
Vascular and Liver Homeostasis in Juvenile Mice Require Endothelial Cyclic AMP-Dependent Protein Kinase A

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
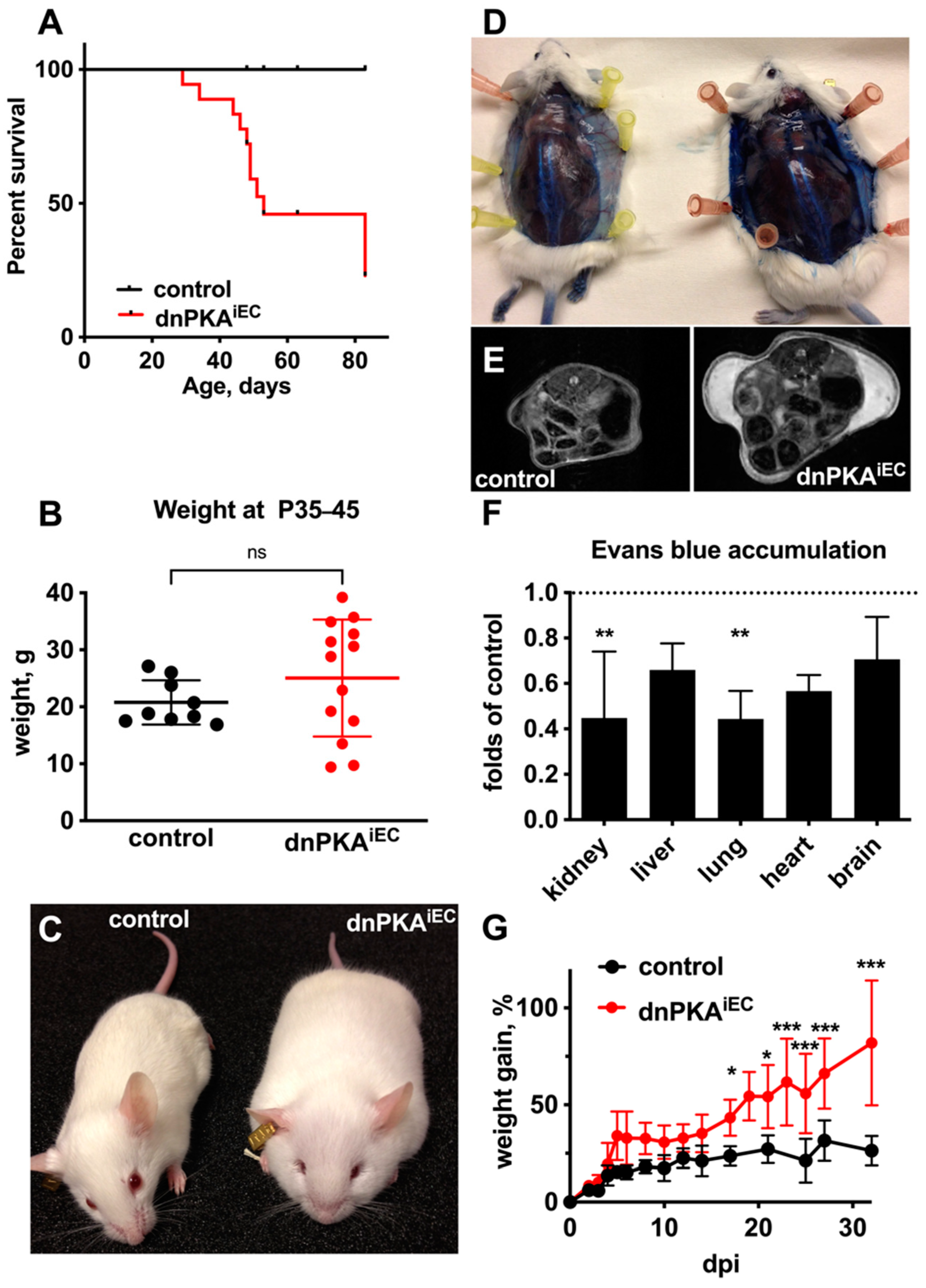
2.1. Inhibition of Endothelial PKA in Mice Results in Edema and Premature Death
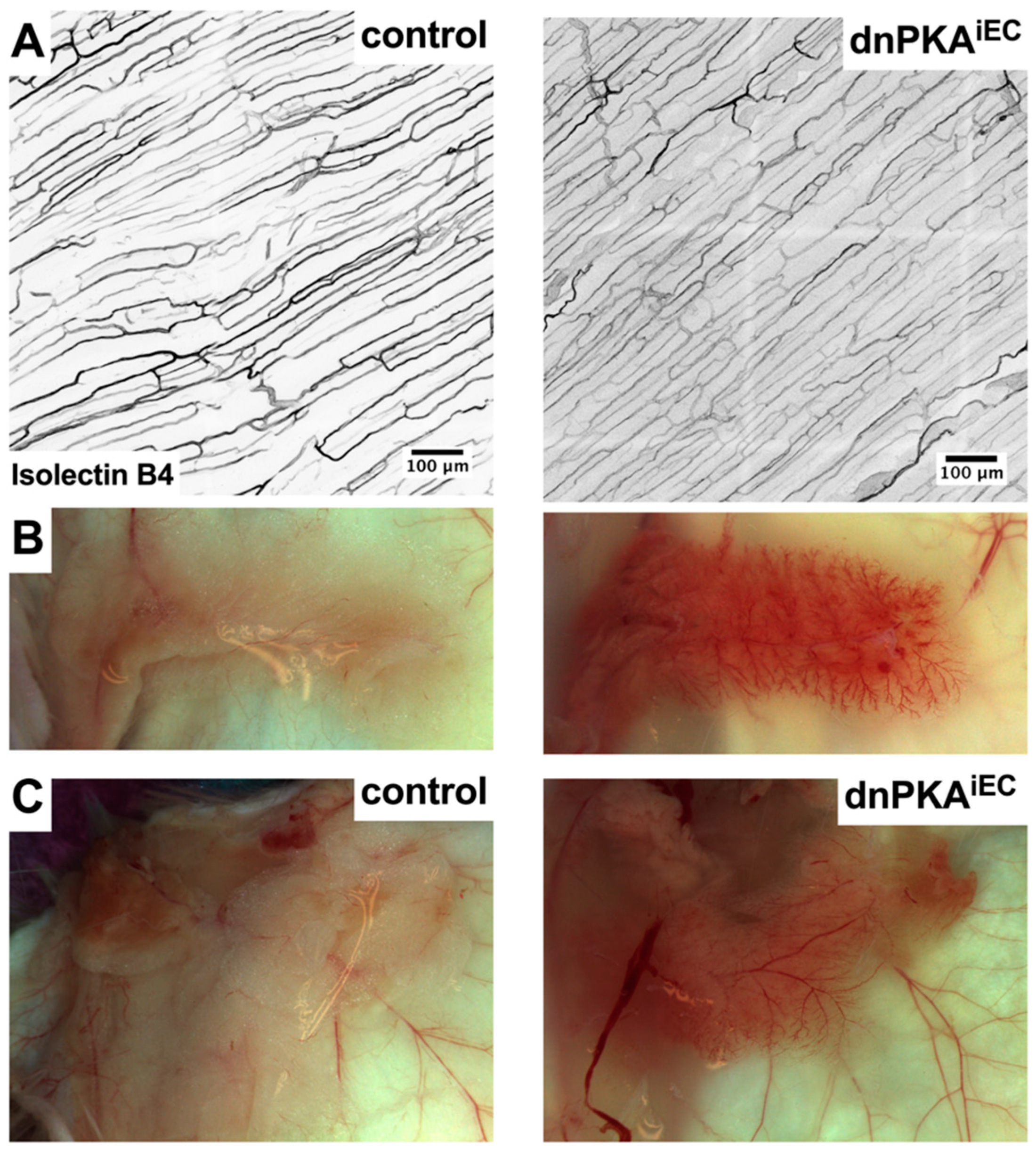
2.2. Differential Response to PKA Inhibition in Different Vascular Beds
2.3. Inhibition of Endothelial PKA in Lymphatics Is Not Sufficient to Induce Edema
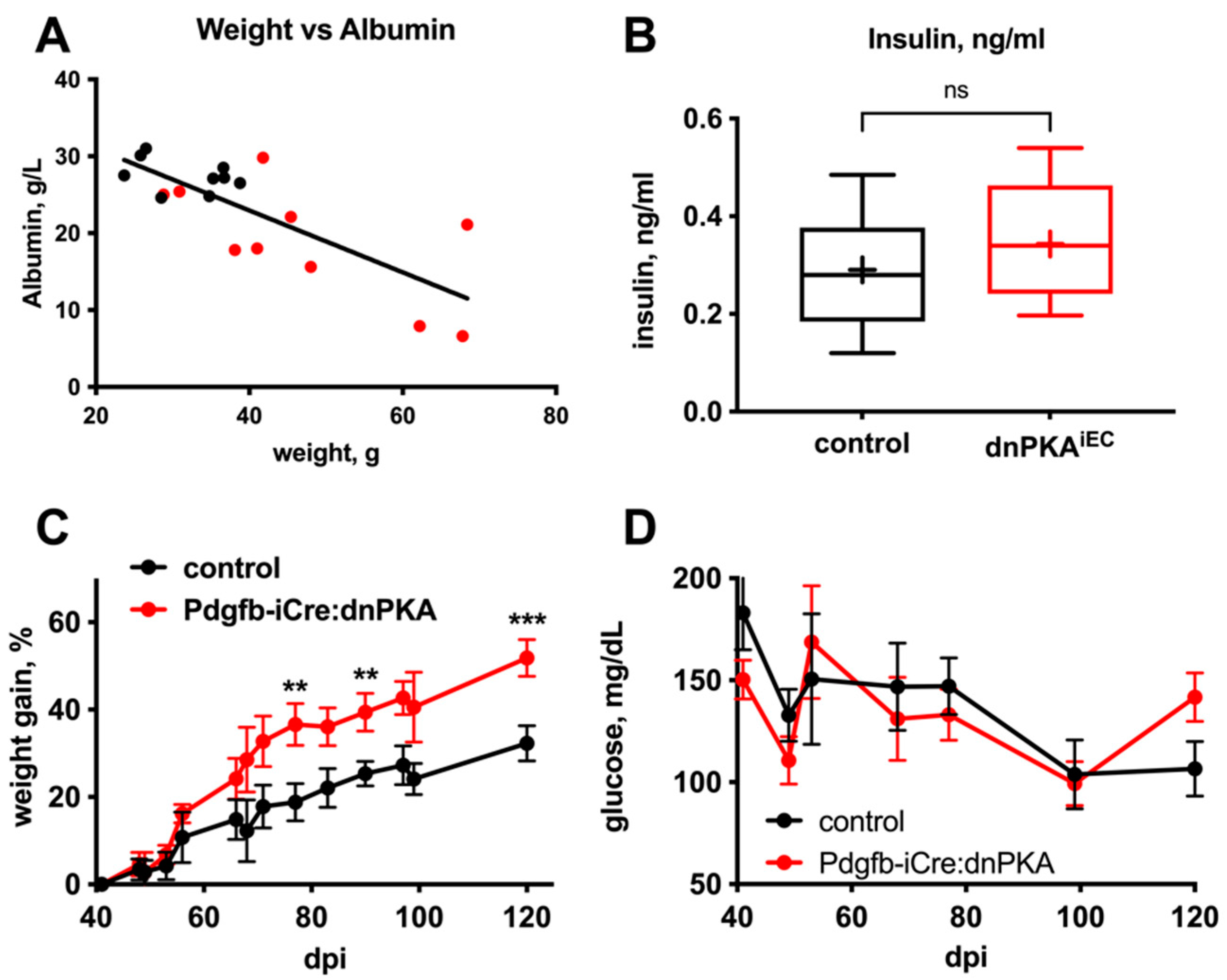
2.4. Changes in Blood Parameters Indicate Hepatic Defects in dnPKAiEC Mice
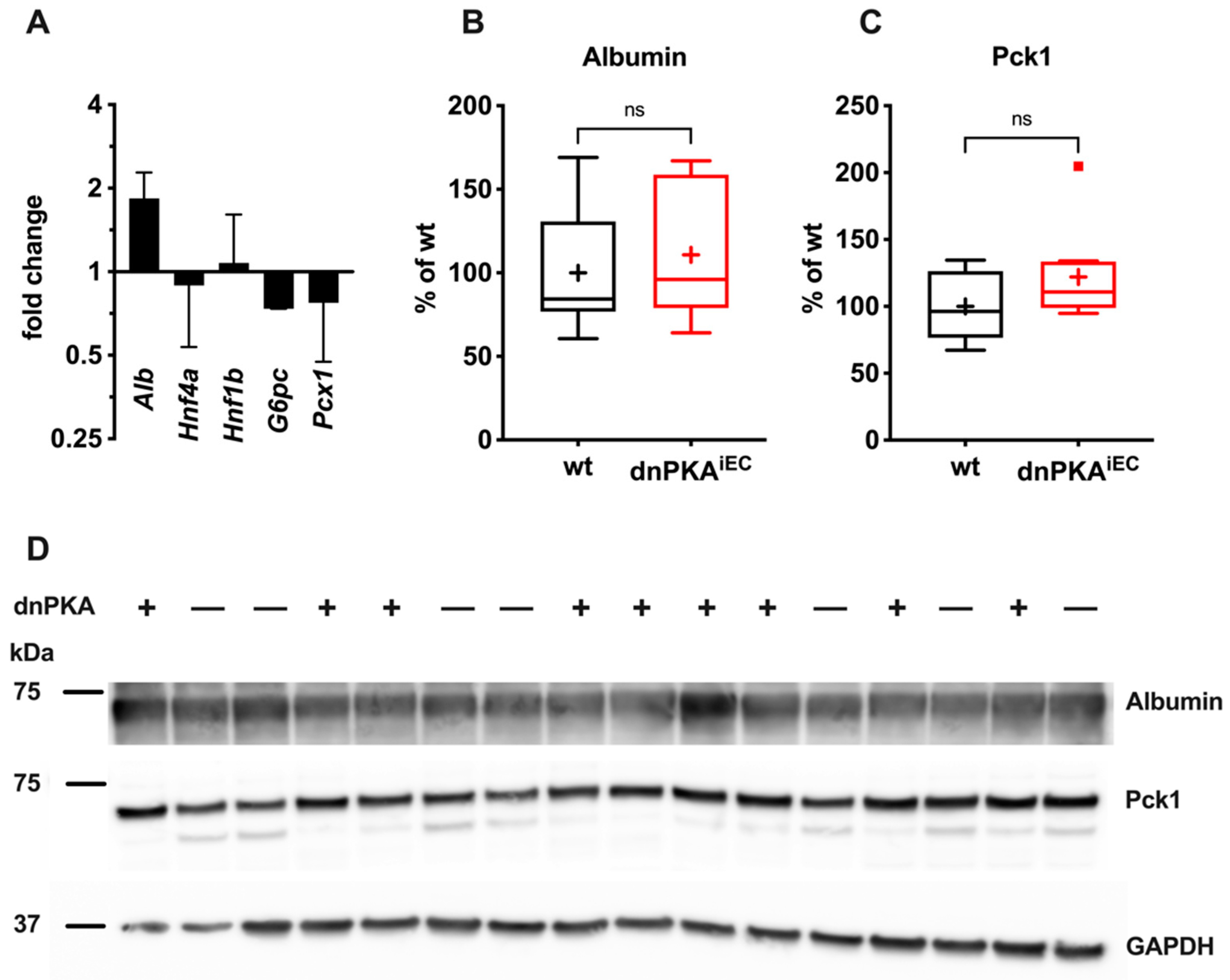
2.5. Gluconeogenesis but Not Albumin Expression Is Perturbed in dnPKAiEC Mice
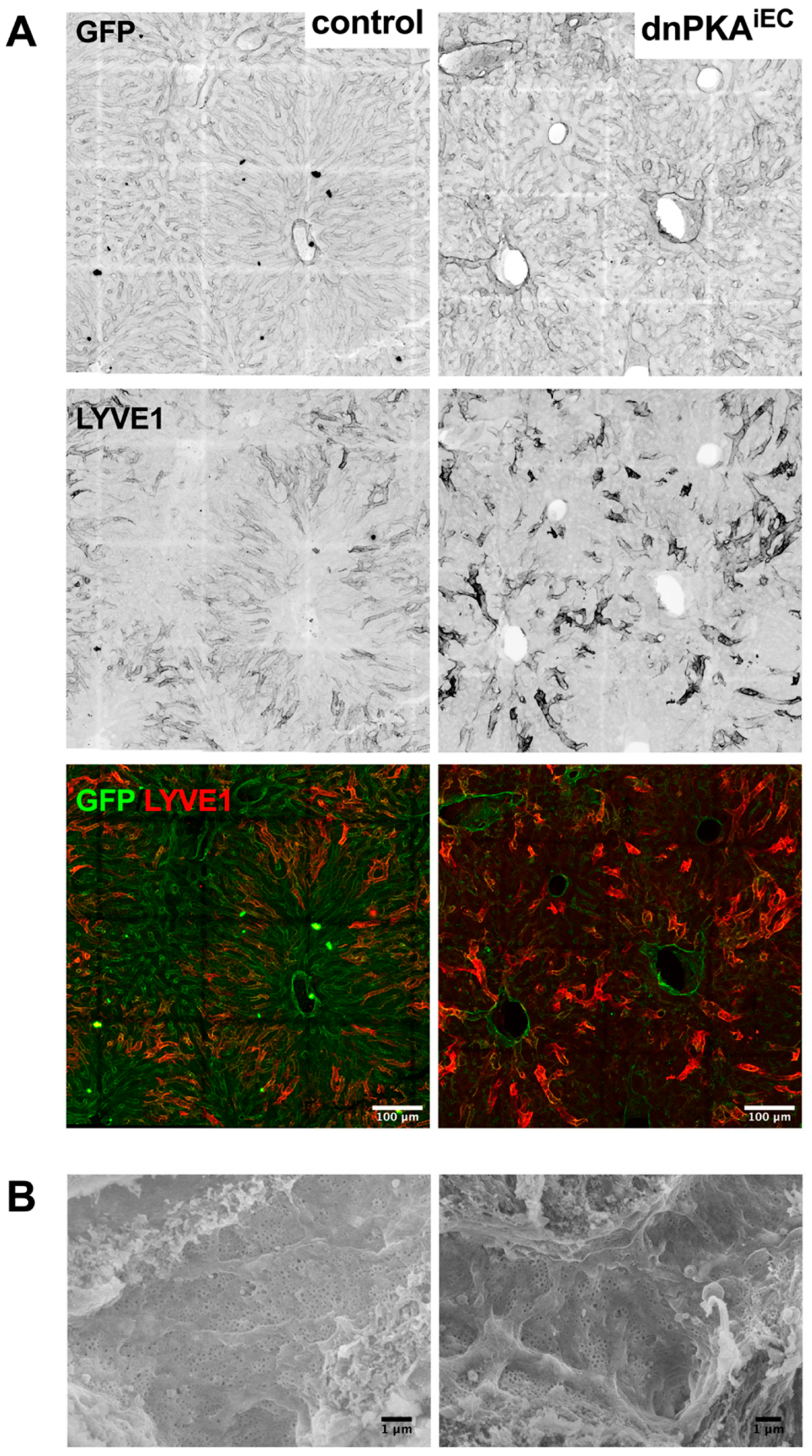
2.6. Inhibition of Endothelial PKA Leads to Disorganization of Hepatic Vasculature
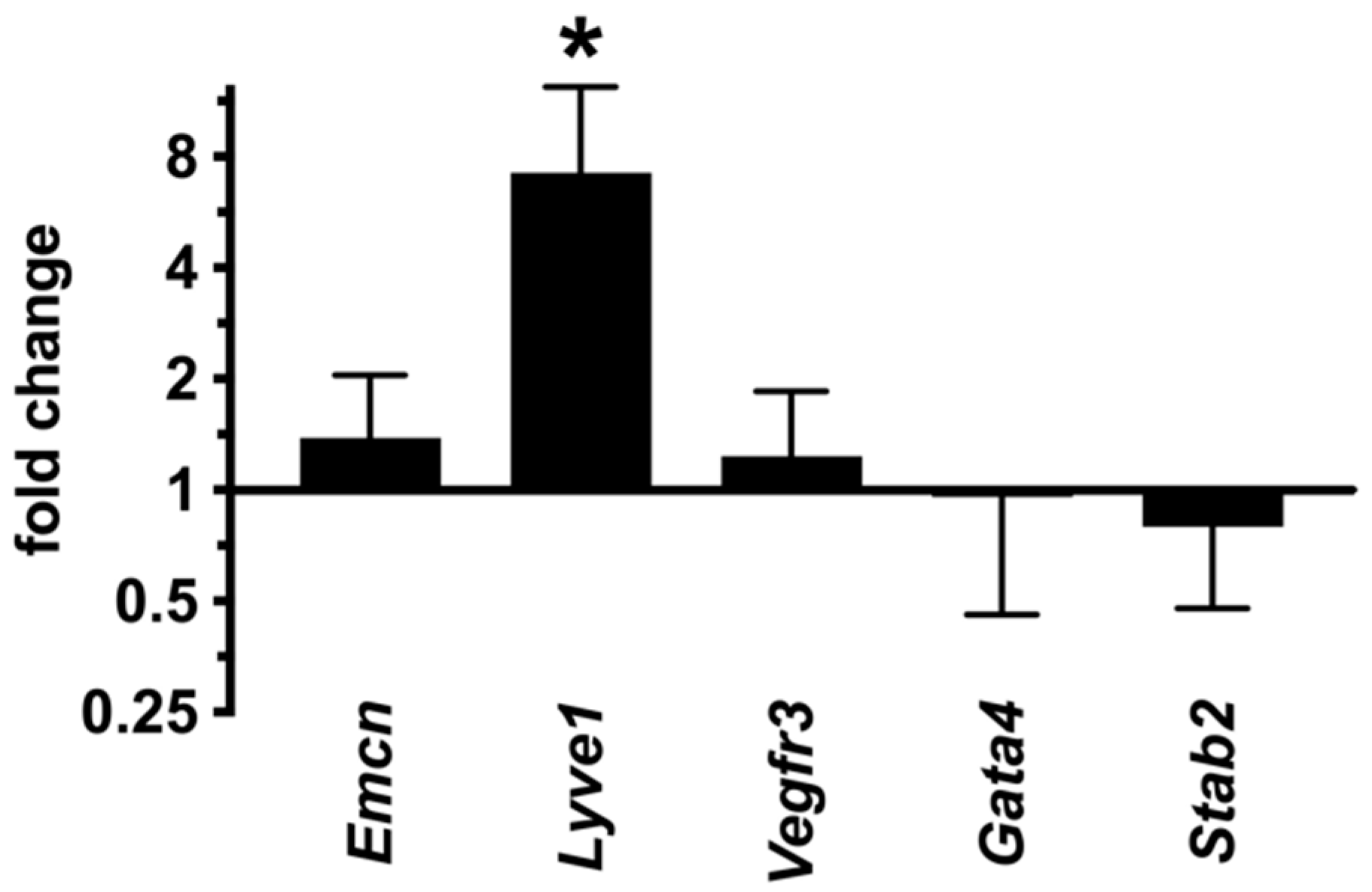
2.7. LSECs Preserve Their Differentiation State in dnPKAiEC Mice
2.8. Inhibition of Endothelial PKA Results in Cell-Autonomous Reactivation of the Angiogenic Program
3. Discussion
4. Materials and Methods
4.1. Animal Experimentation
4.2. Quantitative PCR
4.3. Immunostaining
4.4. Western Blotting
4.5. Scanning Electron Microscopy
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nedvetsky, P.I.; Zhao, X.; Mathivet, T.; Aspalter, I.M.; Stanchi, F.; Metzger, R.J.; Mostov, K.E.; Gerhardt, H. cAMP-dependent protein kinase A (PKA) regulates angiogenesis by modulating tip cell behavior in a Notch-independent manner. Development 2016, 143, 3582–3590. [Google Scholar] [CrossRef] [PubMed]
- Goldfinger, L.E.; Tzima, E.; Stockton, R.; Kiosses, W.B.; Kinbara, K.; Tkachenko, E.; Gutierrez, E.; Groisman, A.; Nguyen, P.; Chien, S.; et al. Localized alpha4 integrin phosphorylation directs shear stress-induced endothelial cell alignment. Circ. Res. 2008, 103, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.-S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Osborne, L.D.; Guilluy, C.; Chen, Z.; O’Brien, E.T.; Reader, J.S.; Burridge, K.; Superfine, R.; Tzima, E. Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat. Commun. 2014, 5, 3984. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.K.; Kusumbe, A.P.; Adams, R.H. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. 2014, 25, 148–157. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Xu, C.; Hasan, S.S.; Schmidt, I.; Rocha, S.F.; Pitulescu, M.E.; Bussmann, J.; Meyen, D.; Raz, E.; Adams, R.H.; Siekmann, A.F. Arteries are formed by vein-derived endothelial tip cells. Nat. Commun. 2014, 5, 5758. [Google Scholar] [CrossRef]
- Lu, Y.; Xiong, Y.; Huo, Y.; Han, J.; Yang, X.; Zhang, R.; Zhu, D.-S.; Klein-Hessling, S.; Li, J.; Zhang, X.; et al. Grb-2-associated binder 1 (Gab1) regulates postnatal ischemic and VEGF-induced angiogenesis through the protein kinase A-endothelial NOS pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 2957–2962. [Google Scholar] [CrossRef]
- Shabb, J.B. Physiological Substrates of cAMP-Dependent Protein Kinase. Chem. Rev. 2001, 101, 2381–2412. [Google Scholar] [CrossRef]
- Bakre, M.M.; Zhu, Y.; Yin, H.; Burton, D.W.; Terkeltaub, R.; Deftos, L.J.; Varner, J.A. Parathyroid hormone-related peptide is a naturally occurring, protein kinase A-dependent angiogenesis inhibitor. Nat. Med. 2002, 8, 995–1003. [Google Scholar] [CrossRef]
- Aslam, M.; Härtel, F.V.; Arshad, M.; Gündüz, D.; Abdallah, Y.; Sauer, H.; Piper, H.M.; Noll, T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: Role of CPI-17. Cardiovasc. Res. 2010, 87, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Garmy-Susini, B.; Avraamides, C.J.; Stoletov, K.; Klemke, R.L.; Varner, J.A. A PKA-Csk-pp60Src signaling pathway regulates the switch between endothelial cell invasion and cell-cell adhesion during vascular sprouting. Blood 2010, 116, 5773–5783. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Pfeil, U.; Gündüz, D.; Rafiq, A.; Kummer, W.; Piper, H.M.; Noll, T. Intermedin (adrenomedullin2) stabilizes the endothelial barrier and antagonizes thrombin-induced barrier failure in endothelial cell monolayers. Br. J. Pharmacol. 2012, 165, 208–222. [Google Scholar] [CrossRef]
- Willis, B.S.; Niswender, C.M.; Su, T.; Amieux, P.S.; McKnight, G.S. Cell-type specific expression of a dominant negative PKA mutation in mice. PLoS ONE 2011, 6, e18772. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakayama, M.; Pitulescu, M.E.; Schmidt, T.S.; Bochenek, M.L.; Sakakibara, A.; Adams, S.; Davy, A.; Deutsch, U.; Lüthi, U.; et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010, 465, 483–486. [Google Scholar] [CrossRef]
- Breier, G.; Breviario, F.; Caveda, L.; Berthier, R.; Schnürch, H.; Gotsch, U.; Vestweber, D.; Risau, W.; Dejana, E. Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood 1996, 87, 630–641. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Dillard, M.E.; Lagutin, O.V.; Lin, F.-J.; Tsai, S.; Tsai, M.-J.; Samokhvalov, I.M.; Oliver, G. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007, 21, 2422–2432. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Corral, I.; Stanczuk, L.; Frye, M.; Ulvmar, M.H.; Diéguez-Hurtado, R.; Olmeda, D.; Mäkinen, T.; Ortega, S. Vegfr3-CreER (T2) mouse, a new genetic tool for targeting the lymphatic system. Angiogenesis 2016, 19, 433–445. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Claxton, S.; Kostourou, V.; Jadeja, S.; Chambon, P.; Hodivala-Dilke, K.; Fruttiger, M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis 2008, 46, 74–80. [Google Scholar] [CrossRef]
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.S.; Vidal, V.; Mertz, M.; Kendall, T.J.; Charlet, A.; Okamoto, H.; Schedl, A. The Angiocrine Factor Rspondin3 Is a Key Determinant of Liver Zonation. Cell Rep. 2015, 13, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Lan, B.Y.-A.; Bedolli, M.; Feng, S.; Hebrok, M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 2006, 43, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Benhamouche, S.; Decaens, T.; Godard, C.; Chambrey, R.; Rickman, D.S.; Moinard, C.; Vasseur-Cognet, M.; Kuo, C.J.; Kahn, A.; Perret, C.; et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell 2006, 10, 759–770. [Google Scholar] [CrossRef]
- Arimoto, J.; Ikura, Y.; Suekane, T.; Nakagawa, M.; Kitabayashi, C.; Iwasa, Y.; Sugioka, K.; Naruko, T.; Arakawa, T.; Ueda, M. Expression of LYVE-1 in sinusoidal endothelium is reduced in chronically inflamed human livers. J. Gastroenterol. 2010, 45, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Incio, J.; Soares, R. Angiogenesis and chronic inflammation: Cause or consequence? Angiogenesis 2007, 10, 149–166. [Google Scholar] [CrossRef]
- Capitao, M.; Soares, R. Angiogenesis and Inflammation Crosstalk in Diabetic Retinopathy. J. Cell. Biochem. 2016, 117, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Farrukh, I.S.; Gurtner, G.H.; Michael, J.R. Pharmacological modification of pulmonary vascular injury: Possible role of cAMP. J. Appl. Physiol. 1987, 62, 47–54. [Google Scholar] [CrossRef]
- Stelzner, T.J.; Weil, J.V.; O’Brien, R.F. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J. Cell. Physiol. 1989, 139, 157–166. [Google Scholar] [CrossRef]
- Kim, S.; Bakre, M.; Yin, H.; Varner, J.A. Inhibition of endothelial cell survival and angiogenesis by protein kinase A. J. Clin. Investig. 2002, 110, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.S.R.; Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 2006, 168, 639–648. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G757-63. [Google Scholar] [CrossRef]
- Maharaj, A.S.R.; D’Amore, P.A. Roles for VEGF in the adult. Microvasc. Res. 2007, 74, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Béchard, D.; Gentina, T.; Delehedde, M.; Scherpereel, A.; Lyon, M.; Aumercier, M.; Vazeux, R.; Richet, C.; Degand, P.; Jude, B.; et al. Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. J. Biol. Chem. 2001, 276, 48341–48349. [Google Scholar] [CrossRef]
- Chanda, D.; Li, T.; Song, K.H.; Kim, Y.H.; Sim, J.; Lee, C.H.; Chiang, J.Y.L.; Choi, H.S. Hepatocyte Growth Factor Family Negatively Regulates Hepatic Gluconeogenesis via Induction of Orphan Nuclear Receptor Small Heterodimer Partner in Primary Hepatocytes. J. Biol. Chem. 2009, 284, 28510–28521. [Google Scholar] [CrossRef] [PubMed]
- Fafalios, A.; Ma, J.; Tan, X.; Stoops, J.; Luo, J.; DeFrances, M.C.; Zarnegar, R. A hepatocyte growth factor receptor (Met)−insulin receptor hybrid governs hepatic glucose metabolism. Nat. Med. 2011, 17, 1577–1584. [Google Scholar] [CrossRef]
- Horton, R.A.; Ceppi, E.D.; Knowles, R.G.; Titheradge, M.A. Inhibition of hepatic gluconeogenesis by nitric oxide: A comparison with endotoxic shock. Biochem. J. 1994, 299, 735–739. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- D’Angelo, G.; Lee, H.; Weiner, R.I. cAMP-dependent protein kinase inhibits the mitogenic action of vascular endothelial growth factor and fibroblast growth factor in capillary endothelial cells by blocking Raf activation. J. Cell. Biochem. 1997, 67, 353–366. [Google Scholar] [CrossRef]
- Zhao, X.; Nedvetsky, P.; Stanchi, F.; Vion, A.-C.; Popp, O.; Zühlke, K.; Dittmar, G.; Klussmann, E.; Gerhardt, H. Endothelial PKA activity regulates angiogenesis by limiting autophagy through phosphorylation of ATG16L1. eLife 2019, 8, e46380. [Google Scholar] [CrossRef] [PubMed]
- Pitulescu, M.E.; Schmidt, I.; Benedito, R.; Adams, R.H. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 2010, 5, 1518–1534. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; O’Reilly, J.N.; Warren, A.; Le Couteur, D.G. A Standardized Method for the Analysis of Liver Sinusoidal Endothelial Cells and Their Fenestrations by Scanning Electron Microscopy. J. Vis. Exp. 2015, 98, e52698. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| Alb | GCAGATGACAGGGCGGAACTTG | CAGCAGCAATGGCAGGCAGAT |
| Hnf4a | GCTGTCCTCGTAGCTTGACC | TTAAGAAGTGCTTCCGGGCT |
| Hnf1b | AGGGAGGTGGTCGATGTCA | TCTGGACTGTCTGGTTGAACT |
| G6pc | GTCTTGTCAGGCATTGCTGTG | CAGGTAGAATCCAAGCGCGA |
| Pcx1 | GGGCGGAGCTAACATCTACC | TATACTCCAGACGCCGGACA |
| Emcn | GGTTCCCAGAACAAGACTGAGA | TGGAATAGGAGGGGGTGGTT |
| Lyve1 | CACTAGGCACCCAGTCCAAG | TCCAACCCATCCATAGCTGC |
| Vegfr3 | CACAATCCCCATGCTCTGGT | TGTGAGGAGGCACATTCACC |
| Gata4 | CCATCTCGCCTCCAGAGT | CTGGAAGACACCCCAATCTC |
| Stab2 | CCAGGGATATCCAGGACGTA | TGTCCAGACGGCTACATCAA |
| Esm1 | GCAAGAGGACAGTGCTGGAT | GGTGCCATAGGGACAGTCTTT |
| Vegfa | AGGAGCCGAGCTCATGGA | GGGACCACTTGGCATGGTG |
| Vegfc | TGTGCTTCTTGTCTCTGGCG | CCTTCAAAAGCCTTGACCTCG |
| Fgf2 | TTCATAGCAAGGTACCGGTTG | AGAAGAGCGACCCACACG |
| Hgf | GCGAATTGGTGTTCTGCCTG | GAGCAGACTGATCCCTAAAGCA |
| Dll4 | GGCAAACTGCAGAACCACAC | TGGCTTCTCACTGTGTAACCG |
| Sele | TCTATTTCCCACGATGCATTT | CTGCCAAAGCCTTCAATCAT |
| Vcam1 | CTGGGAAGCTGGAACGAAGT | GCCAAACACTTGACCGTGAC |
| Icam1 | TTTGAGCTGAGCGAGATCGG | AGAGGTCTCAGCTCCACACT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nedvetsky, P.I.; Cornelissen, I.; Mathivet, T.; Bouleti, C.; Ou, P.; Baatsen, P.; Zhao, X.; Schuit, F.; Stanchi, F.; Mostov, K.E.; et al. Vascular and Liver Homeostasis in Juvenile Mice Require Endothelial Cyclic AMP-Dependent Protein Kinase A. Int. J. Mol. Sci. 2022, 23, 11419. https://doi.org/10.3390/ijms231911419
Nedvetsky PI, Cornelissen I, Mathivet T, Bouleti C, Ou P, Baatsen P, Zhao X, Schuit F, Stanchi F, Mostov KE, et al. Vascular and Liver Homeostasis in Juvenile Mice Require Endothelial Cyclic AMP-Dependent Protein Kinase A. International Journal of Molecular Sciences. 2022; 23(19):11419. https://doi.org/10.3390/ijms231911419
Chicago/Turabian StyleNedvetsky, Pavel I., Ivo Cornelissen, Thomas Mathivet, Claire Bouleti, Phalla Ou, Pieter Baatsen, Xiaocheng Zhao, Frans Schuit, Fabio Stanchi, Keith E. Mostov, and et al. 2022. "Vascular and Liver Homeostasis in Juvenile Mice Require Endothelial Cyclic AMP-Dependent Protein Kinase A" International Journal of Molecular Sciences 23, no. 19: 11419. https://doi.org/10.3390/ijms231911419
APA StyleNedvetsky, P. I., Cornelissen, I., Mathivet, T., Bouleti, C., Ou, P., Baatsen, P., Zhao, X., Schuit, F., Stanchi, F., Mostov, K. E., & Gerhardt, H. (2022). Vascular and Liver Homeostasis in Juvenile Mice Require Endothelial Cyclic AMP-Dependent Protein Kinase A. International Journal of Molecular Sciences, 23(19), 11419. https://doi.org/10.3390/ijms231911419