
Oxidative Stress as a Contributor to Insulin Resistance in the Skeletal Muscles of Mice with Polycystic Ovary Syndrome
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Increased Oxidative Stress in the Whole-Body and Skeletal Muscles of Mice with DHEA-Induced PCOS
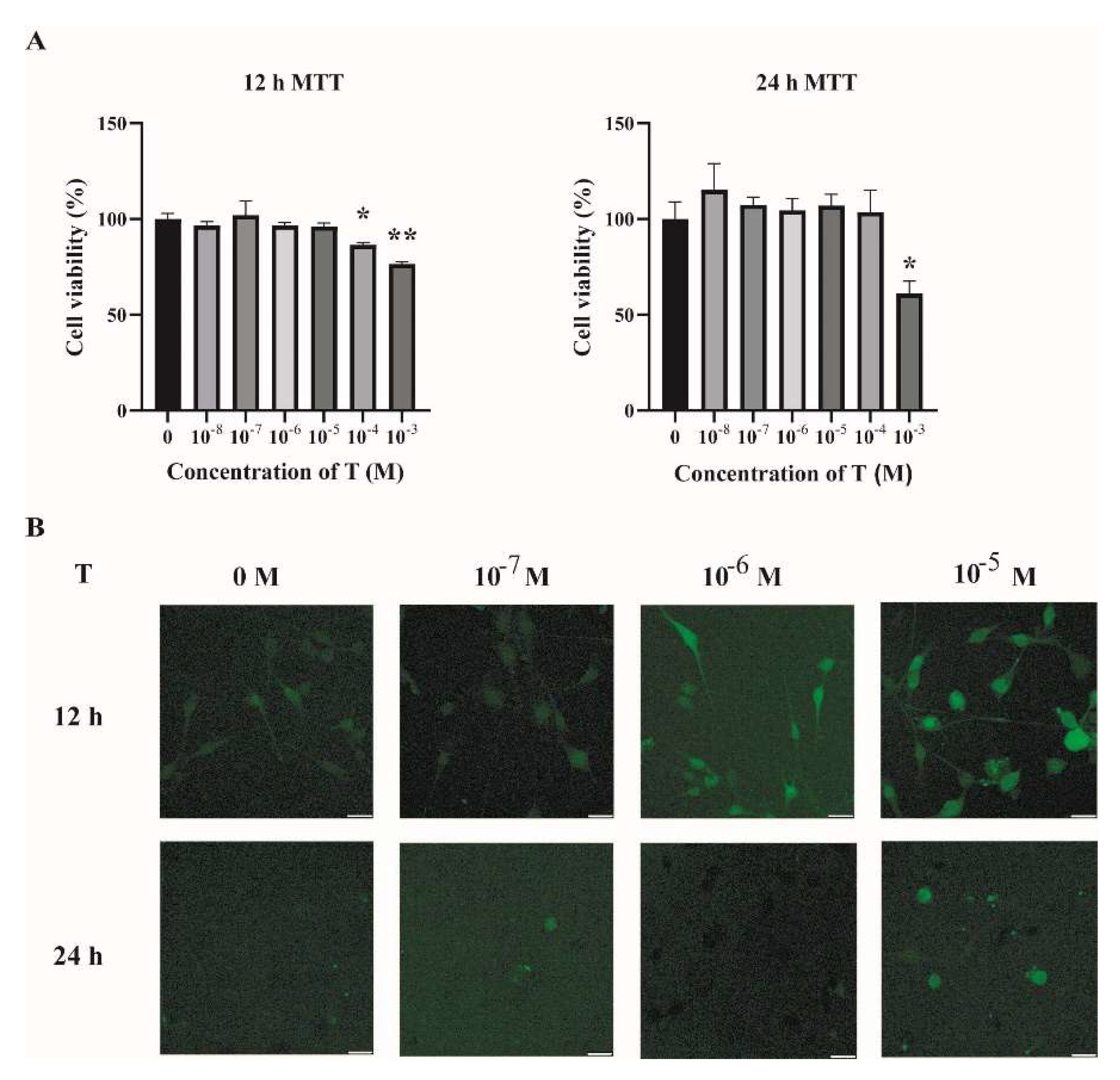
2.2. Increased ROS Levels in Testosterone-Treated C2C12 Cells
2.3. Impaired Mitochondrial Function and Insulin Sensitivity in T-Treated C2C12 Cells
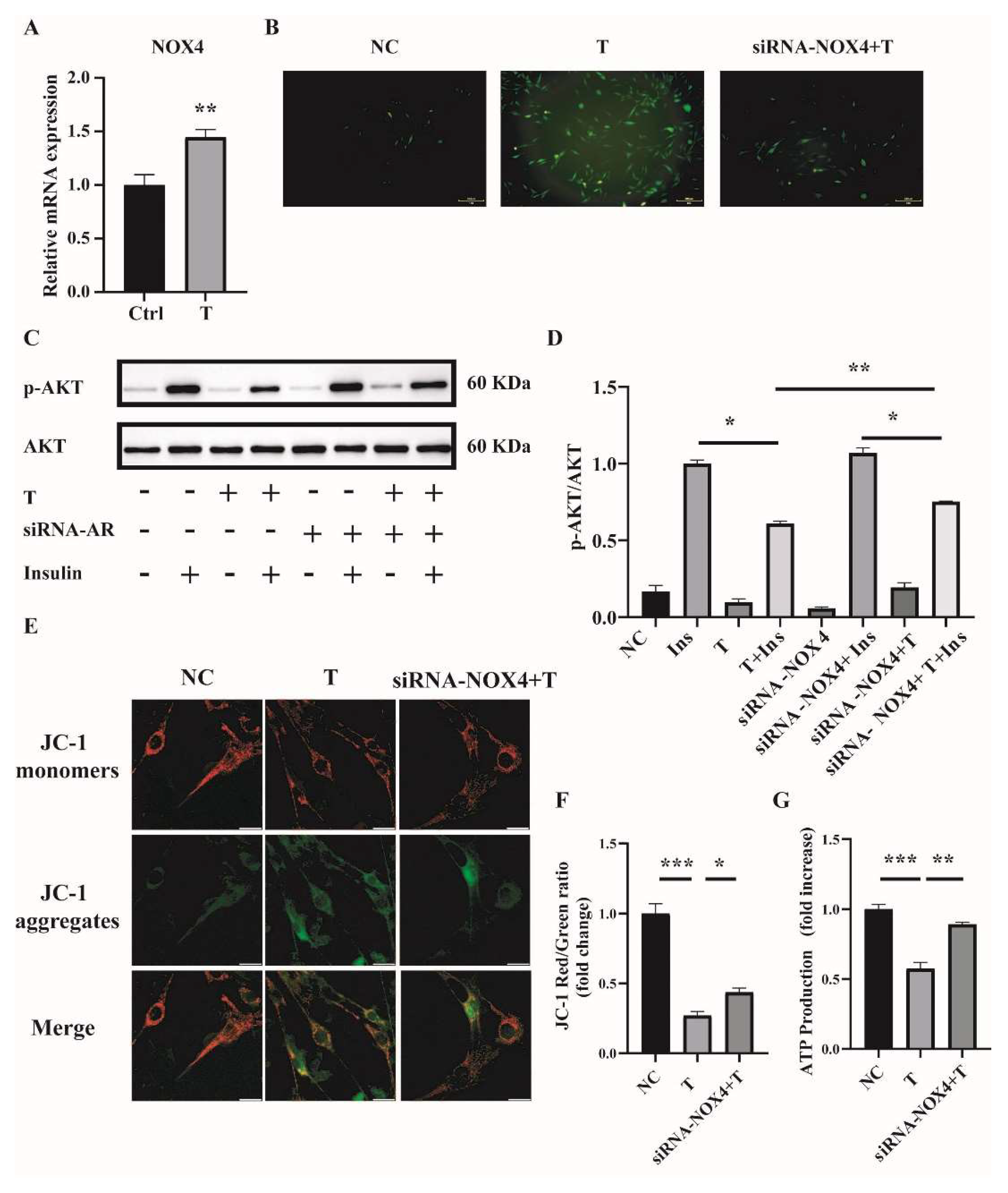
2.4. NOX4 Plays an Important Role in T-Induced ROS Production in C2C12 Cells
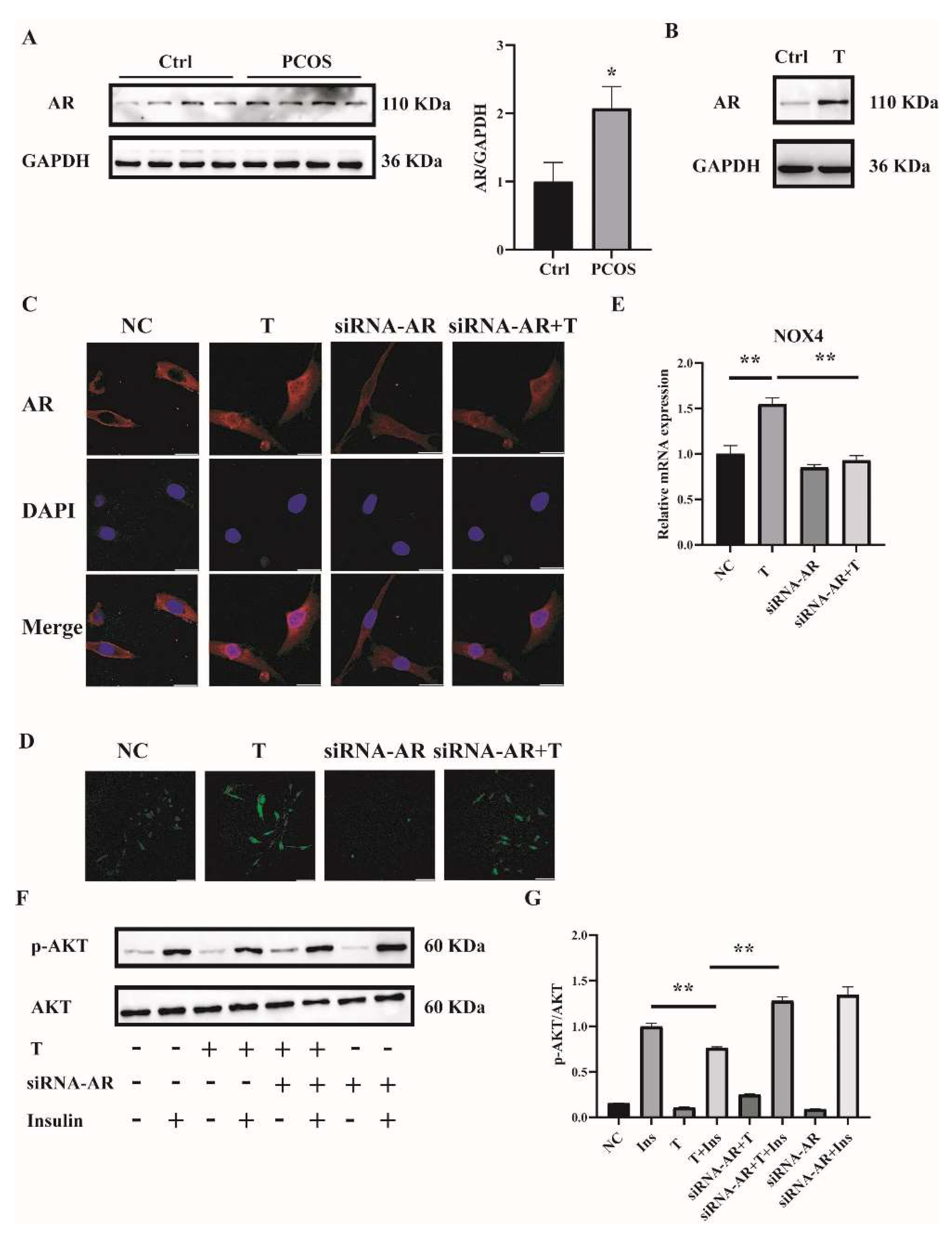
2.5. T Induces ROS in C2C12 Cells by Binding to the Androgen Receptor (AR)
2.6. Reducing ROS Levels Ameliorates Mitochondrial Function and Insulin Resistance in T-Treated C2C12 Cells
2.7. NAC Ameliorates Insulin Resistance in the Skeletal Muscle of DHEA-Induced PCOS Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Tissue Collection
4.3. Oxidative-Redox Metabolome in Serum and Tissues
4.4. Cell Culture and Treatments
4.5. Detection of ROS and Total Superoxide Dismutase (SOD) Activity
4.6. Determination of ATP Content
4.7. Mitochondrial Membrane Potential Determination
4.8. Western Blot Analysis
4.9. Extraction of RNA and Quantitative Real-Time PCR Analysis
4.10. Small Interfering RNA (siRNA) Transfection
4.11. Immunofluorescence Microscopy Analysis
4.12. Insulin-Signaling Assay in Animals and Cultured Cells
4.13. MTT Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Russell, S.J.; Schneider, M.F. Alternative signaling pathways from IGF1 or insulin to AKT activation and FOXO1 nuclear efflux in adult skeletal muscle fibers. J. Biol. Chem. 2020, 295, 15292–15306. [Google Scholar] [CrossRef]
- Nilsson, E.; Benrick, A.; Kokosar, M.; Krook, A.; Lindgren, E.; Källman, T.; Martis, M.M.; Højlund, K.; Ling, C.; Stener-Victorin, E. Transcriptional and Epigenetic Changes Influencing Skeletal Muscle Metabolism in Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 4465–4477. [Google Scholar] [CrossRef]
- Corbould, A.; Zhao, H.; Mirzoeva, S.; Aird, F.; Dunaif, A. Enhanced Mitogenic Signaling in Skeletal Muscle of Women With Polycystic Ovary Syndrome. Diabetes 2006, 55, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Glintborg, D.; Andersen, N.R.; Birk, J.B.; Treebak, J.T.; Frøsig, C.; Beck-Nielsen, H.; Wojtaszewski, J.F. Impaired Insulin-Stimulated Phosphorylation of Akt and AS160 in Skeletal Muscle of Women With Polycystic Ovary Syndrome Is Reversed by Pioglitazone Treatment. Diabetes 2008, 57, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lennon, S.V.; Martin, S.J.; Cotter, T.G. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991, 24, 203. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Bouayed, J.; Bohn, T. Exogenous Antioxidants—Double-Edged Swords in Cellular Redox State: Health Beneficial Effects at Physiologic Doses versus Deleterious Effects at High Doses. Oxid. Med. Cell Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef]
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 2014, 5, 4425. [Google Scholar] [CrossRef]
- Chang, R.J. The reproductive phenotype in polycystic ovary syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 688–695. [Google Scholar] [CrossRef]
- Song, X.; Shen, Q.; Fan, L.; Yu, Q.; Jia, X.; Sun, Y.; Bai, W.; Kang, J. Dehydroepiandrosterone-induced activation of mTORC1 and inhibition of autophagy contribute to skeletal muscle insulin resistance in a mouse model of polycystic ovary syndrome. Oncotarget 2018, 9, 11905–11921. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef]
- Guo, Z.; Yu, Q. Role of mTOR Signaling in Female Reproduction. Front. Endocrinol. 2019, 10, 692. [Google Scholar] [CrossRef]
- Murri, M.; Luque-Ramírez, M.; Insenser, M.; Ojeda, M.; Escobar-Morreale, H.F. Circulating markers of oxidative stress and polycystic ovary syndrome (PCOS): A systematic review and meta-analysis. Hum. Reprod. Updat. 2013, 19, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Rajska, A.; Buszewska-Forajta, M.; Rachoń, D.; Markuszewski, M.J. Metabolomic Insight into Polycystic Ovary Syndrome—An Overview. Int. J. Mol. Sci. 2020, 21, 4853. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Considine, R.V.; A Abdelhadi, O.; Acton, A.J. Oxidative Stress in Response to Saturated Fat Ingestion Is Linked to Insulin Resistance and Hyperandrogenism in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 5360–5371. [Google Scholar] [CrossRef]
- Mujica, L.S.; Bridi, A.; Della Mea, R.; Rissi, V.B.; Guarda, N.D.S.; Moresco, R.N.; Premaor, M.O.; Antoniazzi, A.Q.; Goncalves, P.B.D.; Comim, F.V. Oxidative stress and metabolic markers in pre- and postnatal polycystic ovary syndrome rat protocols. J. Inflamm. Res. 2018, 11, 193–202. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Kleikers, P.W.M.; Radermacher, K.A.; Scheurer, P.; Hermans, J.J.R.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H.H.W. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Experientia 2012, 69, 2327–2343. [Google Scholar] [CrossRef]
- Chen, K.; Kirber, M.; Xiao, H.; Yang, Y.; Keaney, J. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Handayaningsih, A.-E.; Iguchi, G.; Fukuoka, H.; Nishizawa, H.; Takahashi, M.; Yamamoto, M.; Herningtyas, E.-H.; Okimura, Y.; Kaji, H.; Chihara, K.; et al. Reactive Oxygen Species Play an Essential Role in IGF-I Signaling and IGF-I-Induced Myocyte Hypertrophy in C2C12 Myocytes. Endocrinology 2011, 152, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, D.S.; Csikasz, R.I.; Yamamoto, D.L.; Shabalina, I.G.; Wikström, P.; Wilcke, M.; Bengtsson, T. Diphenylene iodonium stimulates glucose uptake in skeletal muscle cells through mitochondrial complex I inhibition and activation of AMP-activated protein kinase. Cell Signal. 2007, 19, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Kuşçu, N.K.; Var, A. Oxidative stress but not endothelial dysfunction exists in non-obese, young group of patients with polycystic ovary syndrome. Acta Obstet. Gynecol. Scand. 2009, 88, 612–617. [Google Scholar] [CrossRef]
- Mohammadi, M. Oxidative stress and polycystic ovary syndrome: A brief review. Int. J. Prev. Med. 2019, 10, 86. [Google Scholar] [CrossRef]
- Dinger, Y.; Akcay, T.; Erdem, T.; Saygi˙li˙, E.I.; Gundogdu, S. DNA damage, DNA susceptibility to oxidation and glutathione level in women with polycystic ovary syndrome. Scand. J. Clin. Lab. Investig. 2005, 65, 721–728. [Google Scholar]
- Sabuncua, T.; Vural, H.; Harma, M.; Harma, M. Oxidative stress in polycystic ovary syndrome and its contribution to the risk of cardiovascular disease. Clin. Biochem. 2001, 34, 407–413. [Google Scholar] [CrossRef]
- McCartney, C.R.; Marshall, J.C. CLINICAL PRACTICE. Polycystic Ovary Syndrome. N. Engl. J. Med. 2016, 375, 54–64. [Google Scholar] [CrossRef]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; A Herzenberg, L. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef]
- Sekhon, L.H.; Gupta, S.; Kim, Y.; Agarwal, A. Female Infertility and Antioxidants. Curr. Womens Health Rev. 2010, 6, 84–95. [Google Scholar] [CrossRef]
- Vida Mokhtari, M.S.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A Review on Various Uses of N-Acetyl Cysteine. Cell J. 2016, 19, 11–17. [Google Scholar]
- Fulghesu, A.M. N-acetyl-cysteine treatment improves insulin sensitivity in women with polycystic ovary syndrome. Fertil. Steril. 2002, 77, 1128–1135. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F. Polycystic ovary syndrome: Definition, aetiology, diagnosis and treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284. [Google Scholar] [CrossRef]
- Foradori, C.; Weiser, M.; Handa, R. Non-genomic actions of androgens. Front. Neuroendocr. 2008, 29, 169–181. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Dominic, P.; Stokes, K.Y. Uncovering sex-specific mechanisms of action of testosterone and redox balance. Redox Biol. 2020, 31, 101490. [Google Scholar] [CrossRef]
- Xing, X.; Zhang, L.; Wu, S.; Ruan, Y.; Hong, L.; Lai, W. Testosterone suppresses oxidative stress via androgen receptor-independent pathway in murine cardiomyocytes. Mol. Med. Rep. 2011, 4, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Pronsato, L.; Boland, R.; Milanesi, L. Non-classical localization of androgen receptor in the C2C12 skeletal muscle cell line. Arch. Biochem. Biophys. 2013, 530, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chignalia, A.Z.; Schuldt, E.Z.; Camargo, L.L.; Montezano, A.C.; Callera, G.E.; Laurindo, F.R.; Lopes, L.R.; Avellar, M.C.W.; Carvalho, M.H.C.; Fortes, Z.B.; et al. Testosterone Induces Vascular Smooth Muscle Cell Migration by NADPH Oxidase and c-Src–Dependent Pathways. Hypertension 2012, 59, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Feraco, A.; Gorini, S.; Armani, A.; Camajani, E.; Rizzo, M.; Caprio, M. Exploring the Role of Skeletal Muscle in Insulin Resistance: Lessons from Cultured Cells to Animal Models. Int. J. Mol. Sci. 2021, 22, 9327. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, F.; Gao, Y.; Pang, Y.; Xie, C.; Jiang, C. An Integrated LC-MS/MS Strategy for Quantifying the Oxidative-Redox Metabolome in Multiple Biological Samples. Anal. Chem. 2020, 92, 8810–8818. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harbor. Protoc. 2018, 469–471. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Q.; Zou, X.; Liu, S.; Wu, H.; Shen, Q.; Kang, J. Oxidative Stress as a Contributor to Insulin Resistance in the Skeletal Muscles of Mice with Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2022, 23, 11384. https://doi.org/10.3390/ijms231911384
Yao Q, Zou X, Liu S, Wu H, Shen Q, Kang J. Oxidative Stress as a Contributor to Insulin Resistance in the Skeletal Muscles of Mice with Polycystic Ovary Syndrome. International Journal of Molecular Sciences. 2022; 23(19):11384. https://doi.org/10.3390/ijms231911384
Chicago/Turabian StyleYao, Qiyang, Xin Zou, Shihe Liu, Haowen Wu, Qiyang Shen, and Jihong Kang. 2022. "Oxidative Stress as a Contributor to Insulin Resistance in the Skeletal Muscles of Mice with Polycystic Ovary Syndrome" International Journal of Molecular Sciences 23, no. 19: 11384. https://doi.org/10.3390/ijms231911384
APA StyleYao, Q., Zou, X., Liu, S., Wu, H., Shen, Q., & Kang, J. (2022). Oxidative Stress as a Contributor to Insulin Resistance in the Skeletal Muscles of Mice with Polycystic Ovary Syndrome. International Journal of Molecular Sciences, 23(19), 11384. https://doi.org/10.3390/ijms231911384