
Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing

 , ,
, , 
Abstract
1. Introduction
2. Oxidative Stress and Mitochondrial DNA Lesions
2.1. Point Mutations and Ribonucleotide Incorporation
2.2. Deletions
2.3. Single-Strand and Double-Strand DNA Breaks
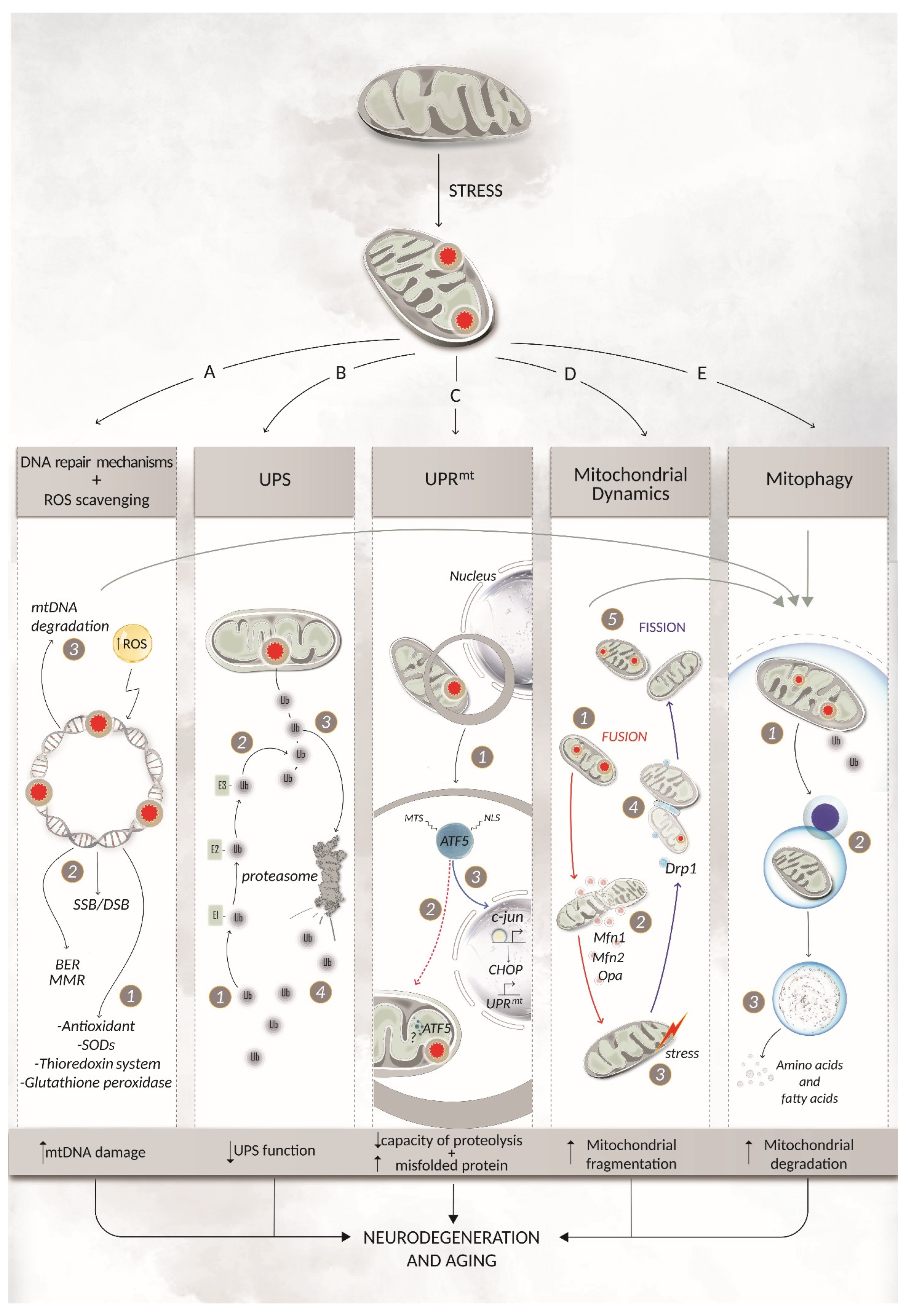
3. Mitochondrial DNA Repair Pathways and Coping Mechanisms
3.1. The Base Excision Repair Pathway
3.2. The Mismatch Repair Pathway and Double-Strand Break Repair Pathways
3.3. mtDNA Degradation
3.4. Mitochondrial Dynamics

{kind=link}
{kind=link}
{kind=link}
| Repair Pathway | Lesion | Enzyme | Enzyme Class | Function | Note | Ref. |
|---|---|---|---|---|---|---|
| BER | Oxidative damage | MUTHY AGG UNG | Hydrolase | Monofunctional glycosylase | [66] | |
| OGG1 NTH | Bifunctional glycosylase (β-elimination) | |||||
| NEIL1 NEIL2 | Bifunctional glycosylase (βδ-elimination) | |||||
| APE1 PNK | Hydrolase | Hydrolysis of phosphate backbone | [67,68] | |||
| Polγ | Transferase | Nucleotide incorporation | [69] | |||
| FEN-1 | Hydrolase | Cleavage of 5′ flap structures | LP-BER | [70] | ||
| ExoG | Removal of 5′-blocking moiety | LP-BER | [71] | |||
| DNA ligase III | Ligase | Nick ligation | ||||
| MMR | Base mismatches | YB-1 | DNA-binding protein | Mismatch sensing and protein recruitment | [39,72] | |
| HR/NHEJ | SSBs and DSBs | No evidence of HR/NHEJ activity in mammalian mitochondria | ||||
| mtDNA degradation | Any lesion | MGME1 | Hydrolase | 5’–3’ exonuclease activity | Involved in degradation of linear DNA after DSBs | [73] |
| Twinkle | Helicase | Unwinding of mtDNA replication fork | Potentially involved in mtDNA degradation | [74,75] | ||
| mtSSB | Single-strand DNA binding protein | Enhancing Twinkle and Polγ activity | [11,59] | |||
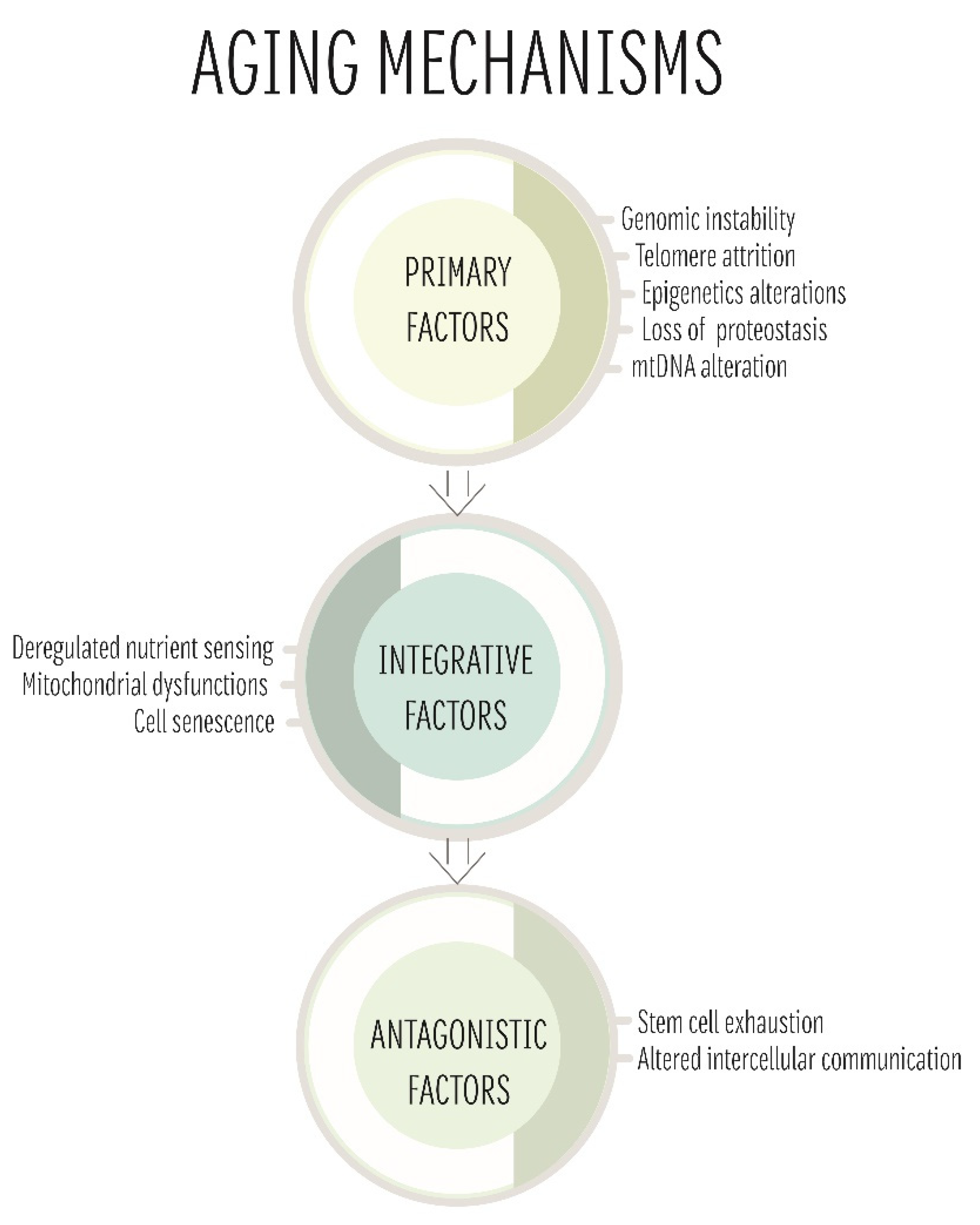
4. The Role of mtDNA in Ageing
4.1. Mitochondrial Free Radical Theory of Ageing
4.2. Clonal Expansion Theory
4.3. The Gradual ROS Response Theory
4.4. Ageing and Mitochondria: One Theory to Rule Them All
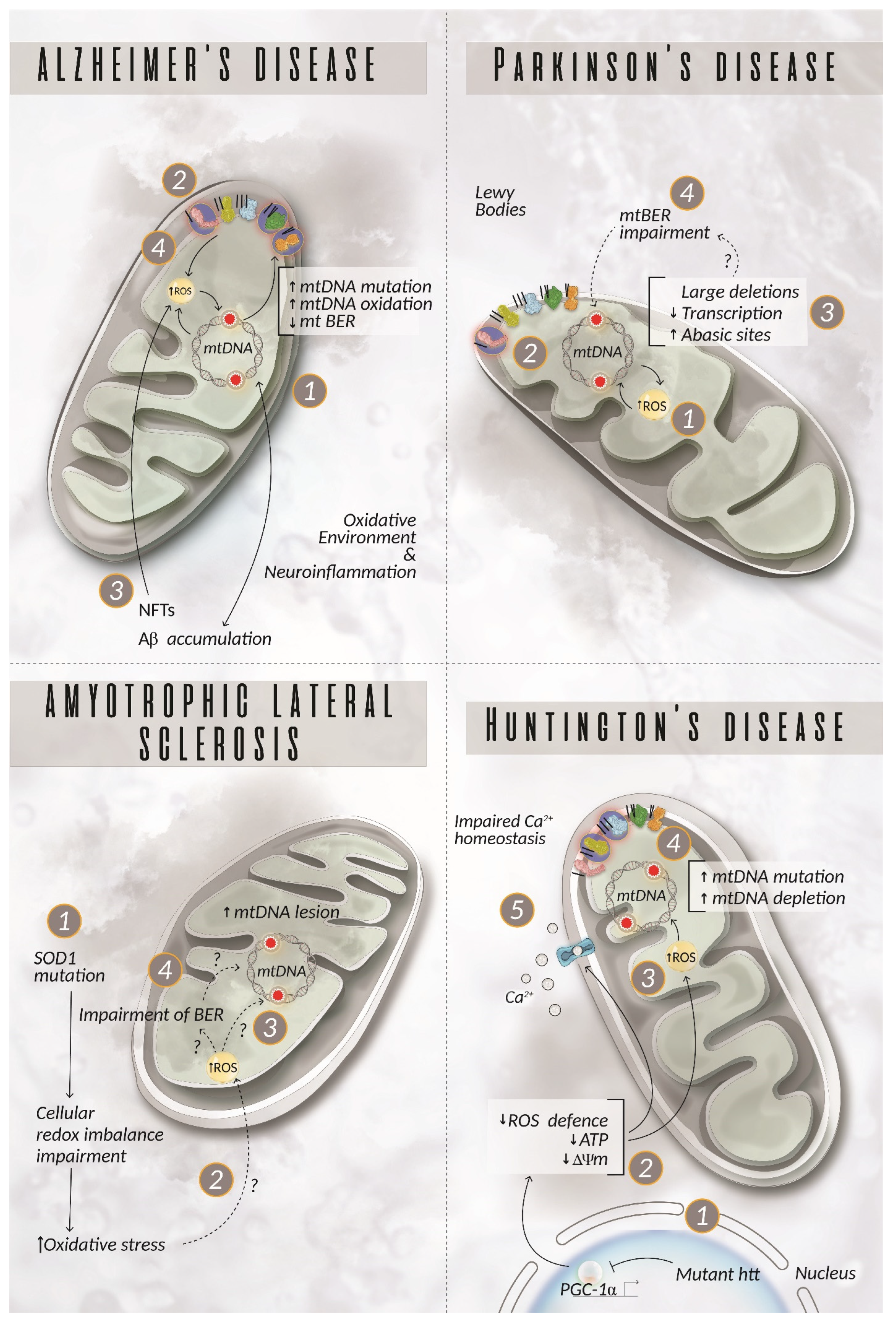
5. The Role of mtDNA in Neurodegenerative Diseases
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Amyotrophic Lateral Sclerosis
5.4. Huntington’s Disease
| Haplogroup AD | Activity | References |
| K and U | Protective effect: neutralises the harmful effect of the APOE ε4 allele | [146] |
| HV | Acts synergistically with the APOE4 allele, significantly associated with the risk of AD | [116] |
| H | Acts synergistically with the APOE4 allele, significantly associated with the risk of AD | [147,148] |
| HV5 | Acts synergistically with the APOE4 allele, significantly associated with the risk of AD | [149] |
| B5a | Genetic susceptibility to AD | [150] |
| L1 and L3 | L1 participants were at a significantly increased risk for developing dementia. L3 participants exhibited higher Aβ42 levels | [151] |
| UK, T | Disparity between studies and no congruent data | [152] |
| UK, K, J, and JT | No validated relevance and no congruent studies | [153] |
| Haplogroup PD | Activity | References |
| A5 | PD-promoting | [154] |
| B5 | Preventive, resistance against PD | [154,155] |
| UKJT and R | Protective: 22% reduction in population-attributable risk for PD | [156,157] |
| J and K | Protective: decreased PD risk | [158,159,160] |
| D | Increased PD risk | [161] |
| B | Decreased PD risk | [161] |
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuro. Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz e Silva, O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Zeng, K.; Yu, X.; Mahaman, Y.A.R.; Wang, J.Z.; Liu, R.; Li, Y.; Wang, X. Defective mitophagy and the etiopathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 32. [Google Scholar] [CrossRef]
- Perrone, L.; Valente, M. The emerging role of metabolism in brain-heart axis: New challenge for the therapy and prevention of Alzheimer Disease. May Thioredoxin Interacting Protein (TXNIP) play a role? Biomolecules 2021, 11, 1652. [Google Scholar] [CrossRef]
- Zhao, L. Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. Enzymes 2019, 45, 311–341. [Google Scholar]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef]
- Allkanjari, K.; Baldock, R.A. Beyond base excision repair: An evolving picture of mitochondrial DNA repair. Biosci. Rep. 2021, 41, BSR20211320. [Google Scholar] [CrossRef]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Oks, O.; Lewin, S.; Goncalves, I.L.; Sapir, A. The UPRmt Protects Caenorhabditis elegans from Mitochondrial Dysfunction by Upregulating Specific Enzymes of the Mevalonate Pathway. Genetics 2018, 209, 457–473. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; de Freitas Germano, J. At the heart of mitochondrial quality control: Many roads to the top. Cell. Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, Y.; Zhang, Y. Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways. J. Biosci. 2020, 45, 84. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef]
- Gauron, C.; Rampon, C.; Bouzaffour, M.; Ipendey, E.; Teillon, J.; Volovitch, M.; Vriz, S. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci. Rep. 2013, 3, 2084. [Google Scholar] [CrossRef]
- Diwanji, N.; Bergmann, A. The beneficial role of extracellular reactive oxygen species in apoptosis-induced compensatory proliferation. Fly 2017, 11, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, G.W.; Fong, C.S.; Alic, N.; Higgins, V.J.; Dawes, I.W. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: Oxidative-stress-response genes. Proc. Natl. Acad. Sci. USA 2004, 101, 6564–6569. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Berglund, A.K.; Navarrete, C.; Engqvist, M.K.; Hoberg, E.; Szilagyi, Z.; Taylor, R.W.; Gustafsson, C.M.; Falkenberg, M.; Clausen, A.R. Nucleotide pools dictate the identity and frequency of ribonucleotide incorporation in mitochondrial DNA. PLoS Genet. 2017, 13, e1006628. [Google Scholar] [CrossRef]
- Wanrooij, P.H.; Chabes, A. Ribonucleotides in mitochondrial DNA. FEBS Lett. 2019, 593, 1554–1565. [Google Scholar] [CrossRef]
- Chen, T.; He, J.; Huang, Y.; Zhao, W. The generation of mitochondrial DNA large-scale deletions in human cells. J. Hum. Genet. 2011, 56, 689–694. [Google Scholar] [CrossRef]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988, 51, 1525. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Dianov, G.L. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie 1999, 81, 155–160. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef]
- Liu, P.; Qian, L.; Sung, J.S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M., 3rd; Shen, B.; Demple, B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef]
- Sun, H.; He, L.; Wu, H.; Pan, F.; Wu, X.; Zhao, J.; Hu, Z.; Sekhar, C.; Li, H.; Zheng, L.; et al. The FEN1 L209P mutation interferes with long-patch base excision repair and induces cellular transformation. Oncogene 2017, 36, 194–207. [Google Scholar] [CrossRef]
- de Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar] [CrossRef]
- Morel, F.; Renoux, M.; Lachaume, P.; Alziari, S. Bleomycin-induced double-strand breaks in mitochondrial DNA of Drosophila cells are repaired. Mutat. Res. 2008, 637, 111–117. [Google Scholar] [CrossRef]
- Ricchetti, M.; Fairhead, C.; Dujon, B. Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature 1999, 402, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; Van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Coffey, G.; Campbell, C. An alternate form of Ku80 is required for DNA end-binding activity in mammalian mitochondria. Nucleic Acids Res. 2000, 28, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase θ. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Yui, R.; Matsuura, E.T. Detection of deletions flanked by short direct repeats in mitochondrial DNA of aging Drosophila. Mutat. Res. 2006, 594, 155–161. [Google Scholar] [CrossRef]
- Phadnis, N.; Sia, R.A.; Sia, E.A. Analysis of repeat-mediated deletions in the mitochondrial genome of Saccharomyces cerevisiae. Genetics 2005, 171, 1549–1559. [Google Scholar] [CrossRef]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol 2019, 29, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef] [PubMed]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef]
- Kurihara, Y.; Kanki, T.; Aoki, Y.; Hirota, Y.; Saigusa, T.; Uchiumi, T.; Kang, D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 2012, 287, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. The “fast” and the “slow” modes of mitochondrial DNA degradation. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 490–498. [Google Scholar] [CrossRef]
- Nissanka, N.; Bacman, S.R.; Plastini, M.J.; Moraes, C.T. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef]
- Peter, B.; Falkenberg, M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes 2020, 11, 408. [Google Scholar] [CrossRef]
- Ruhanen, H.; Borrie, S.; Szabadkai, G.; Tyynismaa, H.; Jones, A.W.; Kang, D.; Taanman, J.W.; Yasukawa, T. Mitochondrial single-stranded DNA binding protein is required for maintenance of mitochondrial DNA and 7S DNA but is not required for mitochondrial nucleoid organisation. Biochim. Biophys. Acta 2010, 1803, 931–939. [Google Scholar] [CrossRef]
- Dan, X.; Babbar, M.; Moore, A.; Wechter, N.; Tian, J.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 2020, 48, 6611–6623. [Google Scholar] [CrossRef]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Liesa, M.; Sebastián, D.; Segalés, J.; Palacín, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fang, H.; Chi, Z.; Wu, Z.; Wei, D.; Mo, D.; Niu, K.; Balajee, A.S.; Hei, T.K.; Nie, L.; et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 2015, 43, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Tam, Z.Y.; Gruber, J.; Halliwell, B.; Gunawan, R. Context-Dependent Role of Mitochondrial Fusion-Fission in Clonal Expansion of mtDNA Mutations. PLoS Comput. Biol. 2015, 11, e1004183. [Google Scholar] [CrossRef]
- Sbai, O.; Djelloul, M.; Auletta, A.; Ieraci, A.; Vascotto, C.; Perrone, L. RAGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Aβ to mitochondria in microglia. Cell Death Dis. 2022, 13, 302. [Google Scholar] [CrossRef]
- Prakash, A.; Doublié, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef]
- Bazzani, V.; Barchiesi, A.; Radecka, D.; Pravisani, R.; Guadagno, A.; Di Loreto, C.; Baccarani, U.; Vascotto, C. Mitochondrial apurinic/apyrimidinic endonuclease 1 enhances mtDNA repair contributing to cell proliferation and mitochondrial integrity in early stages of hepatocellular carcinoma. BMC Cancer 2020, 20, 969. [Google Scholar] [CrossRef]
- Kashkina, E.; Qi, T.; Weinfeld, M.; Young, D. Polynucleotide kinase/phosphatase, Pnk1, is involved in base excision repair in Schizosaccharomyces pombe. DNA Repair 2012, 11, 676–683. [Google Scholar] [CrossRef]
- Pinz, K.G.; Bogenhagen, D.F. The influence of the DNA polymerase gamma accessory subunit on base excision repair by the catalytic subunit. DNA Repair 2006, 5, 121–128. [Google Scholar] [CrossRef]
- Kalifa, L.; Beutner, G.; Phadnis, N.; Sheu, S.-S.; Sia, E.A. Evidence for a Role of FEN1 in Maintaining Mitochondrial DNA Integrity. DNA Repair 2009, 8, 1242–1249. [Google Scholar] [CrossRef]
- Wu, C.-C.; Lin, J.L.J.; Yang-Yen, H.-F.; Yuan, H.S. A unique exonuclease ExoG cleaves between RNA and DNA in mitochondrial DNA replication. Nucleic. Acids Res. 2019, 47, 5405–5419. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Matic, S.; Jiang, M.; Nicholls, T.J.; Uhler, J.P.; Dirksen-Schwanenland, C.; Polosa, P.L.; Simard, M.L.; Li, X.; Atanassov, I.; Rackham, O.; et al. Mice lacking the mitochondrial exonuclease MGME1 accumulate mtDNA deletions without developing progeria. Nat. Commun. 2018, 9, 1202. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Nandakumar, D.; Tang, G.-Q.; Patel, S.S. Human Mitochondrial DNA Helicase TWINKLE Is Both an Unwinding and Annealing Helicase. J. Biol. Chem. 2012, 287, 14545–14556. [Google Scholar] [CrossRef]
- van der Rijt, S.; Molenaars, M.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front. Cell. Dev. Biol. 2020, 8, 594416. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Sanz, A.; Stefanatos, R.K. The mitochondrial free radical theory of aging: A critical view. Curr. Aging Sci. 2008, 1, 10–21. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Nekhaeva, E.; Bodyak, N.B.; Khrapko, K. Mutation and intracellular clonal expansion of mitochondrial genomes: Two synergistic components of the aging process? Mech. Ageing Dev. 2003, 124, 49–53. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Origin and evolution of the free radical theory of aging: A brief personal history, 1954–2009. Biogerontology 2009, 10, 773–781. [Google Scholar] [CrossRef] [PubMed]
- de Grey, A.D. Reactive oxygen species production in the mitochondrial matrix: Implications for the mechanism of mitochondrial mutation accumulation. Rejuvenation Res. 2005, 8, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef]
- Cerielo, A. Oxidative stress and glycemic regulation. Metabolism 2000, 49, 27–29. [Google Scholar] [CrossRef]
- Garbarino, V.R.; Orr, M.E.; Rodriguez, K.A.; Buffenstein, R. Mechanisms of oxidative stress resistance in the brain: Lessons learned from hypoxia tolerant extremophilic vertebrates. Arch. Biochem. Biophys. 2015, 576, 8–16. [Google Scholar] [CrossRef]
- Pikó, L.; Hougham, A.J.; Bulpitt, K.J. Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: Evidence for an increased frequency of deletions/additions with aging. Mech. Ageing Dev. 1988, 43, 279–293. [Google Scholar] [CrossRef]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombès, A.; Fardeau, M.; Oldfors, A. Ageing muscle: Clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Greaves, L.C.; Nooteboom, M.; Elson, J.L.; Tuppen, H.A.; Taylor, G.A.; Commane, D.M.; Arasaradnam, R.P.; Khrapko, K.; Taylor, R.W.; Kirkwood, T.B.; et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 2014, 10, e1004620. [Google Scholar] [CrossRef]
- Posada, D.; Vicens, A. Selective Pressures on Human Cancer Genes along the Evolution of Mammals. Genes 2018, 9, 582. [Google Scholar]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Kennedy, S.R. The Complicated Nature of Somatic mtDNA Mutations in Aging. Front. Aging 2022, 2, 805126. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K. The timing of mitochondrial DNA mutations in aging. Nat. Genet. 2011, 43, 726–727. [Google Scholar] [CrossRef]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef]
- Smith, A.L.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef]
- Wolf, A.M. MtDNA mutations and aging-not a closed case after all? Signal Transduct. Target. Ther. 2021, 6, 56. [Google Scholar] [CrossRef]
- Giaimo, S.; Traulsen, A. The selection force weakens with age because ageing evolves and not vice versa. Nat. Commun. 2022, 13, 686. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifesp.pan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell. Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Perrone, L.; Sbai, O.; Nawroth, P.P.; Bierhaus, A. The Complexity of Sporadic Alzheimer’s Disease Pathogenesis: The Role of RAGE as Therapeutic Target to Promote Neuroprotection by Inhibiting Neurovascular Dysfunction. Int. J. Alzheimer’s Dis. 2012, 2012, 734956–734966. [Google Scholar] [CrossRef]
- Armstrong, R.A. What causes alzheimer’s disease? Folia Neuropathol. 2013, 51, 169–188. [Google Scholar] [CrossRef]
- Hutchin, T.P.; Heath, P.R.; Pearson, R.C.; Sinclair, A.J. Mitochondrial DNA mutations in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1997, 241, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- van der Walt, J.M.; Dementieva, Y.A.; Martin, E.R.; Scott, W.K.; Nicodemus, K.K.; Kroner, C.C.; Welsh-Bohmer, K.A.; Saunders, A.M.; Roses, A.D.; Small, G.W.; et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 2004, 365, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Nardini, M.; Micheli, D.; Rocchi, A.; Nesti, C.; Giglioli, N.J.; Petrozzi, L.; Rossi, C.; Ceravolo, R.; Bacci, A.; et al. Lack of association between mtDNA haplogroups and Alzheimer’s disease in Tuscany. Neurol. Sci. 2007, 28, 142–147. [Google Scholar] [CrossRef]
- Maruszak, A.; Canter, J.A.; Styczyńska, M.; Zekanowski, C.; Barcikowska, M. Mitochondrial haplogroup H and Alzheimer’s disease—Is there a connection? Neurobiol. Aging 2009, 30, 1749–1755. [Google Scholar] [CrossRef]
- Antonyová, V.; Kejík, Z.; Brogyányi, T.; Kaplánek, R.; Pajková, M.; Talianová, V.; Hromádka, R.; Masařík, M.; Sýkora, D.; Mikšátková, L.; et al. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair 2020, 91–92, 102871. [Google Scholar] [CrossRef]
- Di Napoli, M.; Shah, I.M.; Stewart, D.A. Molecular pathways and genetic aspects of Parkinson’s disease: From bench to bedside. Expert Rev. Neurother. 2007, 7, 1693–1729. [Google Scholar] [CrossRef]
- Nicoletti, V.; Palermo, G.; Del Prete, E.; Mancuso, M.; Ceravolo, R. Understanding the Multiple Role of Mitochondria in Parkinson’s Disease and Related Disorders: Lesson From Genetics and Protein-Interaction Network. Front. Cell. Dev. Biol. 2021, 9, 636506. [Google Scholar] [CrossRef]
- Martín-Jiménez, R.; Lurette, O.; Hebert-Chatelain, E. Damage in Mitochondrial DNA Associated with Parkinson’s Disease. DNA Cell Biol. 2020, 39, 1421–1430. [Google Scholar] [CrossRef]
- Bogaerts, V.; Theuns, J.; van Broeckhoven, C. Genetic findings in Parkinson’s disease and translation into treatment: A leading role for mitochondria? Genes Brain Behav. 2008, 7, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Progress Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Larsen, N.B.; Rasmussen, M.; Rasmussen, L.J. Nuclear and mitochondrial DNA repair: Similar pathways? Mitochondrion 2005, 5, 89–108. [Google Scholar] [CrossRef]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; DiMauro, S. Early-onset familial parkinsonism due to POLG mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef]
- Dai, Y.; Clark, J.; Zheng, K.; Kujoth, G.C.; Prolla, T.A.; Simon, D.K. Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice. Neurotoxicol. Teratol. 2014, 46, 62–67. [Google Scholar] [CrossRef]
- Nido, G.S.; Dölle, C.F.; lønes, I.; Tuppen, H.A.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease. Neurobiol. Aging 2018, 63, 120–127. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Montine, T.J.; Smith, M.A.; Siedlak, S.L.; Gu, G.; Robertson, D.; Perry, G. The mitochondrial common deletion in Parkinson’s disease and related movement disorders. Parkinsonism Relat. Disord. 2002, 8, 165–170. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef]
- Murakami, T.; Nagai, M.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Kurata, T.; Takehisa, Y.; Kamiya, T.; Abe, K. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res. 2007, 1150, 182–189. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Kim, J.; Pyzel, A.; Baranova, O.V.; Khattar, N.K.; Larkin, T.M.; Baranov, S.V.; Chen, N.; et al. Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Natl. Acad. Sci. USA 2019, 116, 16593–16602. [Google Scholar] [CrossRef]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Damiano, M.; Diguet, E.; Malgorn, C.; D’Aurelio, M.; Galvan, L.; Petit, F.; Benhaim, L.; Guillermier, M.; Houitte, D.; Dufour, N.; et al. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet. 2013, 22, 3869–3882. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, S.L.; Milanese, C.; Boogaard, M.W.; Buijsen, R.A.M.; Hogenboom, M.; Roos, R.A.C.; Mastroberardino, P.G.; van Roon-Mom, W.M.C.; Aziz, N.A. Bioenergetics in fibroblasts of patients with Huntington disease are associated with age at onset. Neurol. Genet. 2018, 4, e275. [Google Scholar] [CrossRef] [PubMed]
- Victor, M.B.; Richner, M.; Olsen, H.E.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.L.; Yang, X.W.; et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef]
- Carrieri, G.; Bonafè, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum. Genet. 2001, 108, 194–198. [Google Scholar] [CrossRef]
- Maruszak, A.; Safranow, K.; Branicki, W.; Gawęda-Walerych, K.; Pośpiech, E.; Gabryelewicz, T.; Canter, J.A.; Barcikowska, M.; Zekanowski, C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimer’s Dis. 2011, 27, 197–210. [Google Scholar] [CrossRef]
- Coto, E.; Gómez, J.; Alonso, B.; Corao, A.I.; Díaz, M.; Menéndez, M.; Martínez, C.; Calatayud, M.T.; Morís, G.; Álvarez, V. Late-onset Alzheimer’s disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy. Neurogenetics 2011, 12, 345–346. [Google Scholar] [CrossRef]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5, e12037. [Google Scholar] [CrossRef]
- Bi, R.; Zhang, W.; Yu, D.; Li, X.; Wang, H.Z.; Hu, Q.X.; Zhang, C.; Lu, W.; Ni, J.; Fang, Y.; et al. Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer’s disease in Han Chinese. Neurobiol. Aging 2015, 36, e7–e16. [Google Scholar] [CrossRef]
- Tranah, G.J.; Yokoyama, J.S.; Katzman, S.M.; Nalls, M.A.; Newman, A.B.; Harris, T.B.; Cesari, M.; Manini, T.M.; Schork, N.J.; Cummings, S.R.; et al. Mitochondrial DNA sequence associations with dementia and amyloid-β in elderly African Americans. Neurobiol. Aging 2014, 35, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brinton, R.D. Triad of Risk for Late Onset Alzheimer’s: Mitochondrial Haplotype, APOE Genotype and Chromosomal Sex. Front. Aging Neurosci. 2016, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Filosto, M.; Orsucci, D.; Siciliano, G. Mitochondrial DNA sequence variation and neurodegeneration. Hum. Genom. 2008, 3, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Li, T.; Wang, Z.F.; Huang, S.S.; Shao, Z.Q.; Wang, K.; Zhong, H.Q.; Chen, S.F.; Zhang, X.; Zhu, J.H. Mitochondrial DNA variants modulate genetic susceptibility to Parkinson’s disease in Han Chinese. Neurobiol. Dis. 2018, 114, 17–23. [Google Scholar] [CrossRef]
- Liou, C.W.; Chuang, J.H.; Chen, J.B.; Tiao, M.M.; Wang, P.W.; Huang, S.T.; Huang, T.L.; Lee, W.C.; Weng, S.W.; Huang, P.H.; et al. Mitochondrial DNA variants as genetic risk factors for Parkinson disease. Eur. J. Neurol. 2016, 23, 1289–1300. [Google Scholar] [CrossRef]
- Georgiou, A.; Demetriou, C.A.; Heraclides, A.; Christou, Y.P.; Leonidou, E.; Loukaides, P.; Yiasoumi, E.; Panagiotou, D.; Manoli, P.; Thomson, P.; et al. Mitochondrial superclusters influence age of onset of Parkinson’s disease in a gender specific manner in the Cypriot population: A case-control study. PLoS ONE 2017, 12, e0183444. [Google Scholar] [CrossRef]
- Pyle, A.; Foltynie, T.; Tiangyou, W.; Lambert, C.; Keers, S.M.; Allcock, L.M.; Davison, J.; Lewis, S.J.; Perry, R.H.; Barker, R.; et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann. Neurol. 2005, 57, 564–567. [Google Scholar] [CrossRef]
- van der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Haines, J.L.; Koller, W.C.; Lyons, K.; et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef]
- Ghezzi, D.; Marelli, C.; Achilli, A.; Goldwurm, S.; Pezzoli, G.; Barone, P.; Pellecchia, M.T.; Stanzione, P.; Brusa, L.; Bentivoglio, A.R.; et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur. J. Hum. Genet. 2005, 13, 748–752. [Google Scholar] [CrossRef]
- Huerta, C.; Castro, M.G.; Coto, E.; Blázquez, M.; Ribacoba, R.; Guisasola, L.M.; Salvador, C.; Martínez, C.; Lahoz, C.H.; Alvarez, V. Mitochondrial DNA polymorphisms and risk of Parkinson’s disease in Spanish population. J. Neurol. Sci. 2005, 236, 49–54. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chen, W.J.; Lin, X.Z.; Zhang, Q.J.; Cai, J.P.; Liou, C.W.; Wang, N. Mitochondrial DNA haplogroups and the risk of sporadic Parkinson’s disease in Han Chinese. Chin. Med. J. 2015, 128, 1748–1754. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzani, V.; Equisoain Redin, M.; McHale, J.; Perrone, L.; Vascotto, C. Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. Int. J. Mol. Sci. 2022, 23, 11391. https://doi.org/10.3390/ijms231911391
Bazzani V, Equisoain Redin M, McHale J, Perrone L, Vascotto C. Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. International Journal of Molecular Sciences. 2022; 23(19):11391. https://doi.org/10.3390/ijms231911391
Chicago/Turabian StyleBazzani, Veronica, Mara Equisoain Redin, Joshua McHale, Lorena Perrone, and Carlo Vascotto. 2022. "Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing" International Journal of Molecular Sciences 23, no. 19: 11391. https://doi.org/10.3390/ijms231911391
APA StyleBazzani, V., Equisoain Redin, M., McHale, J., Perrone, L., & Vascotto, C. (2022). Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. International Journal of Molecular Sciences, 23(19), 11391. https://doi.org/10.3390/ijms231911391