

Enhancing Effects of the Cyano Group on the C-X∙∙∙N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX∙∙∙NMe3, (n = 0–3; X = H, Cl, Br, I)



Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
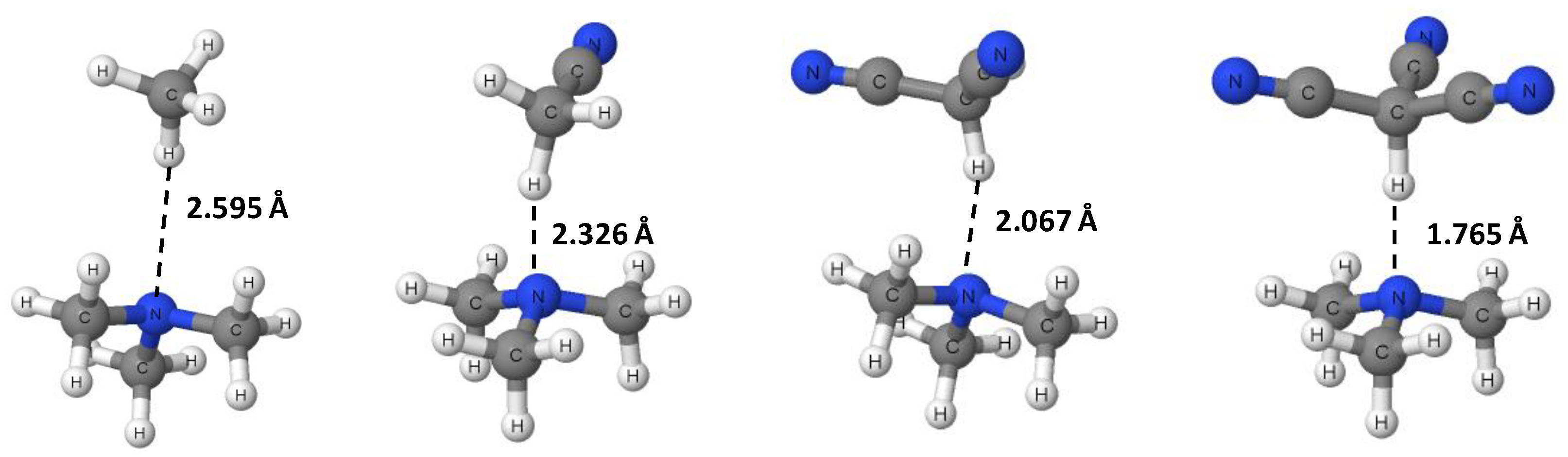
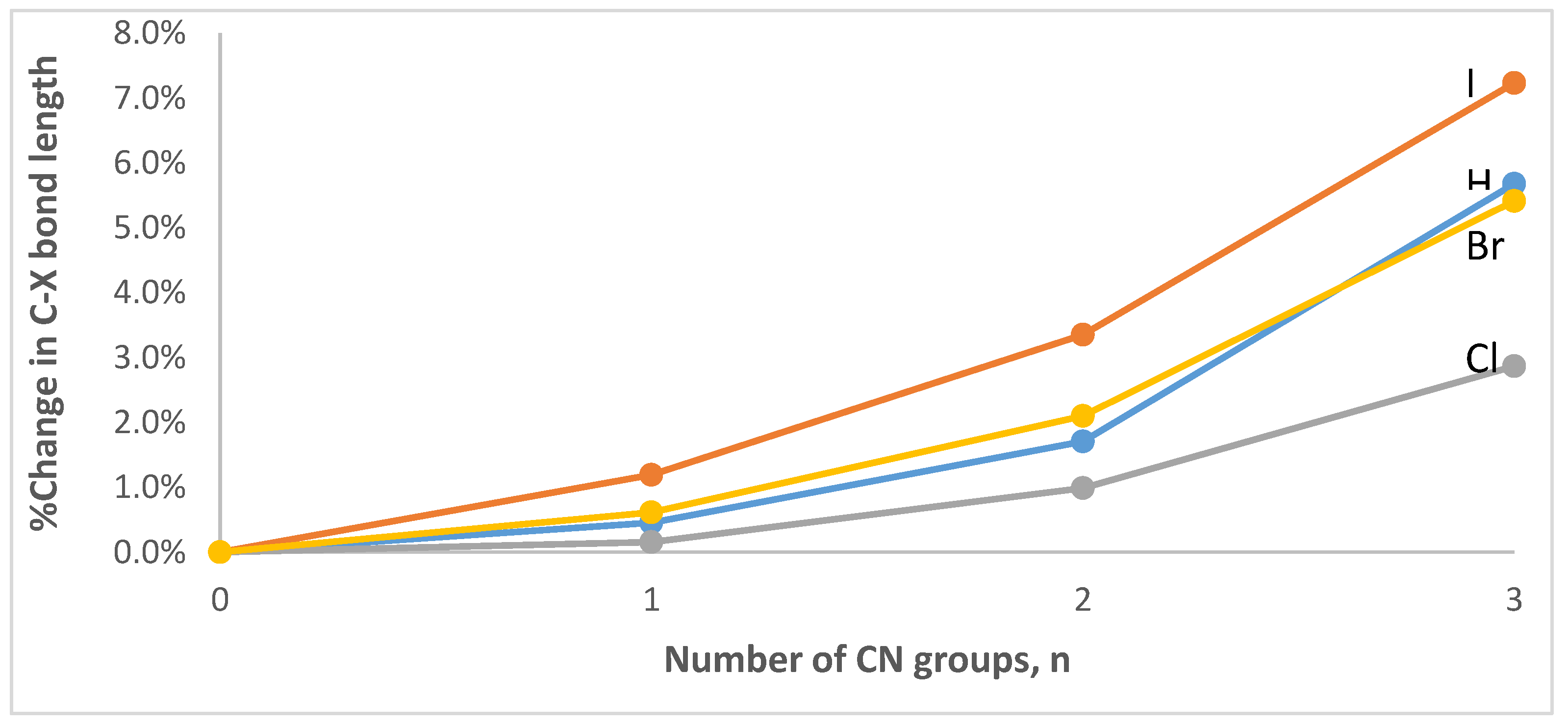
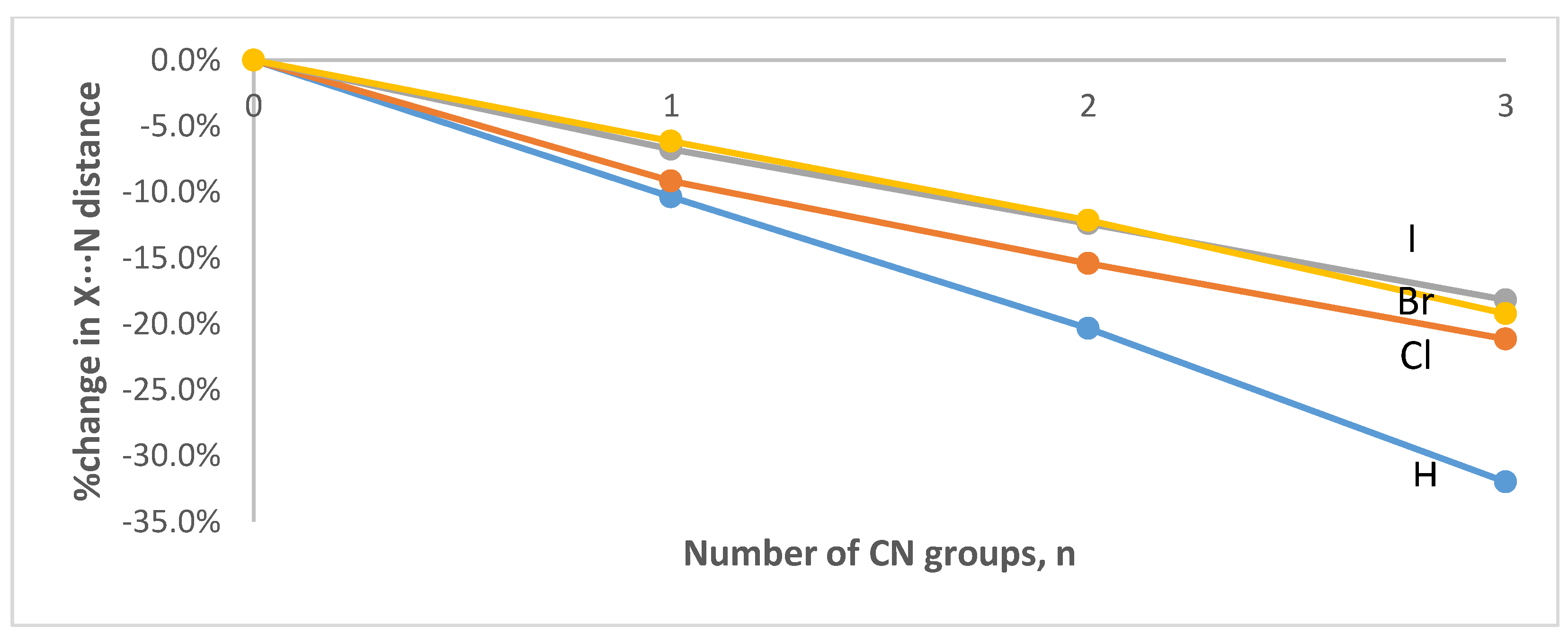
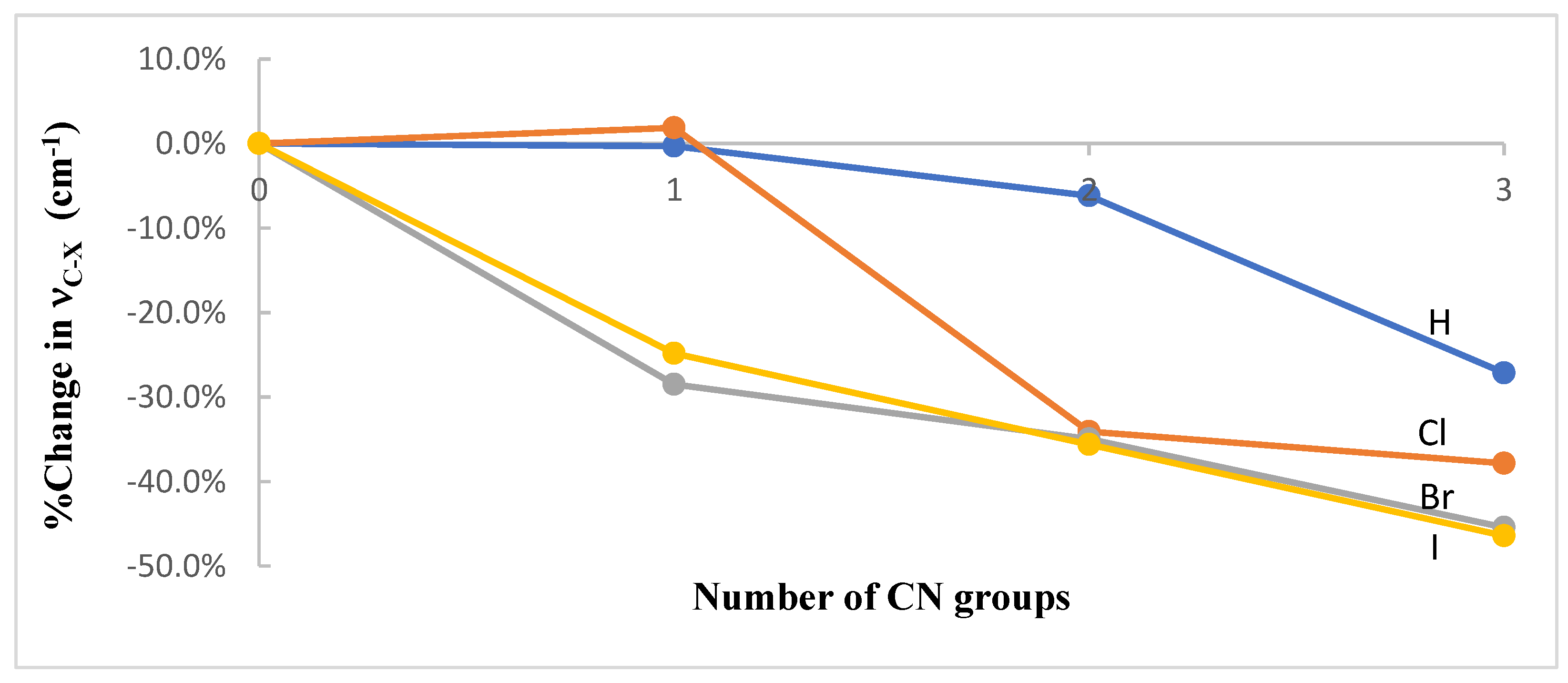
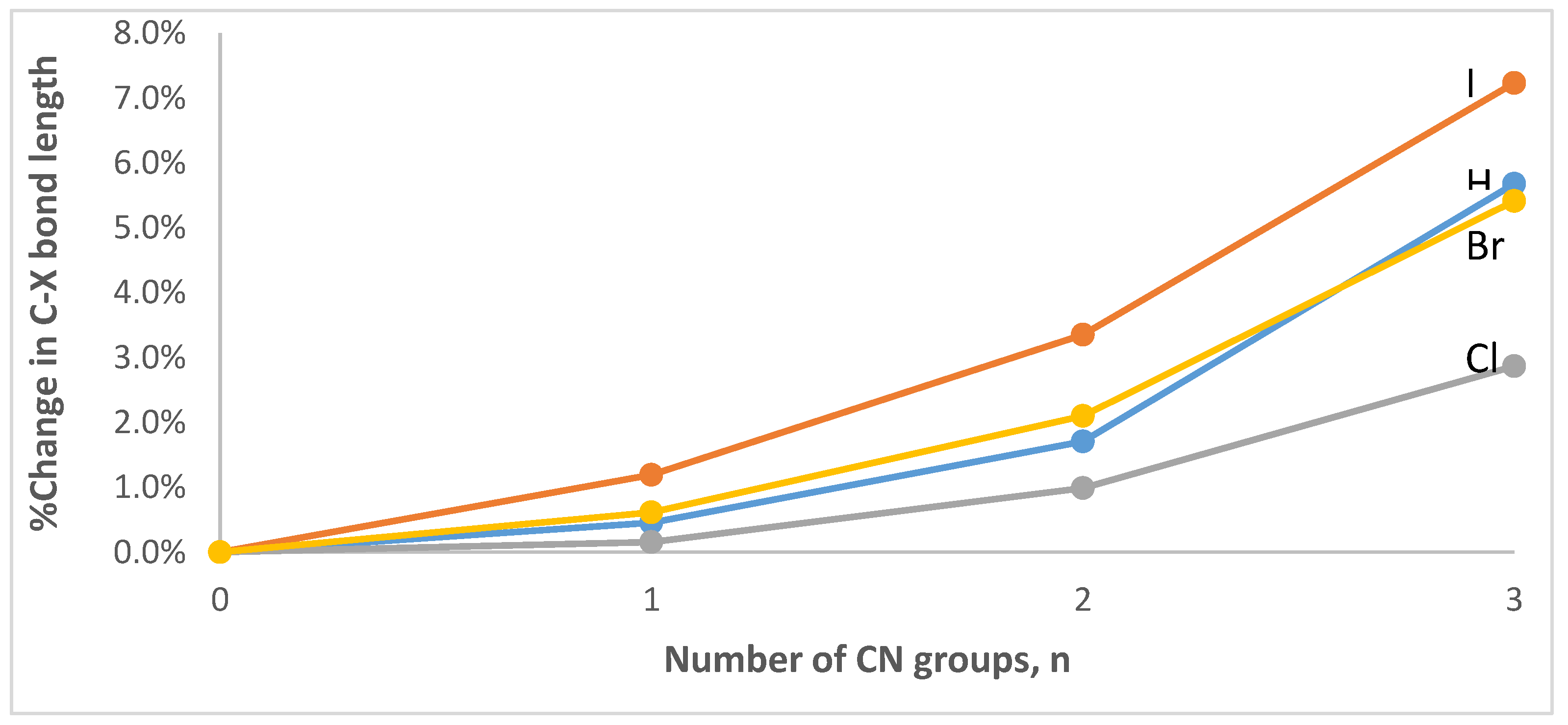
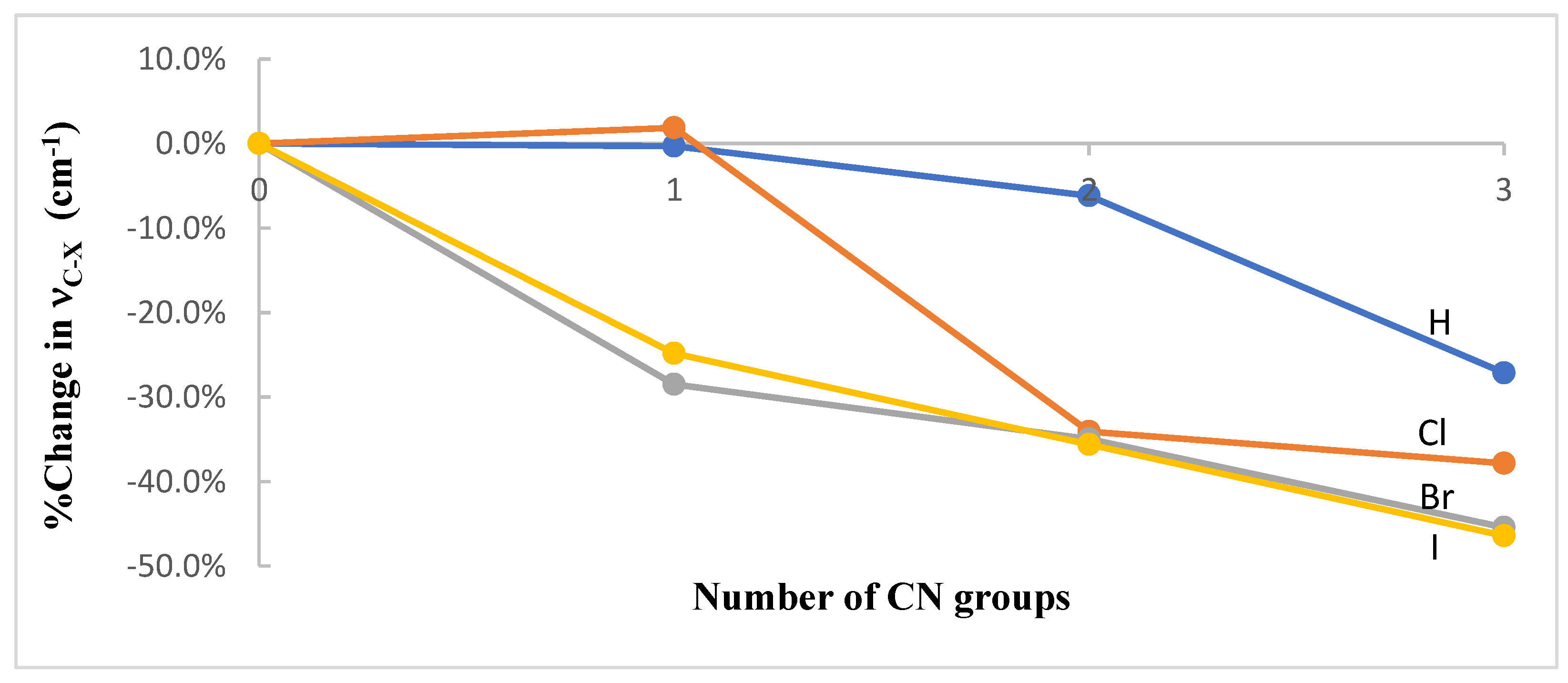
3.1. Geometries and Vibrational Frequencies
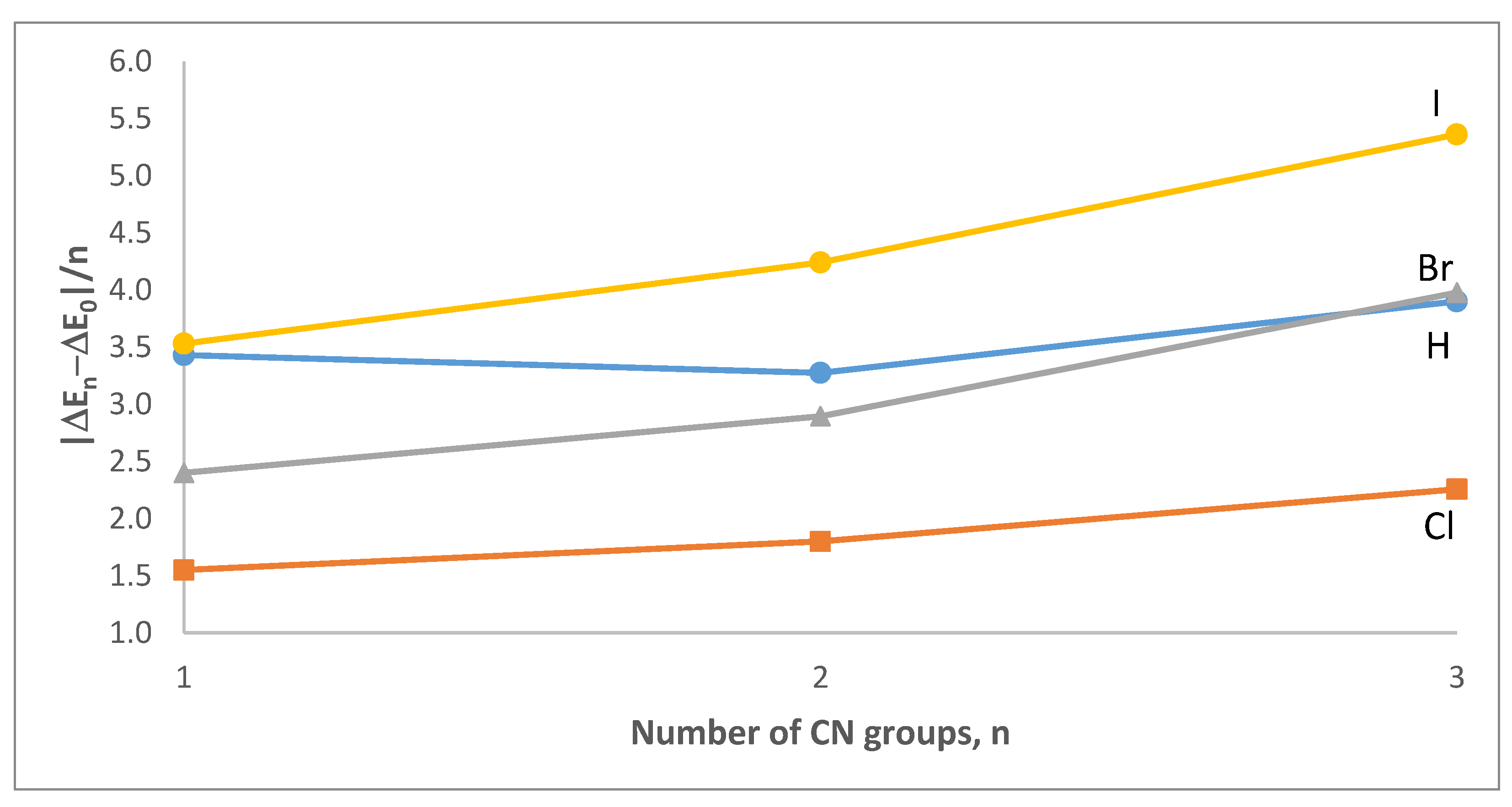
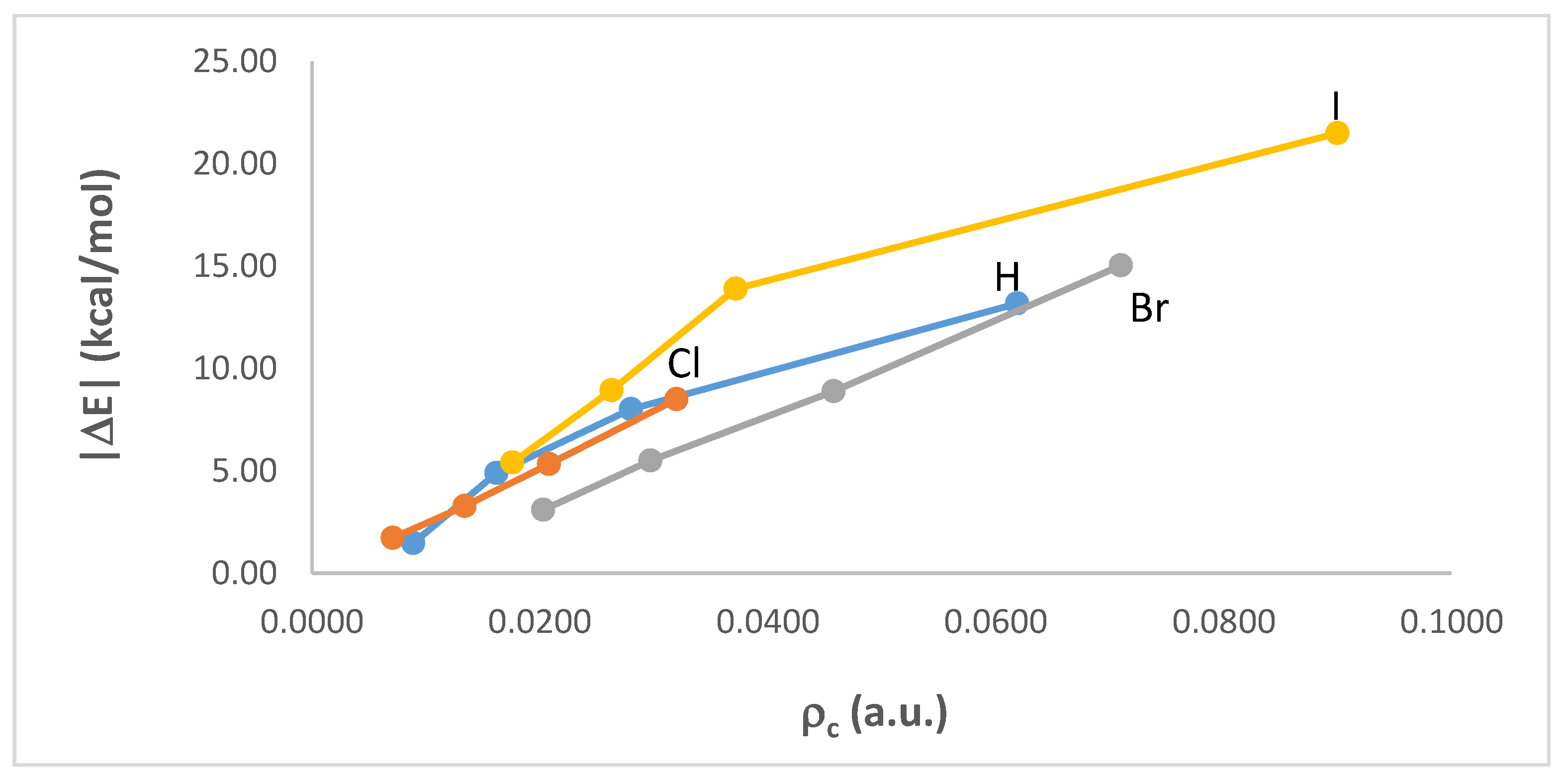
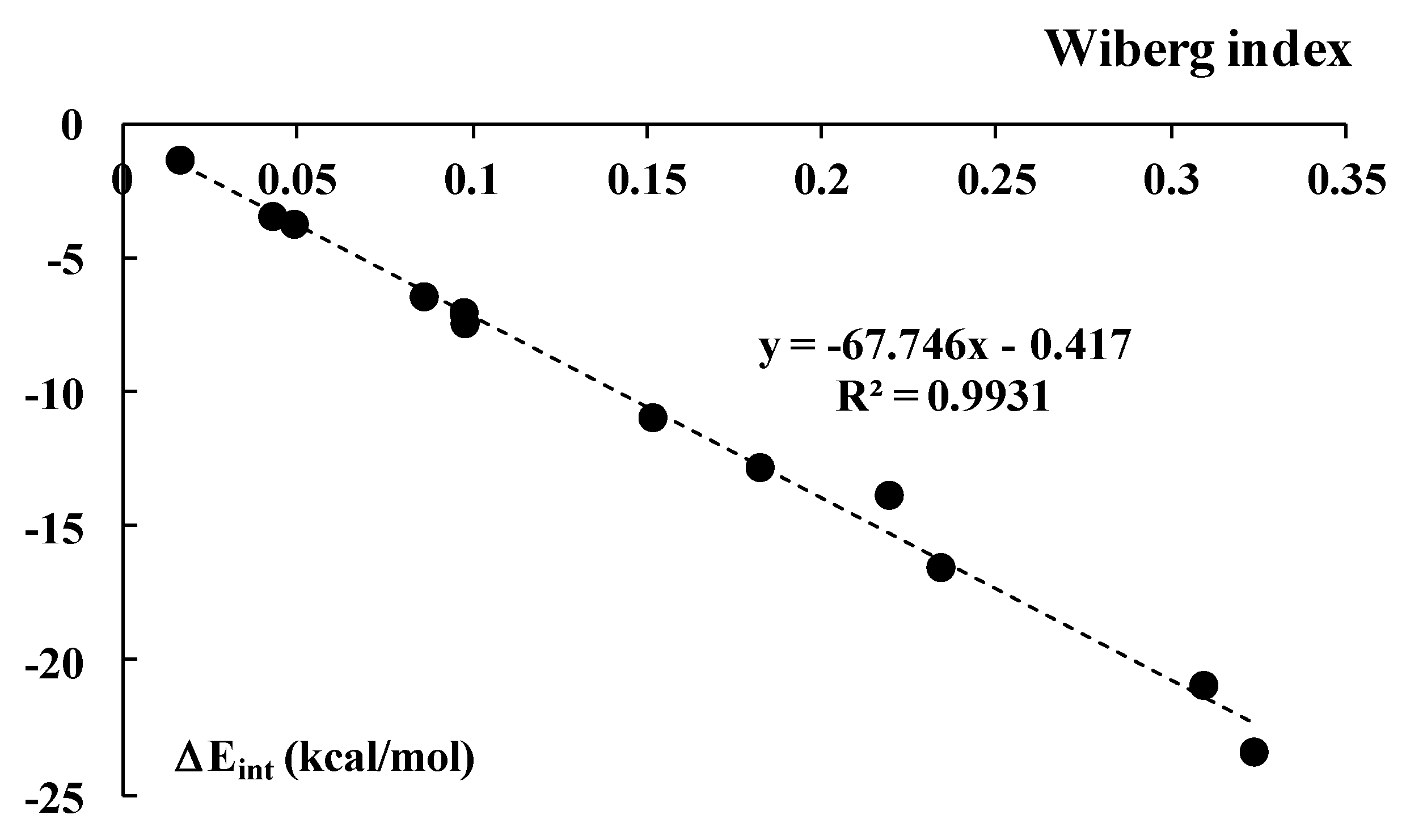
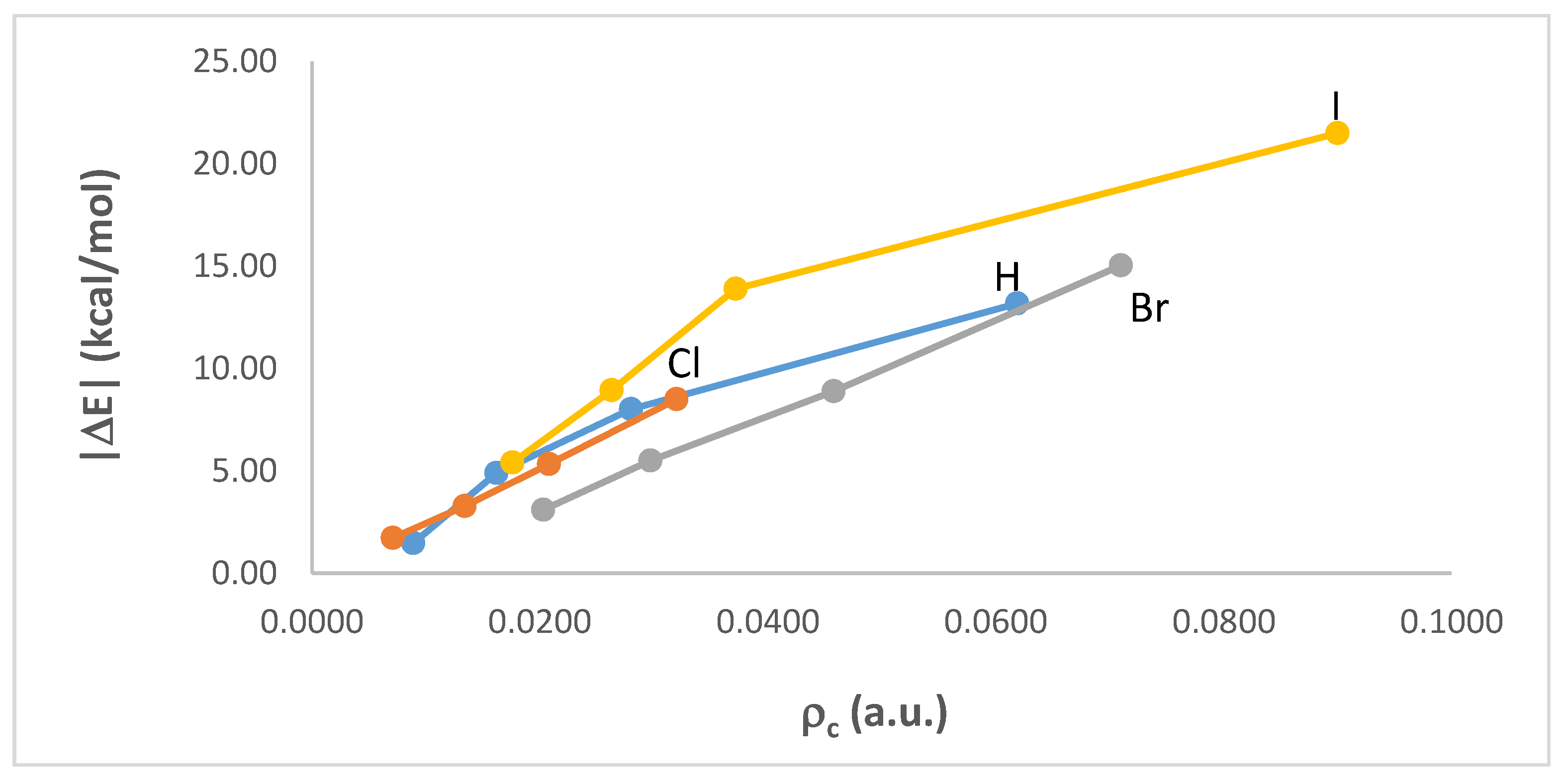
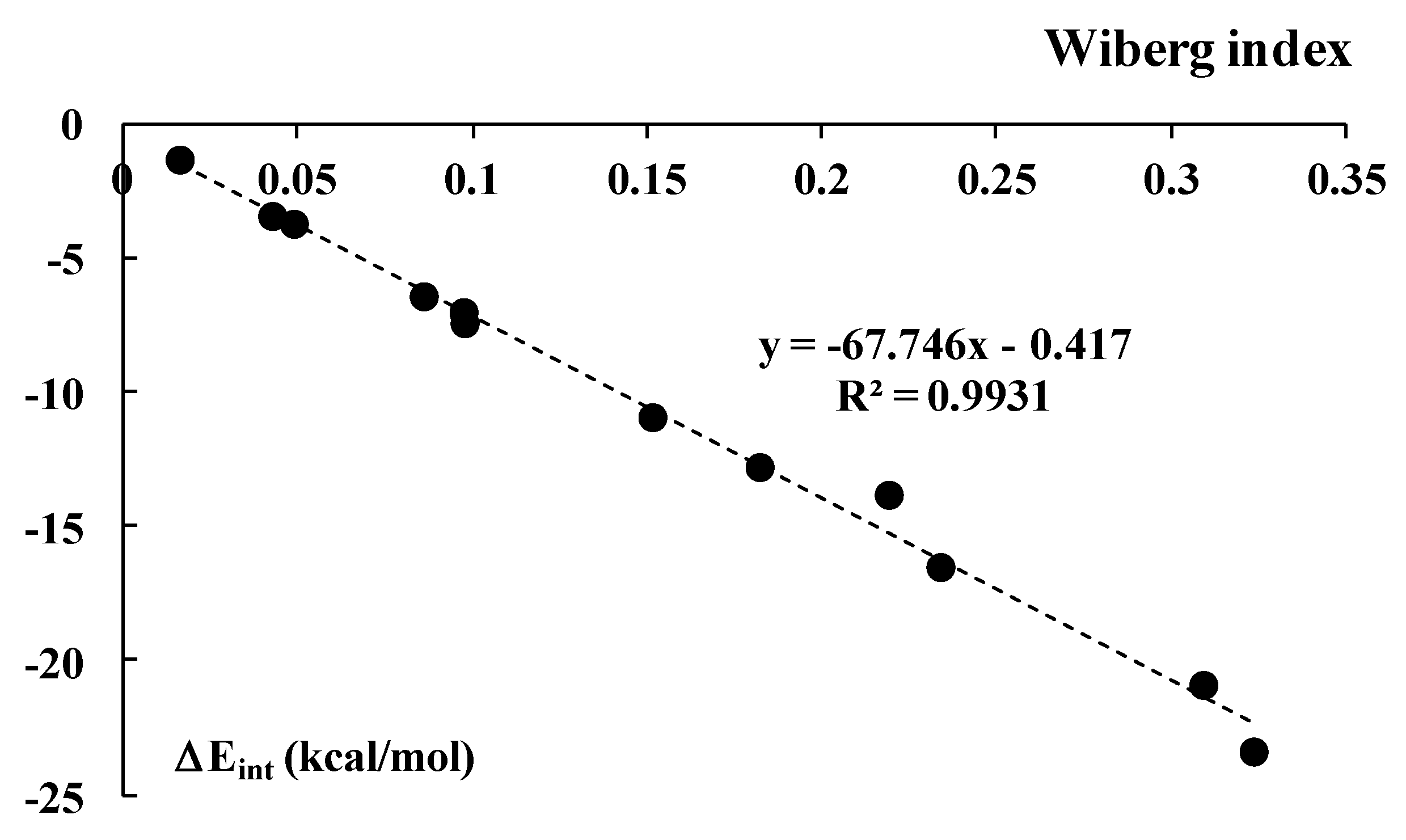
3.2. Intermolecular Interaction Energies and AIM Analyses
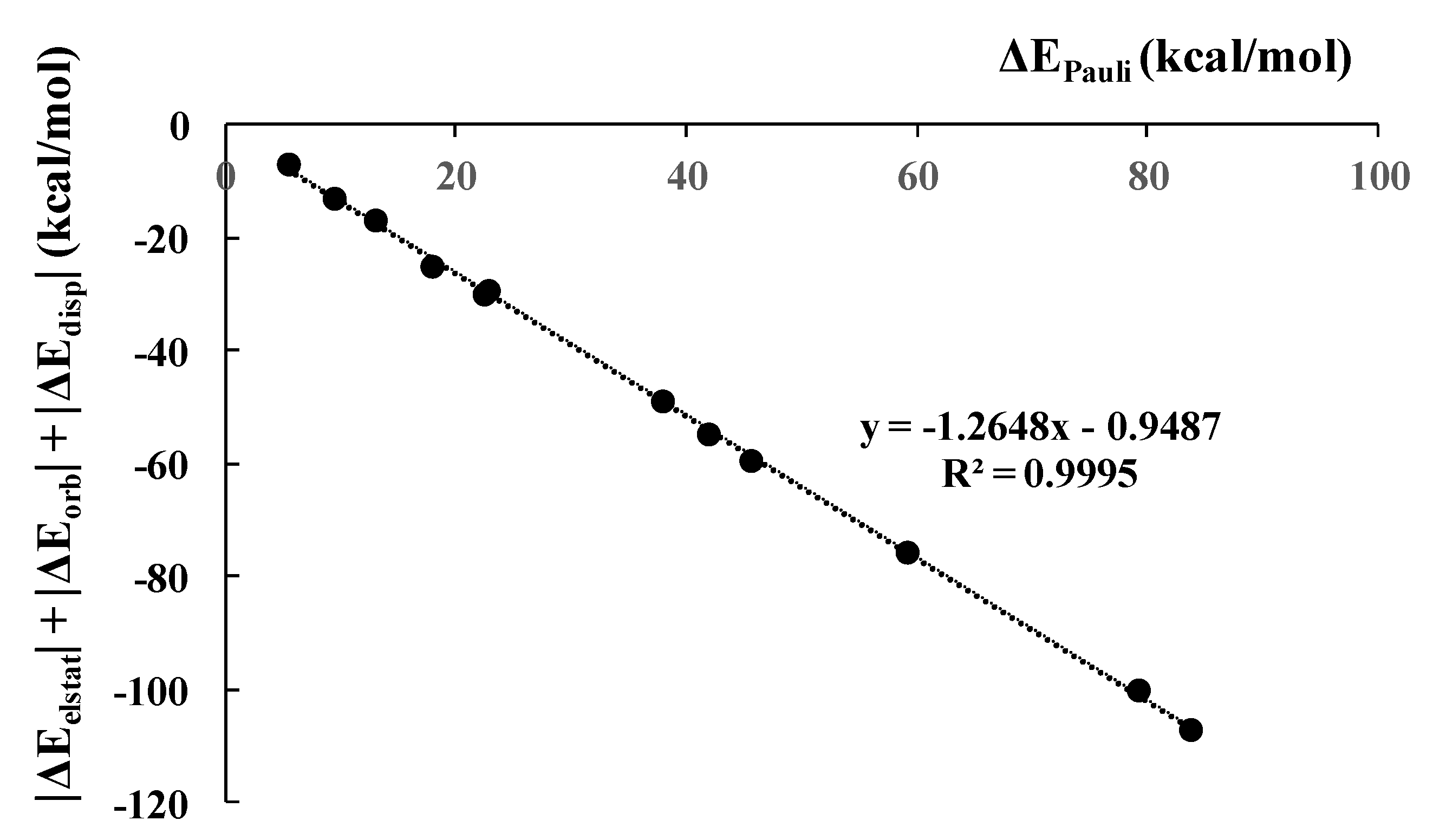
3.3. Energy Decomposition Analysis
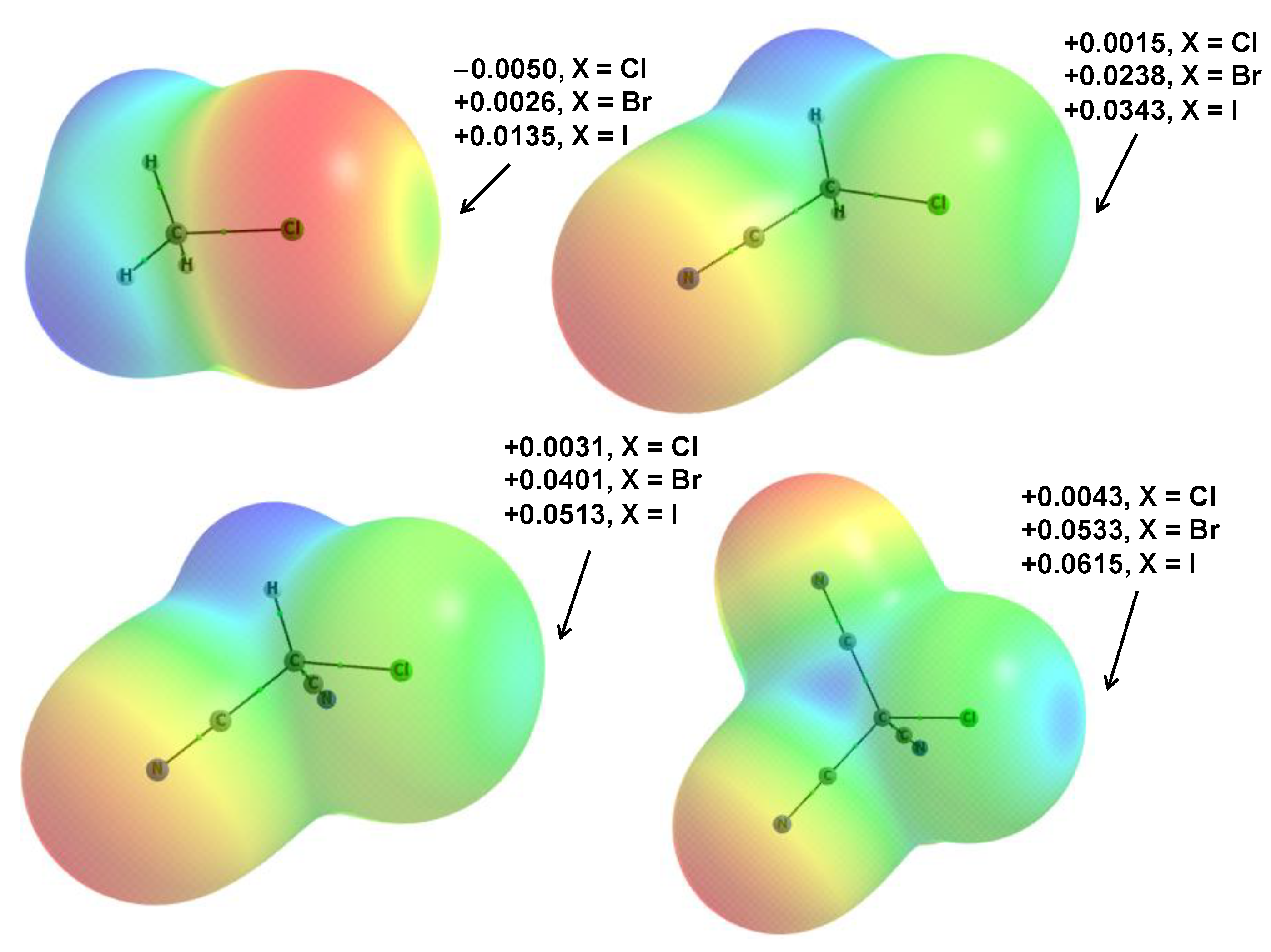
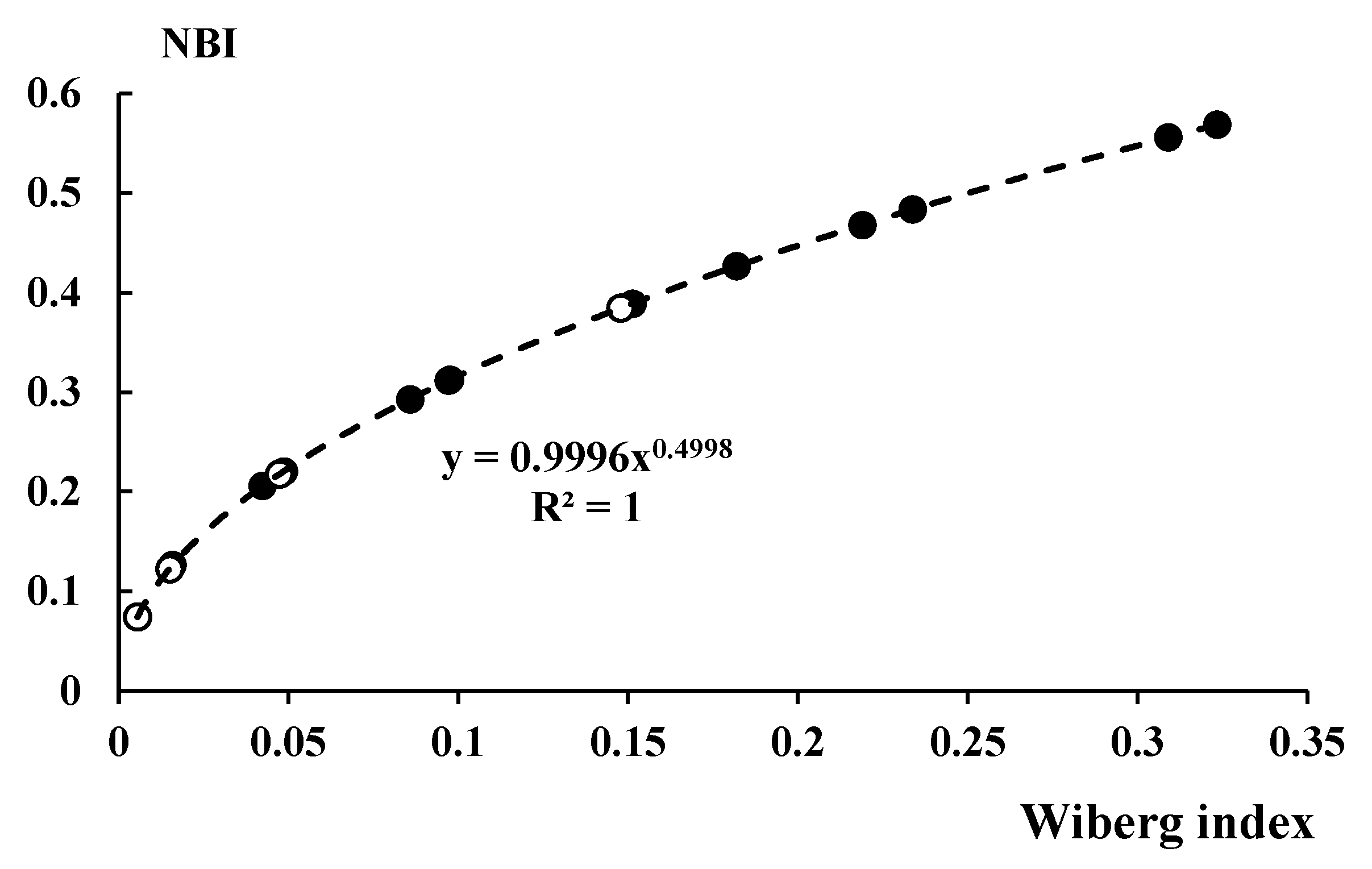
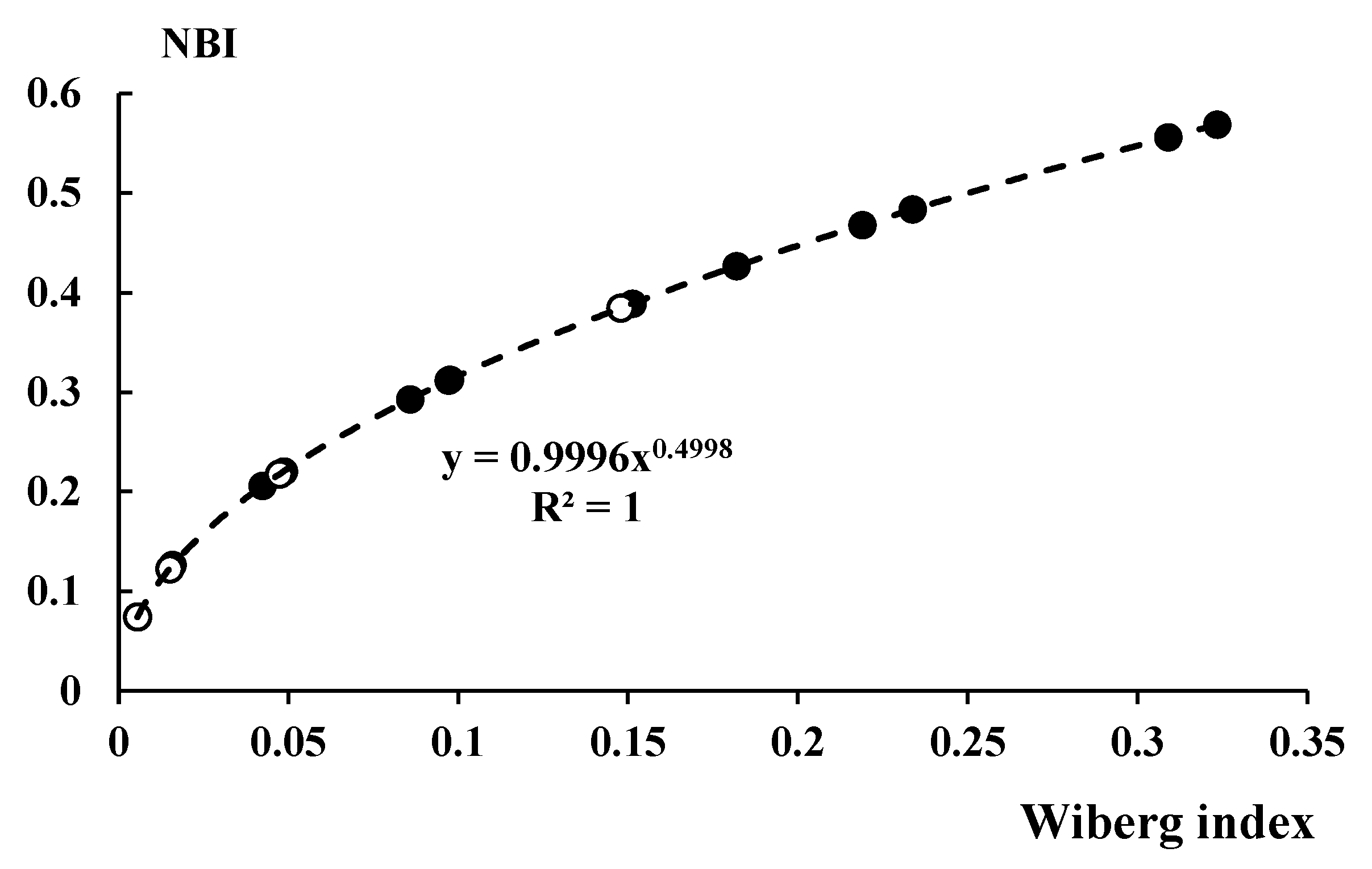
3.4. NBO Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Scheiner, S. New ideas from an Old Concept: The Hydrogen Bond. Biochem 2019, 4, 6–9. [Google Scholar] [CrossRef]
- Scheiner, S. The Hydrogen Bond: A Hundred Years and Counting. J. Indian Inst. Sci. 2020, 100, 61–76. [Google Scholar] [CrossRef]
- Grabowski, S.J. Understanding Hydrogen Bonds: Theoretical and Experimental Views; The Royal Society of Chemistry: Cambridge, UK, 2021. [Google Scholar]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. The Halogen Bond: An Interim Perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Sahik, S. On the Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Costa, P.J. The Halogen Bond: Nature and Applications. Phys. Sci. Rev. 2017, 2, 2017-0136. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen Bond Tunability I: The Effects of Aromatic Fluorine Substitution on the Strengths of Halogen-Bonding Interactions Involving Chlorine, Bromine, and Iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef]
- Akorta, I.; Legon, A.C. Nucleophilicities of Lewis Bases B and Electrophilicities of Lewis Acids A Determined from the Dissociation Energies of Complexes B· · ·A Involving Hydrogen Bonds, Tetrel Bonds, Pnictogen Bonds, Chalcogen Bonds and Halogen Bonds. Molecules 2017, 10, 1786. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen Bonding Based Recognition Processes: A World Parallel to Hydrogen Bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and Halogen Bonds are Ruled by the Same Mechanisms. Phys.Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef]
- Walters, L.P.; Bickelhaupt, F.M. Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen Bonding vs. Halogen Bonding: The Solvent Decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef]
- Grabowski, S.J. QTAIM Characteristics of Halogen Bond and Related Interactions. J. Phys. Chem. A 2012, 116, 1838–1845. [Google Scholar] [CrossRef]
- Quiñonero, D.; Frontera, A. Hydrogen Bond versus Halogen Bond in HXOn (X = F, Cl, Br, and I) Complexes with Lewis Bases. Inorganics 2019, 7, 9. [Google Scholar] [CrossRef]
- Shields, Z.P.; Murray, J.S.; Politzer, P. Directional Tendencies of Halogen and Hydrogen Bonds. Int. J. Quantum Chem. 2010, 110, 2823–2832. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Arunan, E. Why Are Hydrogen Bonds Directional? J. Chem. Sci. 2016, 128, 1571–1577. [Google Scholar] [CrossRef]
- Saccone, M.; Cavallo, G.; Metrangolo, P.; Pace, A.; Pibiri, I.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bond Directionality Translates Tecton Geometry into Self-Assembled Architecture Geometry. CrystEngComm 2013, 15, 3102–3105. [Google Scholar] [CrossRef]
- Riley, K.E. Critical Comparison of R-X∙∙∙Y and R-H∙∙∙Y Directionality in Halogen and Hydrogen Bonds using Modern Computational Chemistry Methods. Chem. Phys. Lett. 2020, 744, 137221. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Frontera, A. Substituent Effects in Multivalent Halogen Bonding Complexes: A Combined Theoretical and Crystallographic Study. Molecules 2018, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Macaveiua, L.; Politzer, P. Factors Affecting the Strengths of σ-Hole Electrostatic Potentials. J. Chem. Theory Comput. 2014, 5, 590–596. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Hasb, A.A.M. Polarization Plays the Key Role in Halogen Bonding: A Point-of-Charge-Based Quantum Mechanical Study. Theor. Chem. Acc. 2019, 138, 1–12. [Google Scholar] [CrossRef]
- Brinck, T.; Borrfors, A.N. Electrostatics and Polarization Determine the Strength of the Halogen Bond: A Red card for Charge Transfer. J. Mol. Model. 2019, 25, 125. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The Intrinsic Strength of the Halogen Bond: Electrostatic and Covalent Contributions Described by Coupled Cluster Theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. The Relative Roles of Electrostatics and Dispersion in the Stabilization of Halogen Bonds. Phys. Chem. Chem. Phys. 2013, 15, 17742–17751. [Google Scholar] [CrossRef]
- Orlova, A.P.; Jasien, P.G. Halogen Bonding in Self-Assembling Systems: A Comparison of Intra- and Interchain Binding Energies. Comput. Theor. Chem. 2018, 1139, 63–69. [Google Scholar] [CrossRef]
- Nepal, B.; Scheiner, S. NX···Y Halogen Bonds. Comparison with NH···Y H-Bonds and CX···Y Halogen Bonds. Phys. Chem. Chem. Phys. 2016, 18, 18015–18023. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Murray, J.S.; Politzer, P.; Concha, M.C.; Hobza, P. Br···O Complexes as Probes of Factors Affecting Halogen Bonding: Interactions of Bromobenzenes and Bromopyrimidines with Acetone. J. Chem. Theory Comput. 2009, 5, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Zeng, Y.; Li, X.; Zheng, S.; Meng, L. Enhancing Effects of Electron-Withdrawing Groups and Metallic Ions on Halogen Bonding in the YC6F4X···C2H8N2 (X = Cl, Br, I., Y = F, CN, NO2, LiNC+, NaNC+) Complex. J. Phys. Chem. A 2013, 117, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimia, A.; Razmazma, H.; Delarami, H.S. The Nature of Halogen Bonds in [N∙∙∙X∙∙∙N]+ Complexes: A Theoretical Study. Phys. Chem. Res. 2016, 4, 1–15. [Google Scholar]
- Tian, W.; Huang, X.; Li, Q.; Li, W.; Cheng, J.; Gong, B. Effect of Superalkali Substituents on the Strengths and Properties of Hydrogen and Halogen bonds. J. Mol. Model. 2013, 19, 1311–1318. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, A.-Y.; Cao, L.-J. Electronic Properties of the Halogen Bonds Z3CX···Y- Between Halide Anions and Methyl Halides. Struct. Chem. 2012, 23, 627–636. [Google Scholar] [CrossRef]
- Franchini, D.; Forni, A.; Genoni, A.; Pieraccini, S.; Gandini, E.; Sironi, M. The Origin of the σ-Hole in Halogen Atoms: A Valence Bond Perspective. ChemistryOpen 2020, 9, 445–450. [Google Scholar] [CrossRef]
- Scheiner, S.; Grabowski, S.J.; Kar, T. Influence of Hybridization and Substitution upon the Properties of the CH··O Hydrogen Bond. J. Phys. Chem. A 2001, 105, 10607–10612. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Rege, P.D.; Malkina, O.L.; Goroff, N.S. The Effect of Lewis Bases on the 13C NMR of Iodoalkynes. J. Am. Chem. Soc. 2002, 124, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Perkins, C.; Libri, S.; Adams, H.; Brammer, L. Diiodoacetylene: Compact, Strong Ditopic Halogen Bond Donor. CrystEngComm 2012, 14, 3033–3038. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Yurieva, A.G.; Poleshchuk, O.K.; Filimonov, V.D. Comparative Analysis of a Full-Electron Basis Set and Pseudopotential for the Iodine Atom in DFT Quantum-Chemical Calculations of Iodine-Containing Compounds. J. Struct. Chem. 2008, 49, 548–552. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. 3. The Atoms Aluminum through Argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron through Neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on the Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-D Group 16−18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Todd, A.; Keith, T.K. AIMAll (Version 11.08.23); Overland Park, KS, USA. Gristmill Software. 2011. Available online: aim.tkgristmill.com (accessed on 22 September 2022).
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Watts, J.D.; Barlett, R.J. Triple Excitations in Coupled-Cluster Theory: Energies and Analytical Derivatives. Int. J. Quantum Chem. 1993, 48, 51–66. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Baseggio, O.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; et al. ADF2019, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands; Available online: http://www.scm.com (accessed on 22 September 2022).
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3, and CNCH3 as σ Donors and π Acceptors. A Theoretical Study by the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Velde, G.T.E.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type Basis Sets for the Elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Reed, E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C. Valency and Bonding, A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Wiberg, K. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: https://nbo6.chem.wisc.edu/ (accessed on 22 September 2022).
- Zou, J.-W.; Lu, Y.-X.; Yu, Q.-S.; Zhang, H.-X.; Jiang, Y.-J. Halogen Bonding: An AIM Analysis of the Weak Interactions. Chin. J. Chem. 2006, 24, 1709–1715. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the Evaluation of the Local Kinetic, Potential and Total Energy Densities in Closed-Shell Interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X-H∙∙∙F-Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 11, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys.Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Carrol, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Mayer, I. Bond Order and Valence Indices: A Personal Account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | C-H (Å) | H∙∙∙N (Å) | C-H∙∙∙N (°) |
|---|---|---|---|
| ωB97X-D/aug-cc-pVTZ | |||
| 0 | 1.089 | 2.595 | 151.2 |
| 1 | 1.094 | 2.326 | 151.0 |
| 2 | 1.108 | 2.067 | 157.2 |
| 3 | 1.151 | 1.765 | 180.0 |
| MP2/aug-cc-pVTZ | |||
| 0 | 1.088 | 2.569 | 155.8 |
| 1 | 1.092 | 2.370 | 145.1 |
| 2 | 1.106 | 2.066 | 153.2 |
| 3 | 1.159 | 1.711 | 180.0 |
| MP2/aug-cc-pVDZ | |||
| 0 | 1.099 | 2.522 | 158.3 |
| 1 | 1.104 | 2.292 | 151.8 |
| 2 | 1.118 | 2.043 | 154.5 |
| 3 | 1.172 | 1.693 | 180.0 |
| n | C-Cl (Å) | Cl∙∙∙N (Å) | C-Cl∙∙∙N (°) |
|---|---|---|---|
| ωB97X-D/aug-cc-pVTZ | |||
| 0 | 1.787 | 3.321 | 168.6 |
| 1 | 1.789 | 3.018 | 175.5 |
| 2 | 1.804 | 2.810 | 178.9 |
| 3 | 1.838 | 2.620 | 180.0 |
| MP2/aug-cc-pVTZ | |||
| 0 | 1.781 | 3.115 | 169.1 |
| 1 | 1.785 | 2.896 | 171.1 |
| 2 | 1.804 | 2.679 | 177.5 |
| 3 | 1.876 | 2.385 | 180.0 |
| MP2/aug-cc-pVDZ | |||
| 0 | 1.798 | 3.116 | 169.5 |
| 1 | 1.803 | 2.903 | 171.7 |
| 2 | 1.823 | 2.700 | 177.8 |
| 3 | 1.888 | 2.439 | 180.0 |
| n | C-Br (Å) | Br∙∙∙N (Å) | C-Br∙∙∙N (°) |
|---|---|---|---|
| ωB97X-D/aug-cc-pVTZ | |||
| 0 | 1.941 | 3.102 | 179.9 |
| 1 | 1.953 | 2.911 | 179.2 |
| 2 | 1.981 | 2.725 | 179.7 |
| 3 | 2.046 | 2.506 | 180.0 |
| MP2/aug-cc-pVTZ | |||
| 0 | 1.933 | 2.896 | 179.5 |
| 1 | 1.951 | 2.711 | 178.7 |
| 2 | 1.997 | 2.505 | 179.3 |
| 3 | 2.099 | 2.302 | 180.0 |
| MP2/aug-cc-pVDZ | |||
| 0 | 1.951 | 2.925 | 180.0 |
| 1 | 1.969 | 2.747 | 179.2 |
| 2 | 2.013 | 2.548 | 179.5 |
| 3 | 2.106 | 2.347 | 180.0 |
| n | C-I (Å) | I∙∙∙N (Å) | C-I∙∙∙N (°) |
|---|---|---|---|
| ωB97X-D/aug-cc-pVTZ | |||
| 0 | 2.145 | 3.099 | 179.9 |
| 1 | 2.171 | 2.890 | 180.0 |
| 2 | 2.217 | 2.715 | 180.0 |
| 3 | 2.300 | 2.536 | 180.0 |
| MP2/aug-cc-pVTZ | |||
| 0 | 2.138 | 2.875 | 179.9 |
| 1 | 2.170 | 2.697 | 179.7 |
| 2 | 2.222 | 2.543 | 179.5 |
| 3 | 2.292 | 2.424 | 180.0 |
| MP2/aug-cc-pVDZ | |||
| 0 | 2.164 | 2.889 | 179.9 |
| 1 | 2.198 | 2.710 | 179.8 |
| 2 | 2.248 | 2.565 | 179.7 |
| 3 | 2.310 | 2.457 | 180.0 |
| n | νC-H | νC-Cl | νC-Br | νC-I |
|---|---|---|---|---|
| ωB97X-D/aug-cc-pVTZ | ||||
| 0 | 3033 | 748 | 632 | 556 |
| 1 | 3024 | 762 | 452 | 418 |
| 2 | 2846 | 493 | 411 | 358 |
| 3 | 2210 | 465 | 345 | 298 |
| MP2/aug-cc-pVDZ | ||||
| 0 | 3045 | 747 | 624 | 542 |
| 1 | 3034 | 747 | 427 | 389 |
| 2 | 2854 | 473 | 374 | 340 |
| 3 | 2160 | 387 | 308 | 302 |
| n | H | Cl | Br | I |
|---|---|---|---|---|
| 0 | −1.47 | −1.73 | −3.10 | −5.41 |
| 1 | −4.90 | −3.28 | −5.50 | −8.94 |
| 2 | −8.02 | −5.33 | −8.89 | −13.89 |
| 3 | −13.17 | −8.50 | −15.03 | −21.49 |
| n | H | I |
|---|---|---|
| 0 | −1.46 | −4.30 |
| 1 | −4.69 | −7.53 |
| 2 | −7.49 | −12.19 |
| 3 | −12.21 | −18.93 |
| n | ρc | Gc | Vc | Hc | |Gc/Vc| |
|---|---|---|---|---|---|
| CH3−n(CN)nH∙∙∙NMe3 | |||||
| 0 | 0.0089 | 0.0058 | −0.0048 | 0.0010 | 1.21 |
| 1 | 0.0162 | 0.0106 | −0.0095 | 0.0011 | 1.12 |
| 2 | 0.0280 | 0.0183 | −0.0198 | −0.0015 | 0.92 |
| 3 | 0.0550 | 0.0322 | −0.0485 | −0.0163 | 0.66 |
| CH3−n(CN)nCl∙∙∙NMe3 | |||||
| 0 | 0.0071 | 0.0049 | −0.0037 | 0.0012 | 1.32 |
| 1 | 0.0134 | 0.0097 | −0.0081 | 0.0016 | 1.20 |
| 2 | 0.0208 | 0.0153 | −0.0141 | 0.0012 | 1.09 |
| 3 | 0.0320 | 0.0230 | −0.0240 | −0.0010 | 0.96 |
| CH3−n(CN)nBr∙∙∙NMe3 | |||||
| 0 | 0.0137 | 0.0093 | −0.0081 | 0.0012 | 1.15 |
| 1 | 0.0200 | 0.0138 | −0.0130 | 0.0008 | 1.06 |
| 2 | 0.0296 | 0.0200 | −0.0209 | −0.0009 | 0.96 |
| 3 | 0.0473 | 0.0302 | −0.0372 | −0.0070 | 0.81 |
| CH3−n(CN)nI∙∙∙NMe3 | |||||
| 0 | 0.0176 | 0.0110 | −0.0104 | 0.0006 | 1.06 |
| 1 | 0.0263 | 0.0166 | −0.0175 | −0.0009 | 0.95 |
| 2 | 0.0372 | 0.0231 | −0.0275 | −0.0044 | 0.84 |
| 3 | 0.0900 | 0.0407 | −0.0786 | −0.0379 | 0.52 |
| CN Groups | H | Cl | Br | I |
|---|---|---|---|---|
| 1 | 0.0073 | 0.0063 | 0.0063 | 0.0087 |
| 2 | 0.0096 | 0.0069 | 0.0080 | 0.0098 |
| 3 | 0.0154 | 0.0083 | 0.0112 | 0.0241 |
| Lewis Acid Unit | ΔEint | ΔEPauli | ΔEelstat | ΔEorb | ΔEdisp | ΔEelstat/ΔEorb |
|---|---|---|---|---|---|---|
| CH3H | −1.76 | 3.56 | −1.91 | −1.12 | −2.29 | 1.7 |
| CH2CNH | −5.60 | 7.87 | −6.56 | −3.05 | −3.85 | 2.2 |
| CH(CN)2H | −9.52 | 14.13 | −11.47 | −7.22 | −4.96 | 1.6 |
| C(CN)3H | −17.09 | 33.66 | −23.78 | −21.54 | −5.44 | 1.1 |
| CH3Cl | −1.31 | 5.41 | −2.65 | −1.81 | −2.26 | 1.5 |
| CH2CNCl | −3.41 | 9.38 | −6.21 | −4.12 | −2.47 | 1.5 |
| CH(CN)2Cl | −6.99 | 17.86 | −12.42 | −9.75 | −2.68 | 1.3 |
| C(CN)3Cl | −13.79 | 45.54 | −28.65 | −27.26 | −3.41 | 1.1 |
| CH3Br | −3.70 | 12.95 | −8.76 | −5.00 | −2.89 | 1.8 |
| CH2CNBr | −7.40 | 22.39 | −15.97 | −10.41 | −3.42 | 1.5 |
| CH(CN)2Br | −12.76 | 41.85 | −28.96 | −21.75 | −3.91 | 1.3 |
| C(CN)3Br | −20.87 | 79.17 | −51.76 | −44.13 | −4.14 | 1.2 |
| CH3I | −6.40 | 22.76 | −16.37 | −8.74 | −4.05 | 1.9 |
| CH2CNHI | −10.90 | 37.87 | −27.52 | −16.75 | −4.49 | 1.6 |
| CH(CN)2I | −16.48 | 59.10 | −42.28 | −28.61 | −4.69 | 1.5 |
| C(CN)3I | −23.35 | 83.70 | −58.90 | −43.38 | −4.77 | 1.4 |
| Lewis Acid Unit | Q(X) | Q(N) | WBI | NBI | n→σ* |
|---|---|---|---|---|---|
| CH3H | 0.230 | −0.348 | 0.0055 | 0.0743 | 0.69 |
| CH2CNH | 0.283 | −0.363 | 0.0151 | 0.1227 | 2.49 |
| CH(CN)2H | 0.320 | −0.367 | 0.0474 | 0.2177 | 8.07 |
| C(CN)3H | 0.344 | −0.344 | 0.1479 | 0.3845 | 29.15 |
| CH3Cl | −0.082 | −0.339 | 0.0159 | 0.1263 | 0.89 |
| CH2CNCl | −0.026 | −0.336 | 0.0424 | 0.2059 | 2.22 |
| CH(CN)2Cl | 0.004 | −0.315 | 0.0972 | 0.3118 | 5.02 |
| C(CN)3Cl | −0.012 | −0.260 | 0.2190 | 0.4680 | 13.65 |
| CH3Br | −0.039 | −0.335 | 0.0486 | 0.2204 | 3.78 |
| CH2CNBr | 0.013 | −0.325 | 0.0975 | 0.3123 | 6.99 |
| CH(CN)2Br | 0.040 | −0.296 | 0.1820 | 0.4267 | 13.55 |
| C(CN)3Br | 0.059 | −0.250 | 0.3091 | 0.5560 | 27.98 |
| CH3I | 0.039 | −0.337 | 0.0858 | 0.2929 | 6.51 |
| CH2CNHI | 0.097 | −0.326 | 0.1513 | 0.3890 | 11.84 |
| CH(CN)2I | 0.143 | −0.308 | 0.2338 | 0.4835 | 19.74 |
| C(CN)3I | 0.187 | −0.290 | 0.3235 | 0.5688 | 30.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra, R.D.; Grabowski, S.J. Enhancing Effects of the Cyano Group on the C-X∙∙∙N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX∙∙∙NMe3, (n = 0–3; X = H, Cl, Br, I). Int. J. Mol. Sci. 2022, 23, 11289. https://doi.org/10.3390/ijms231911289
Parra RD, Grabowski SJ. Enhancing Effects of the Cyano Group on the C-X∙∙∙N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX∙∙∙NMe3, (n = 0–3; X = H, Cl, Br, I). International Journal of Molecular Sciences. 2022; 23(19):11289. https://doi.org/10.3390/ijms231911289
Chicago/Turabian StyleParra, Rubén D., and Sławomir J. Grabowski. 2022. "Enhancing Effects of the Cyano Group on the C-X∙∙∙N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX∙∙∙NMe3, (n = 0–3; X = H, Cl, Br, I)" International Journal of Molecular Sciences 23, no. 19: 11289. https://doi.org/10.3390/ijms231911289
APA StyleParra, R. D., & Grabowski, S. J. (2022). Enhancing Effects of the Cyano Group on the C-X∙∙∙N Hydrogen or Halogen Bond in Complexes of X-Cyanomethanes with Trimethyl Amine: CH3−n(CN)nX∙∙∙NMe3, (n = 0–3; X = H, Cl, Br, I). International Journal of Molecular Sciences, 23(19), 11289. https://doi.org/10.3390/ijms231911289