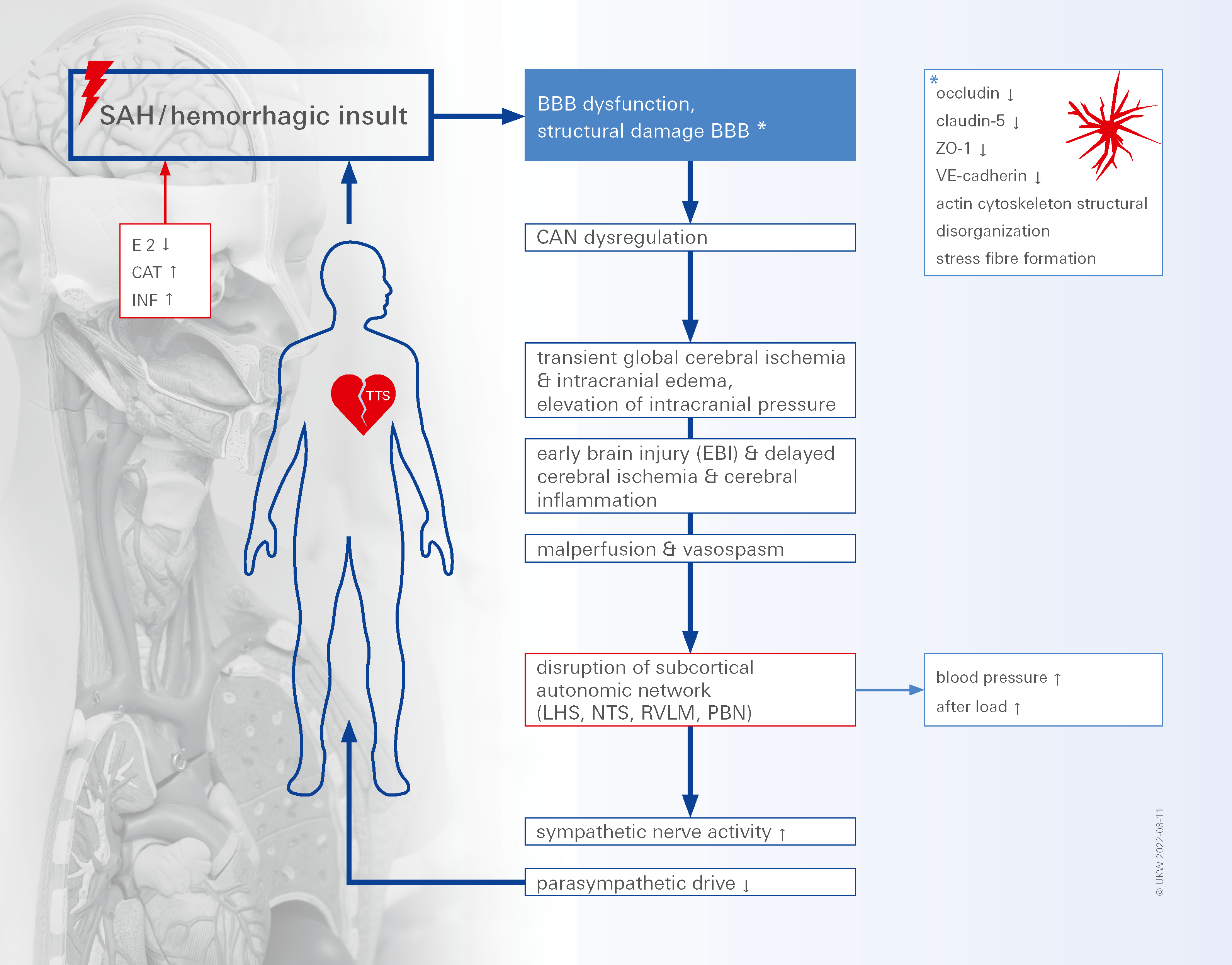
1. Introduction
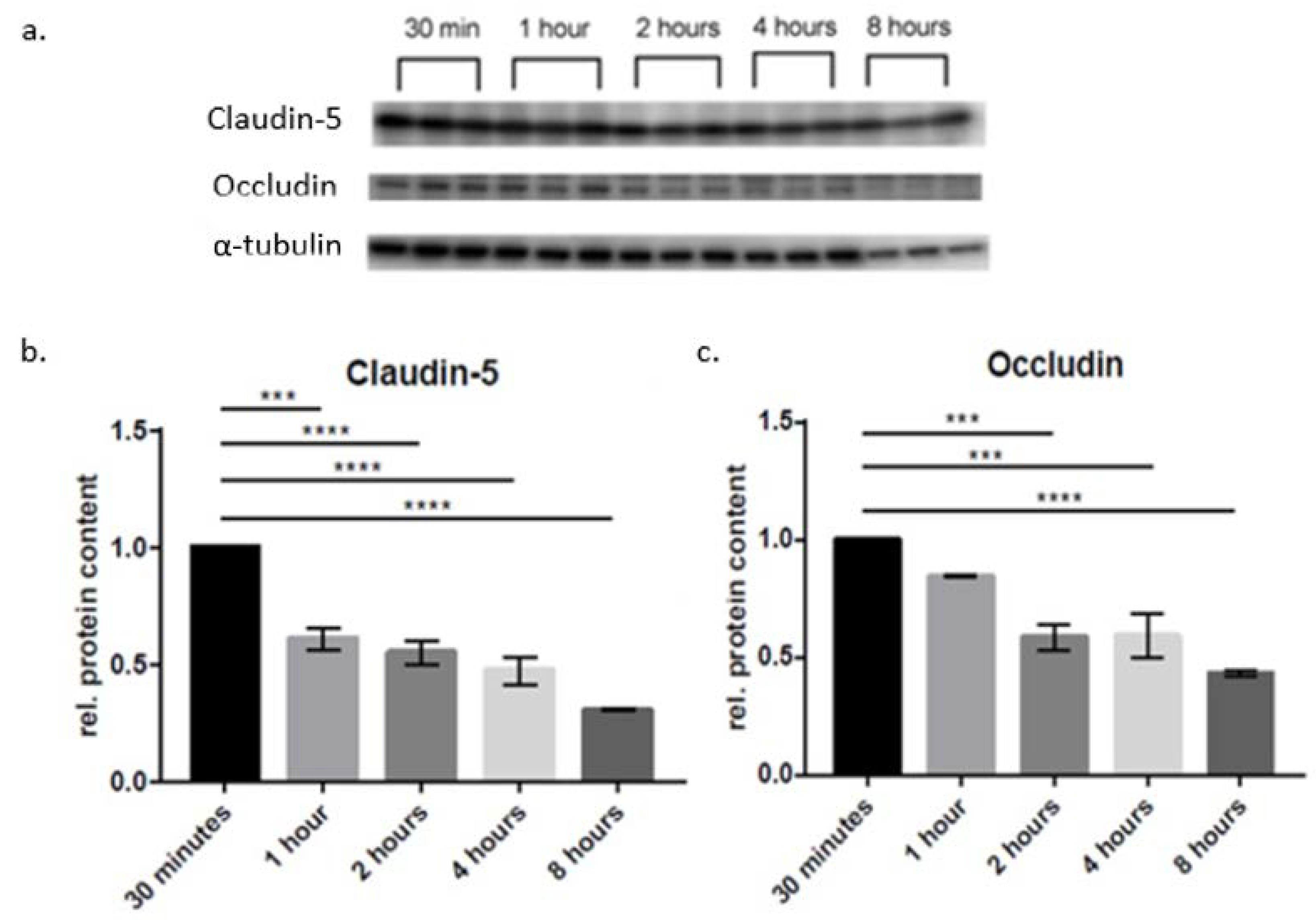
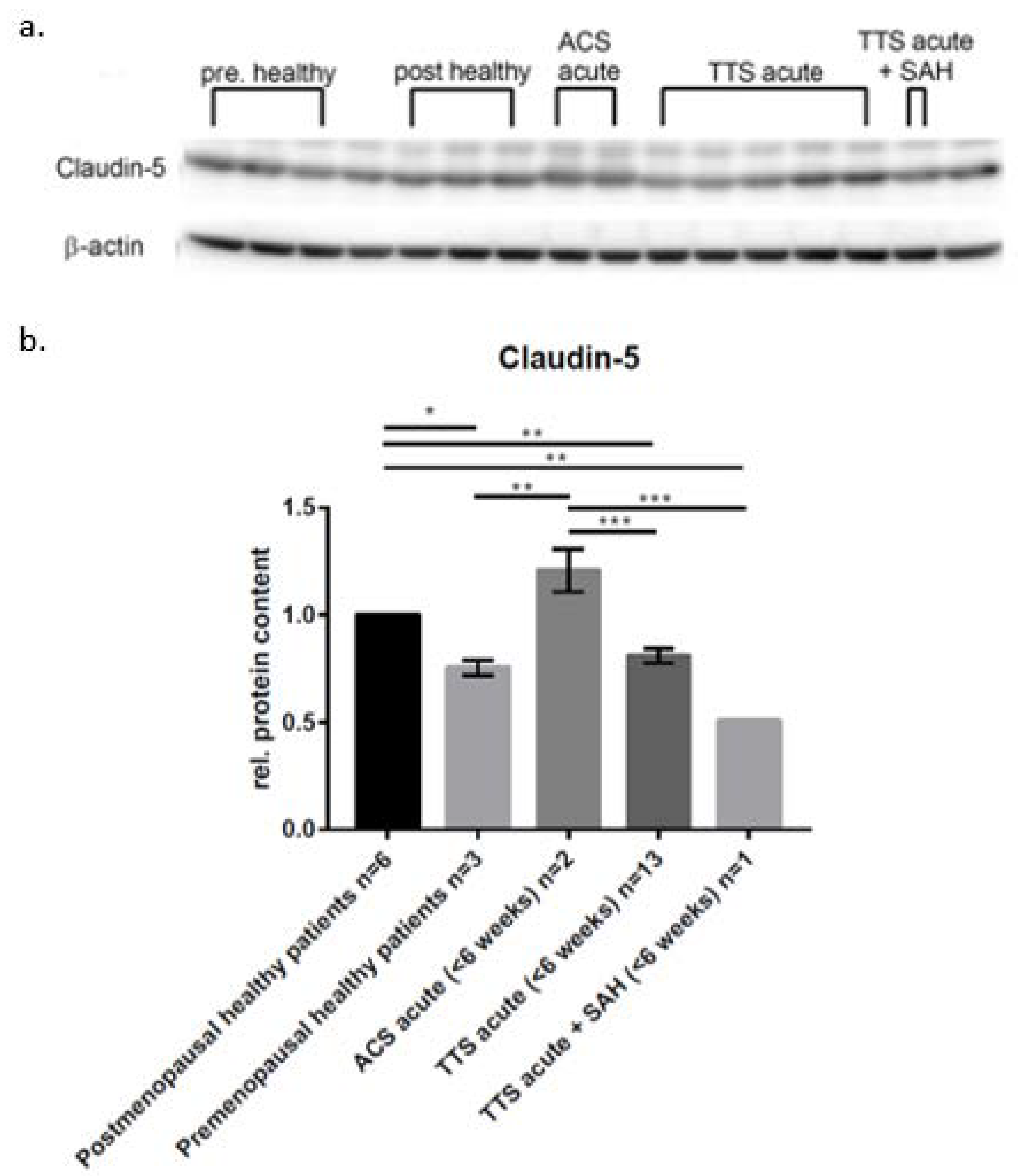
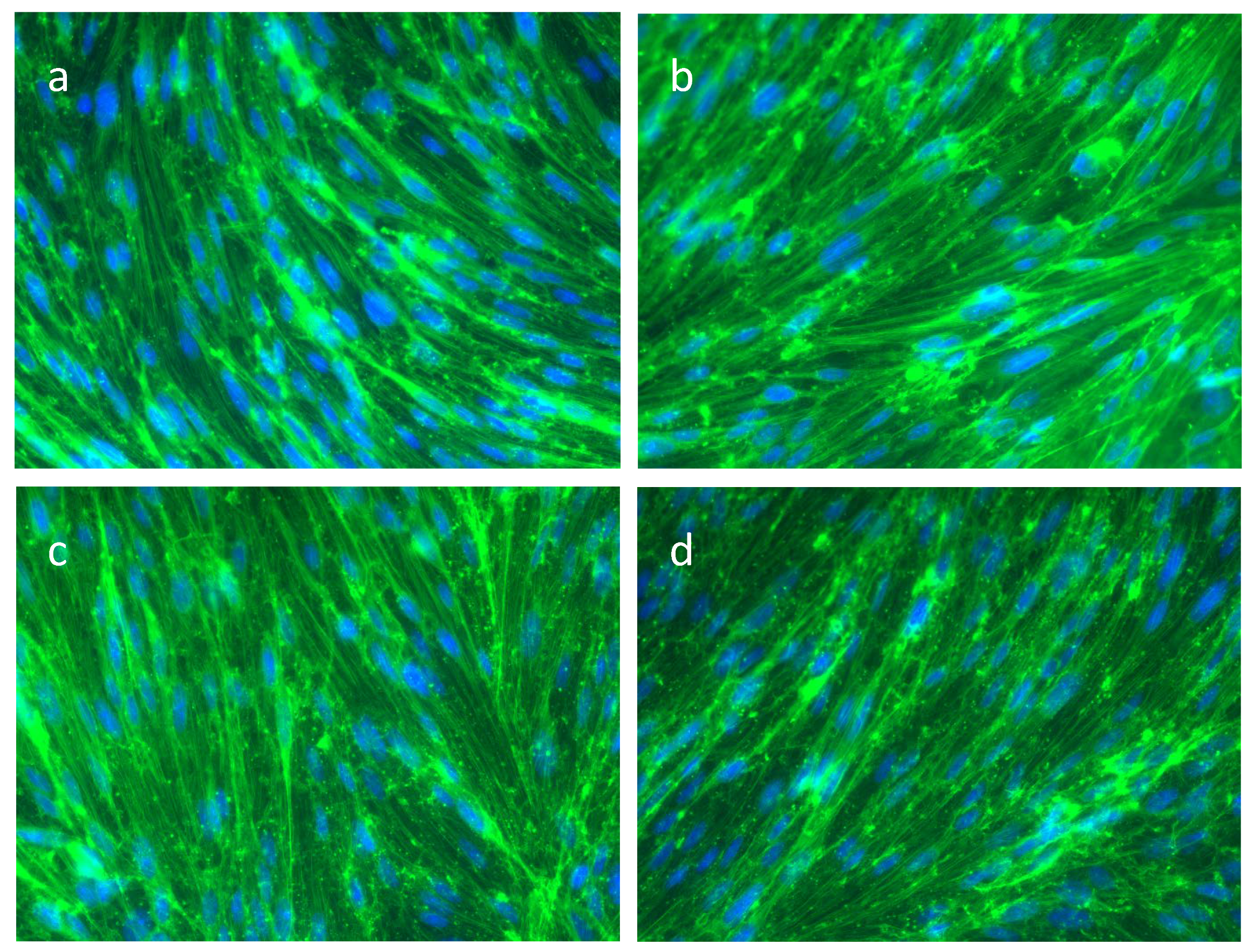
2. Results
3. Discussion
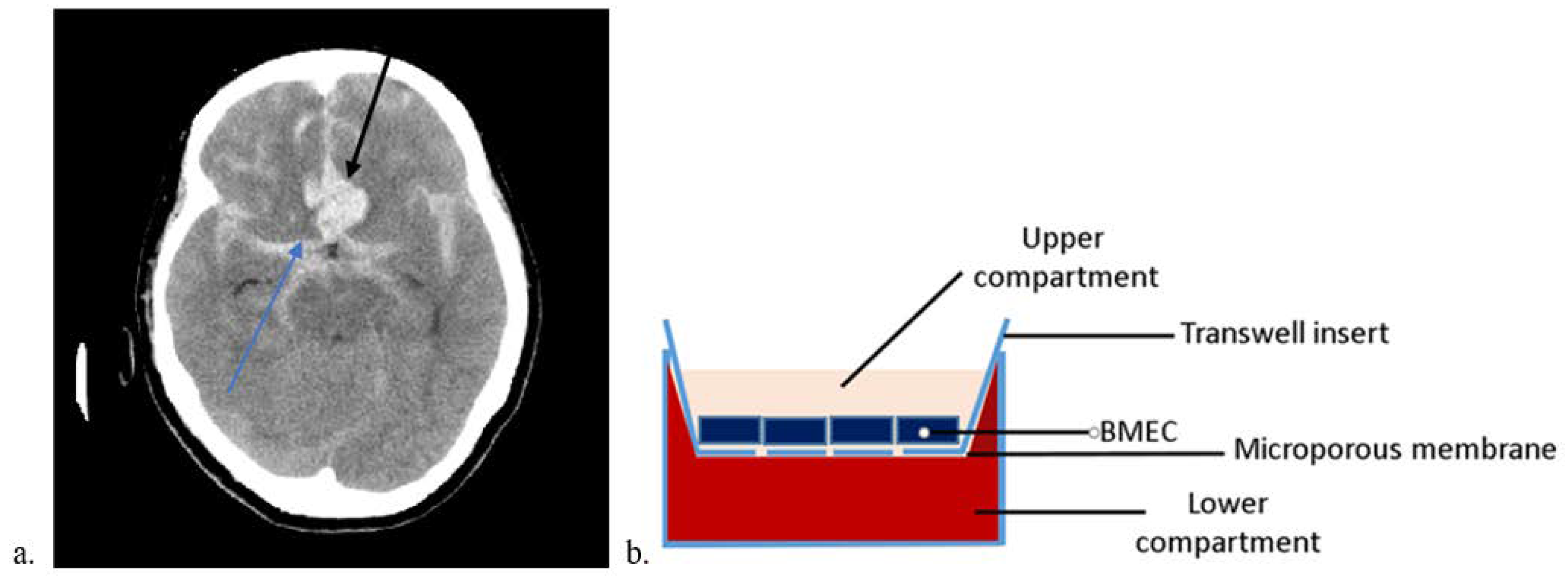
3.1. SAH In Vitro Modeling
3.2. The Actin Cytoskeleton in SAH
3.3. Cardiac Dysfunction following SAH
3.4. Limitations of Our Study
4. Materials and Methods
4.1. Cell Culture
4.2. Human Patient Serum
4.3. Ethics Approval and Consent to Participate
4.4. Western Blot
4.5. Real-Time qPCR
4.6. Actin-Phalloidin Staining
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Macdonald, R.L.; Schweizer, T.A. Spontaneous subarachnoid haemorrhage. Lancet 2017, 389, 655–666. [Google Scholar] [CrossRef]
- Lawton, M.T.; Vates, G.E. Subarachnoid Hemorrhage. N. Engl. J. Med. 2017, 377, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Juvela, S.; Unterberg, A.; Jung, C.; Forsting, M.; Rinkel, G.; European Stroke, O. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cereb. Dis. 2013, 35, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Diringer, M.N.; Bleck, T.P.; Claude Hemphill, J., 3rd; Menon, D.; Shutter, L.; Vespa, P.; Bruder, N.; Connolly, E.S., Jr.; Citerio, G.; Gress, D.; et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: Recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit. Care 2011, 15, 211–240. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.S., Jr.; Rabinstein, A.A.; Carhuapoma, J.R.; Derdeyn, C.P.; Dion, J.; Higashida, R.T.; Hoh, B.L.; Kirkness, C.J.; Naidech, A.M.; Ogilvy, C.S.; et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke 2012, 43, 1711–1737. [Google Scholar] [CrossRef]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Kamp, M.A.; Heiroth, H.J.; Beseoglu, K.; Turowski, B.; Steiger, H.J.; Hanggi, D. Early CT perfusion measurement after aneurysmal subarachnoid hemorrhage: A screening method to predict outcome? Acta Neurochir. Suppl. 2012, 114, 329–332. [Google Scholar] [PubMed]
- Schubert, G.A.; Seiz, M.; Hegewald, A.A.; Manville, J.; Thome, C. Acute hypoperfusion immediately after subarachnoid hemorrhage: A xenon contrast-enhanced CT study. J. Neurotrauma 2009, 26, 2225–2231. [Google Scholar] [CrossRef]
- Keep, R.F.; Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Antonetti, D.A.; Hua, Y.; Xi, G. Brain endothelial cell junctions after cerebral hemorrhage: Changes, mechanisms and therapeutic targets. J. Cereb. Blood Flow Metab. 2018, 38, 1255–1275. [Google Scholar] [CrossRef]
- Elgendy, A.Y.; Elgendy, I.Y.; Mansoor, H.; Mahmoud, A.N. Clinical presentations and outcomes of Takotsubo syndrome in the setting of subarachnoid hemorrhage: A systematic review and meta-analysis. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 236–245. [Google Scholar] [CrossRef]
- Nagai, M.; Forster, C.Y.; Dote, K. Sex Hormone-Specific Neuroanatomy of Takotsubo Syndrome: Is the Insular Cortex a Moderator? Biomolecules 2022, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Hunt, W.E.; Hess, R.M. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J. Neurosurg. 1968, 28, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Almeida, A.G.; Santos, M.O.; Ferro, J.M. Neurological complications of cardiomyopathies. Handb. Clin. Neurol. 2021, 177, 91–109. [Google Scholar] [PubMed]
- Lee, V.H.; Oh, J.K.; Mulvagh, S.L.; Wijdicks, E.F. Mechanisms in neurogenic stress cardiomyopathy after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2006, 5, 243–249. [Google Scholar] [CrossRef]
- Chen, Z.; Venkat, P.; Seyfried, D.; Chopp, M.; Yan, T.; Chen, J. Brain-Heart Interaction: Cardiac Complications After Stroke. Circ. Res. 2017, 121, 451–468. [Google Scholar] [CrossRef]
- Levine, B.D.; Giller, C.A.; Lane, L.D.; Buckey, J.C.; Blomqvist, C.G. Cerebral versus systemic hemodynamics during graded orthostatic stress in humans. Circulation 1994, 90, 298–306. [Google Scholar] [CrossRef]
- van Lieshout, J.J.; Pott, F.; Madsen, P.L.; van Goudoever, J.; Secher, N.H. Muscle tensing during standing: Effects on cerebral tissue oxygenation and cerebral artery blood velocity. Stroke 2001, 32, 1546–1551. [Google Scholar] [CrossRef]
- Gruhn, N.; Larsen, F.S.; Boesgaard, S.; Knudsen, G.M.; Mortensen, S.A.; Thomsen, G.; Aldershvile, J. Cerebral blood flow in patients with chronic heart failure before and after heart transplantation. Stroke 2001, 32, 2530–2533. [Google Scholar] [CrossRef]
- Massaro, A.R.; Dutra, A.P.; Almeida, D.R.; Diniz, R.V.; Malheiros, S.M. Transcranial Doppler assessment of cerebral blood flow: Effect of cardiac transplantation. Neurology 2006, 66, 124–126. [Google Scholar] [CrossRef]
- van den Hurk, K.; Reijmer, Y.D.; van den Berg, E.; Alssema, M.; Nijpels, G.; Kostense, P.J.; Stehouwer, C.D.; Paulus, W.J.; Kamp, O.; Dekker, J.M.; et al. Heart failure and cognitive function in the general population: The Hoorn Study. Eur. J. Heart Fail. 2011, 13, 1362–1369. [Google Scholar] [CrossRef]
- Eggermont, L.H.; de Boer, K.; Muller, M.; Jaschke, A.C.; Kamp, O.; Scherder, E.J. Cardiac disease and cognitive impairment: A systematic review. Heart (Br. Card. Soc.) 2012, 98, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Neulen, A.; Molitor, M.; Kosterhon, M.; Pantel, T.; Holzbach, E.; Rudi, W.S.; Karbach, S.H.; Wenzel, P.; Ringel, F.; Thal, S.C. Correlation of cardiac function and cerebral perfusion in a murine model of subarachnoid hemorrhage. Sci. Rep. 2021, 11, 3317. [Google Scholar] [CrossRef]
- Papanikolaou, J.; Makris, D.; Karakitsos, D.; Saranteas, T.; Karabinis, A.; Kostopanagiotou, G.; Zakynthinos, E. Cardiac and central vascular functional alterations in the acute phase of aneurysmal subarachnoid hemorrhage. Crit. Care Med. 2012, 40, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.; Muse, J.; Taussky, P. Takotsubo Syndrome in Neurologic Disease. World Neurosurg. 2021, 149, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ghadri, J.R.; Wittstein, I.S.; Prasad, A.; Sharkey, S.; Dote, K.; Akashi, Y.J.; Cammann, V.L.; Crea, F.; Galiuto, L.; Desmet, W.; et al. International Expert Consensus Document on Takotsubo Syndrome (Part II): Diagnostic Workup, Outcome, and Management. Eur. Heart J. 2018, 39, 2047–2062. [Google Scholar] [CrossRef]
- Nagai, M.; Kobayashi, Y.; Kobatake, H.; Dote, K.; Kato, M.; Oda, N.; Kunita, E.; Kagawa, E.; Yamane, A.; Osawa, A.; et al. Happy heart syndrome: A case of Takotsubo syndrome with left internal carotid artery occlusion. Clin. Auton. Res. Off. J. Clin. Auton. Res. Soc. 2020, 30, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Osawa, A.; Nagai, M.; Dote, K.; Kato, M.; Oda, N.; Kunita, E.; Kagawa, E.; Yamane, A.; Kobatake, H.; Shiota, H.; et al. A mid-ventricular variant of Takotsubo syndrome: Was it triggered by insular cortex damage? ESC Heart Fail. 2021, 8, 3408–3412. [Google Scholar] [CrossRef]
- Silwedel, C.; Forster, C. Differential susceptibility of cerebral and cerebellar murine brain microvascular endothelial cells to loss of barrier properties in response to inflammatory stimuli. J. Neuroimmunol. 2006, 179, 37–45. [Google Scholar] [CrossRef]
- Abd, T.T.; Hayek, S.; Cheng, J.-W.; Samuels, O.; Lerakis, S. Takotsubo’s Cardiomyopathy is Associated with Severe Subarachnoid Hemorrhage: Retrospective Analysis of 1,251 patients. Circulation 2018, 126, A13973. [Google Scholar]
- Ittner, C.; Burek, M.; Stork, S.; Nagai, M.; Forster, C.Y. Increased Catecholamine Levels and Inflammatory Mediators Alter Barrier Properties of Brain Microvascular Endothelial Cells in vitro. Front. Cardiovasc. Med. 2020, 7, 73. [Google Scholar] [CrossRef]
- Frydland, M.; Moller, J.E.; Lindholm, M.G.; Hansen, R.; Wiberg, S.; Lerche Helgestad, O.K.; Thomsen, J.H.; Goetze, J.P.; Engstrom, T.; Frikke-Schmidt, R.; et al. Biomarkers predictive of late cardiogenic shock development in patients with suspected ST-elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Forster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, A.V.; Stamatovic, S.M.; Phillips, C.M.; Martinez-Revollar, G.; Keep, R.F. Modeling blood-brain barrier pathology in cerebrovascular disease in vitro: Current and future paradigms. Fluids Barriers CNS 2020, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Tso, M.K.; Macdonald, R.L. Subarachnoid hemorrhage: A review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 2014, 5, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, G.; Ma, T.; Li, J.; Wang, P.; Liu, L.; Yu, B.; Liu, Y.; Xue, Y. Blood-brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metab. Brain Dis. 2015, 30, 597–603. [Google Scholar] [CrossRef]
- Russin, J.J.; Montagne, A.; D’Amore, F.; He, S.; Shiroishi, M.S.; Rennert, R.C.; Depetris, J.; Zlokovic, B.V.; Mack, W.J. Permeability imaging as a predictor of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2018, 38, 973–979. [Google Scholar] [CrossRef]
- Kooijman, E.; Nijboer, C.H.; van Velthoven, C.T.; Kavelaars, A.; Kesecioglu, J.; Heijnen, C.J. The rodent endovascular puncture model of subarachnoid hemorrhage: Mechanisms of brain damage and therapeutic strategies. J. Neuroinflamm. 2014, 11, 2. [Google Scholar] [CrossRef]
- Claassen, J.; Carhuapoma, J.R.; Kreiter, K.T.; Du, E.Y.; Connolly, E.S.; Mayer, S.A. Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke 2002, 33, 1225–1232. [Google Scholar] [CrossRef]
- Freeman, N.L.; Field, J. Mammalian homolog of the yeast cyclase associated protein, CAP/Srv2p, regulates actin filament assembly. Cell Motil. Cytoskelet. 2000, 45, 106–120. [Google Scholar] [CrossRef]
- Forster, C.; Kahles, T.; Kietz, S.; Drenckhahn, D. Dexamethasone induces the expression of metalloproteinase inhibitor TIMP-1 in the murine cerebral vascular endothelial cell line cEND. J. Physiol. 2007, 580 Pt 3, 937–949. [Google Scholar] [CrossRef]
- Blecharz, K.G.; Drenckhahn, D.; Forster, C.Y. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J. Cereb. Blood Flow Metab. 2008, 28, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef] [PubMed]
- Lidington, D.; Wan, H.; Bolz, S.S. Cerebral Autoregulation in Subarachnoid Hemorrhage. Front. Neurol. 2021, 12, 688362. [Google Scholar] [CrossRef] [PubMed]
- Luh, C.; Feiler, S.; Frauenknecht, K.; Meyer, S.; Lubomirov, L.T.; Neulen, A.; Thal, S.C. The Contractile Apparatus Is Essential for the Integrity of the Blood-Brain Barrier After Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2019, 10, 534–545. [Google Scholar] [CrossRef]
- Satoh, S.; Ikegaki, I.; Kawasaki, K.; Asano, T.; Shibuya, M. Pleiotropic effects of the rho-kinase inhibitor fasudil after subarachnoid hemorrhage: A review of preclinical and clinical studies. Curr. Vasc. Pharmacol. 2014, 12, 758–765. [Google Scholar] [CrossRef]
- Gibson, C.L.; Srivastava, K.; Sprigg, N.; Bath, P.M.; Bayraktutan, U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J. Neurochem. 2014, 129, 816–826. [Google Scholar] [CrossRef]
- Nagai, M.; Dote, K.; Kato, M. Central autonomic network and Takotsubo cardiomyopathy: How left insular cortex interact? Eur. Heart J. 2019, 40, 3061. [Google Scholar] [CrossRef]
- Templin, C.; Hanggi, J.; Klein, C.; Topka, M.S.; Hiestand, T.; Levinson, R.A.; Jurisic, S.; Luscher, T.F.; Ghadri, J.R.; Jancke, L. Altered limbic and autonomic processing supports brain-heart axis in Takotsubo syndrome. Eur. Heart J. 2019, 40, 1183–1187. [Google Scholar] [CrossRef]
- Nagai, M.; Forster, C.Y.; Dote, K. Right insular cortex atrophy in Takotsubo syndrome: A possible pathogenesis of increased sympathetic nervous system activity? Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2021, 110, 601–602. [Google Scholar] [CrossRef]
- Frontera, J.A.; Parra, A.; Shimbo, D.; Fernandez, A.; Schmidt, J.M.; Peter, P.; Claassen, J.; Wartenberg, K.E.; Rincon, F.; Badjatia, N.; et al. Cardiac arrhythmias after subarachnoid hemorrhage: Risk factors and impact on outcome. Cerebrovasc. Dis. 2008, 26, 71–78. [Google Scholar] [CrossRef]
- Eric Nyam, T.T.; Ho, C.H.; Chio, C.C.; Lim, S.W.; Wang, J.J.; Chang, C.H.; Kuo, J.R.; Wang, C.C. Traumatic brain injury increases the risk of major adverse cardiovascular and cerebrovascular events: A 13-year, population-based study. World Neurosurg. 2019, 122, e740–e753. [Google Scholar] [CrossRef] [PubMed]
- Ay, H.; Koroshetz, W.J.; Benner, T.; Vangel, M.G.; Melinosky, C.; Arsava, E.M.; Ayata, C.; Zhu, M.; Schwamm, L.H.; Sorensen, A.G. Neuroanatomic correlates of stroke-related myocardial injury. Neurology 2006, 66, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosyula, S.; Verghese, D.; Desai, V.K.; Sundaragiri, P.R.; Miller, V.M. Sex differences in acute cardiovascular care: A review and needs assessment. Cardiovasc. Res. 2022, 118, 667–685. [Google Scholar] [CrossRef]
- Nagai, M.; Forster, C.Y.; Dote, K.; Shimokawa, H. Sex hormones in heart failure revisited? Eur. J. Heart Fail. 2019, 21, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53 (Suppl. S6), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Marins, F.R.; Limborco-Filho, M.; D’Abreu, B.F.; Machado de Almeida, P.W.; Gavioli, M.; Xavier, C.H.; Oppenheimer, S.M.; Guatimosim, S.; Fontes, M.A.P. Autonomic and cardiovascular consequences resulting from experimental hemorrhagic stroke in the left or right intermediate insular cortex in rats. Auton. Neurosci. Basic Clin. 2020, 227, 102695. [Google Scholar] [CrossRef]
- Yamasaki, T.; Hayashi, K.; Shibata, Y.; Furuta, T.; Yamamoto, K.; Uchimura, M.; Fujiwara, Y.; Nakagawa, F.; Kambara, M.; Yoshikane, T.; et al. Takotsubo cardiomyopathy following mechanical thrombectomy for acute ischemic stroke: Illustrative case. J. Neurosurg. Case Lessons 2021, 2, CASE21372. [Google Scholar] [CrossRef]
- Sander, D.; Klingelhofer, J. Changes of circadian blood pressure patterns and cardiovascular parameters indicate lateralization of sympathetic activation following hemispheric brain infarction. J. Neurol. 1995, 242, 313–318. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Occludin | ZO-1 | VE-Cadherin | |
|---|---|---|---|
| Postmenopausal healthy patients, n = 6 | 1 | 1 | 1 |
| Premenopausal healthy patients, n = 6 | 0.96 ± 0.156 | 0.82 ± 0.209 | 1.07 ± 0.281 |
| ACS acute (<6 weeks), n = 2 | 1.1 ± 0.083 | 1.12 ± 0.042 | 1.4 ± 0.181 |
| TTS acute (<6 weeks), n = 13 | 0.78 ± 0.057 | 0.77 ± 0.041 | 0.69 ± 0.082 |
| TTS acute + SAH (<6 weeks), n = 1 | 0.69 | 0.76 | 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thal, S.C.; Smetak, M.; Hayashi, K.; Förster, C.Y. Hemorrhagic Cerebral Insults and Secondary Takotsubo Syndrome: Findings in a Novel In Vitro Model Using Human Blood Samples. Int. J. Mol. Sci. 2022, 23, 11557. https://doi.org/10.3390/ijms231911557
Thal SC, Smetak M, Hayashi K, Förster CY. Hemorrhagic Cerebral Insults and Secondary Takotsubo Syndrome: Findings in a Novel In Vitro Model Using Human Blood Samples. International Journal of Molecular Sciences. 2022; 23(19):11557. https://doi.org/10.3390/ijms231911557
Chicago/Turabian StyleThal, Serge C., Manuel Smetak, Kentaro Hayashi, and Carola Y. Förster. 2022. "Hemorrhagic Cerebral Insults and Secondary Takotsubo Syndrome: Findings in a Novel In Vitro Model Using Human Blood Samples" International Journal of Molecular Sciences 23, no. 19: 11557. https://doi.org/10.3390/ijms231911557
APA StyleThal, S. C., Smetak, M., Hayashi, K., & Förster, C. Y. (2022). Hemorrhagic Cerebral Insults and Secondary Takotsubo Syndrome: Findings in a Novel In Vitro Model Using Human Blood Samples. International Journal of Molecular Sciences, 23(19), 11557. https://doi.org/10.3390/ijms231911557