
O-GlcNAcylation in Renal (Patho)Physiology

 ,
,  , , and
, , and 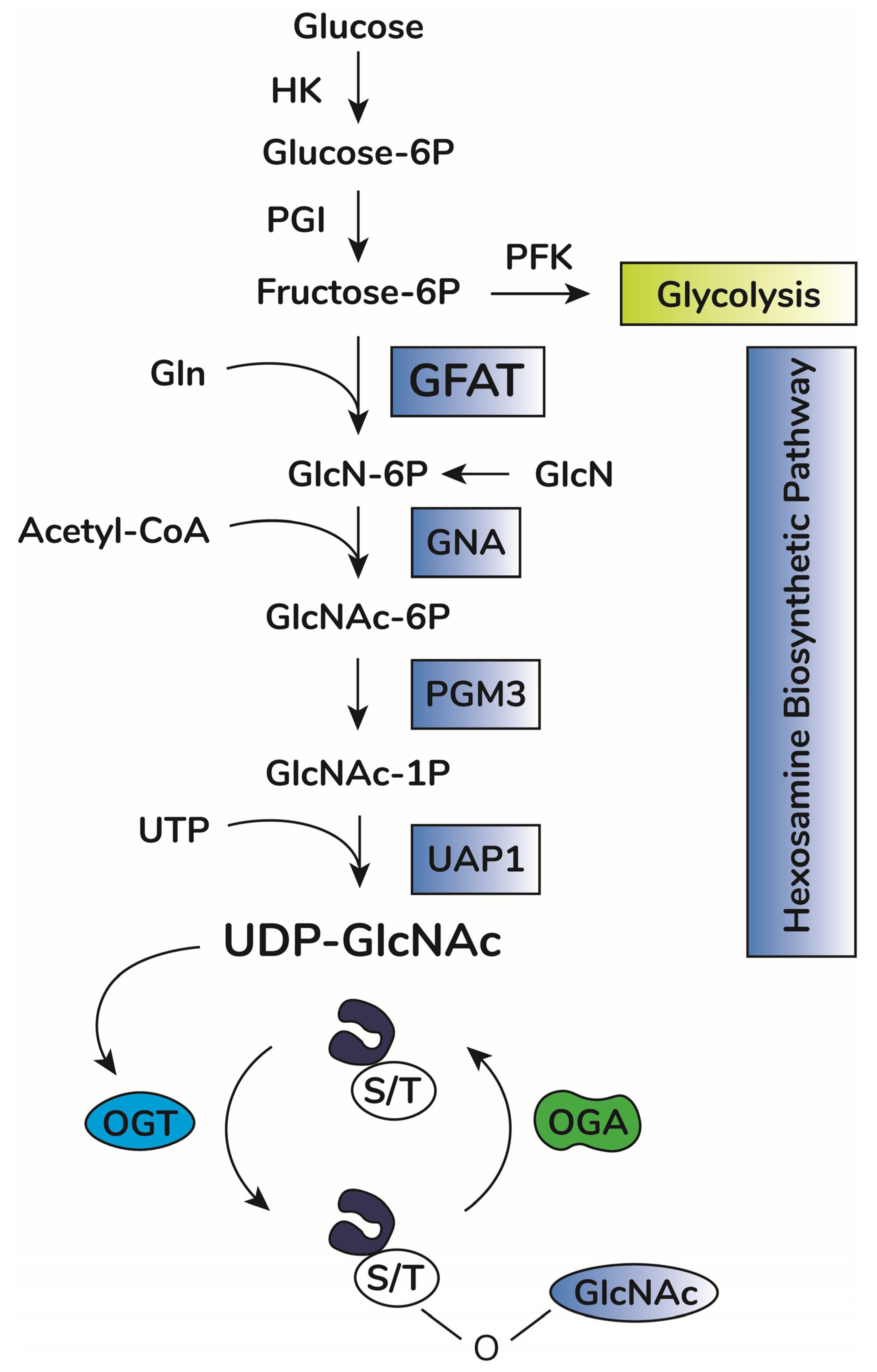{kind=link}
{kind=link}
Abstract
1. Introduction
2. O-GlcNAcylation
3. O-GlcNAcylation in Renal Physiology
4. O-GlcNAcylation in Renal Pathophysiology
4.1. Kidney Diseases Classification
4.2. Short Sweetness Seems Good–Renal Protective Effects of Acute Elevations in O-GlcNAcylation
4.3. Too Sweet to Handle–Prolonged Changes in O-GlcNAcylation Are Involved in Development of Kidney Diseases
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoenig, M.P.; Zeidel, M.L. Homeostasis, the milieu intérieur, and the wisdom of the nephron. Clin. J. Am. Soc. Nephrol. 2014, 9, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. Suppl. 2011, 79, S1–S6. [Google Scholar] [CrossRef]
- Bröer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008, 88, 249–286. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Legouis, D.; Faivre, A.; Cippà, P.E.; de Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant. 2020, 28, gfaa302. [Google Scholar] [CrossRef]
- Shih, H.M.; Wu, C.J.; Lin, S.L. Physiology and pathophysiology of renal erythropoietin-producing cells. J. Formos. Med. Assoc. 2018, 117, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Okusa, M.D. Crosstalk between the nervous system and the kidney. Kidney Int. 2020, 97, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Noel, S.; Pluznick, J.L.; Hamad, A.R.A.; Rabb, H. Gut Microbiota-Kidney Cross-Talk in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 107–116. [Google Scholar] [CrossRef]
- Virzì, G.; Day, S.; de Cal, M.; Vescovo, G.; Ronco, C. Heart-kidney crosstalk and role of humoral signaling in critical illness. Crit. Care 2014, 18, 201. [Google Scholar] [CrossRef]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Elshahat, S.; Cockwell, P.; Maxwell, A.P.; Griffin, M.; O’Brien, T.; O’Neill, C. The impact of chronic kidney disease on developed countries from a health economics perspective: A systematic scoping review. PLoS ONE 2020, 15, e0230512. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.L.; Schlackow, I.; Gray, A.; Emberson, J.; Herrington, W.; Staplin, N.; Reith, C.; Howard, K.; Landray, M.J.; Cass, A.; et al. Impact of CKD on Household Income. Kidney Int. Rep. 2017, 3, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Blantz, R.C. Glomerulotubular balance, tubuloglomerular feedback, and salt homeostasis. J. Am. Soc. Nephrol. 2008, 19, 2272–2275. [Google Scholar] [CrossRef]
- Silva-Aguiar, R.P.; Peruchetti, D.B.; Florentino, L.S.; Takiya, C.M.; Marzolo, M.P.; Dias, W.B.; Pinheiro, A.A.S.; Caruso-Neves, C. Albumin Expands Albumin Reabsorption Capacity in Proximal Tubule Epithelial Cells through a Positive Feedback Loop between AKT and Megalin. Int. J. Mol. Sci. 2022, 23, 848. [Google Scholar] [CrossRef]
- Wagner, M.C.; Campos-Bilderback, S.B.; Chowdhury, M.; Flores, B.; Lai, X.; Myslinski, J.; Pandit, S.; Sandoval, R.M.; Wean, S.E.; Wei, Y.; et al. Proximal Tubules Have the Capacity to Regulate Uptake of Albumin. J. Am. Soc. Nephrol. 2016, 27, 482–494. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Millar, A.H.; Heazlewood, J.L.; Giglione, C.; Holdsworth, M.J.; Bachmair, A.; Schulze, W.X. The Scope, Functions, and Dynamics of Posttranslational Protein Modifications. Annu. Rev. Plant. Biol. 2019, 70, 119–151. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell. Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Facundo, H.T.; Zafir, A.; Jones, S.P. O-GlcNAc signaling in the cardiovascular system. Circ. Res. 2010, 107, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.N.; Collins, H.E.; Wende, A.R.; Chatham, J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017, 45, 545–553. [Google Scholar] [CrossRef]
- Ma, X.; Li, H.; He, Y.; Hao, J. The emerging link between O-GlcNAcylation and neurological disorders. Cell. Mol. Life Sci. 2017, 74, 3667–3686. [Google Scholar] [CrossRef]
- Akan, I.; Olivier-Van Stichelen, S.; Bond, M.R.; Hanover, J.A. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2018, 144, 7–34. [Google Scholar] [CrossRef]
- Baudoin, L.; Issad, T. O-GlcNAcylation and Inflammation: A Vast Territory to Explore. Front. Endocrinol. 2015, 5, 235. [Google Scholar] [CrossRef]
- Chang, Y.H.; Weng, C.L.; Lin, K.I. O-GlcNAcylation and its role in the immune system. J. Biomed. Sci. 2020, 27, 57. [Google Scholar] [CrossRef]
- Biwi, J.; Biot, C.; Guerardel, Y.; Vercoutter-Edouart, A.S.; Lefebvre, T. The Many Ways by Which O-GlcNAcylation May Orchestrate the Diversity of Complex Glycosylations. Molecules 2018, 23, 2858. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Konzman, D.; Abramowitz, L.K.; Steenackers, A.; Mukherjee, M.M.; Na, H.J.; Hanover, J.A. O-GlcNAc: Regulator of Signaling and Epigenetics Linked to X-linked Intellectual Disability. Front. Genet. 2020, 11, 605263. [Google Scholar] [CrossRef]
- Love, D.C.; Krause, M.W.; Hanover, J.A. O-GlcNAc cycling: Emerging roles in development and epigenetics. Semin. Cell Dev. Biol. 2010, 21, 646–654. [Google Scholar] [CrossRef]
- Roth, C.; Chan, S.; Offen, W.A.; Hemsworth, G.R.; Willems, L.I.; King, D.T.; Varghese, V.; Britton, R.; Vocadlo, D.J.; Davies, G.J. Structural and functional insight into human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 610–612. [Google Scholar] [CrossRef]
- Meek, R.W.; Blaza, J.N.; Busmann, J.A.; Alteen, M.G.; Vocadlo, D.J.; Davies, G.J. Cryo-EM structure provides insights into the dimer arrangement of the O-linked β-N-acetylglucosamine transferase OGT. Nat. Commun. 2021, 12, 6508. [Google Scholar] [CrossRef]
- Webster, D.M.; Teo, C.F.; Sun, Y.; Wloga, D.; Gay, S.; Klonowski, K.D.; Wells, L.; Dougan, S.T. O-GlcNAc modifications regulate cell survival and epiboly during zebrafish development. BMC Dev. Biol. 2009, 9, 28. [Google Scholar] [CrossRef]
- O’Donnell, N.; Zachara, N.E.; Hart, G.W.; Marth, J.D. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol. 2004, 24, 1680–1690. [Google Scholar] [CrossRef]
- Muha, V.; Authier, F.; Szoke-Kovacs, Z.; Johnson, S.; Gallagher, J.; McNeilly, A.; McCrimmon, R.J.; Teboul, L.; van Aalten, D.M.F. Loss of O-GlcNAcase catalytic activity leads to defects in mouse embryogenesis. J. Biol. Chem. 2021, 296, 100439. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Ong, Q.; Han, W.; Yang, X. O-GlcNAc as an Integrator of Signaling Pathways. Front. Endocrinol. 2018, 9, 599. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef]
- Liu, J.; Pang, Y.; Chang, T.; Bounelis, P.; Chatham, J.C.; Marchase, R.B. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J. Mol. Cell. Cardiol. 2006, 40, 303–312. [Google Scholar] [CrossRef]
- Walter, L.A.; Lin, Y.H.; Halbrook, C.J.; Chuh, K.N.; He, L.; Pedowitz, N.J.; Batt, A.R.; Brennan, C.K.; Stiles, B.L.; Lyssiotis, C.A.; et al. Inhibiting the Hexosamine Biosynthetic Pathway Lowers O-GlcNAcylation Levels and Sensitizes Cancer to Environmental Stress. Biochemistry 2020, 59, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Pan, X.; Shi, M.; Wu, Q.; Huang, T.; Jiang, H.; Li, W.; Zhang, J. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis. 2018, 9, 485. [Google Scholar] [CrossRef]
- Vasconcelos-Dos-Santos, A.; Loponte, H.F.; Mantuano, N.R.; Oliveira, I.A.; de Paula, I.F.; Teixeira, L.K.; de-Freitas-Junior, J.C.; Gondim, K.C.; Heise, N.; Mohana-Borges, R.; et al. Hyperglycemia exacerbates colon cancer malignancy through hexosamine biosynthetic pathway. Oncogenesis 2017, 6, e306. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H.; Virkamäki, A.; Daniels, M.C.; McClain, D.; Gottschalk, W.K. Insulin and glucosamine infusions increase O-linked N-acetyl-glucosamine in skeletal muscle proteins in vivo. Metabolism 1998, 47, 449–455. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Champattanachai, V.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-κB signaling. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H515–H523. [Google Scholar] [CrossRef]
- Jiménez-Castillo, V.; Illescas-Barbosa, D.; Zenteno, E.; Ávila-Curiel, B.X.; Castañeda-Patlán, M.C.; Robles-Flores, M.; De Oca, D.M.; Pérez-Campos, E.; Torres-Rivera, A.; Bouaboud, A.; et al. Increased O-GlcNAcylation promotes IGF-1 receptor/PhosphatidyI Inositol-3 kinase/Akt pathway in cervical cancer cells. Sci. Rep. 2022, 12, 4464. [Google Scholar] [CrossRef] [PubMed]
- Hardivillé, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Steenackers, A.; Olivier-Van Stichelen, S.; Baldini, S.F.; Dehennaut, V.; Toillon, R.A.; Le Bourhis, X.; El Yazidi-Belkoura, I.; Lefebvre, T. Silencing the Nucleocytoplasmic O-GlcNAc Transferase Reduces Proliferation, Adhesion, and Migration of Cancer and Fetal Human Colon Cell Lines. Front. Endocrinol. 2016, 7, 46. [Google Scholar] [CrossRef]
- Andres, L.M.; Blong, I.W.; Evans, A.C.; Rumachik, N.G.; Yamaguchi, T.; Pham, N.D.; Thompson, P.; Kohler, J.J.; Bertozzi, C.R. Chemical Modulation of Protein O-GlcNAcylation via OGT Inhibition Promotes Human Neural Cell Differentiation. ACS Chem. Biol. 2017, 12, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Kneass, Z.T.; Marchase, R.B. Protein O-GlcNAc modulates motility-associated signaling intermediates in neutrophils. J. Biol. Chem. 2005, 280, 14579–14585. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.W.; Love, D.C.; Ghosh, S.K.; Wang, P.; Yun, S.; Fukushige, T.; Hanover, J.A. Nutrient-Driven O-GlcNAcylation at Promoters Impacts Genome-Wide RNA Pol II Distribution. Front. Endocrinol. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.G.; Potter, S.C.; Joiner, C.M.; Fei, G.Q.; Nabet, B.; Sonnett, M.; Zachara, N.E.; Gray, N.S.; Paulo, J.A.; Walker, S. Mammalian cell proliferation requires noncatalytic functions of O-GlcNAc transferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2016778118. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Van der Laarse, S.A.M.; Leney, A.C.; Heck, A.J.R. Crosstalk between phosphorylation and O-GlcNAcylation: Friend or foe. FEBS J. 2018, 285, 3152–3167. [Google Scholar] [CrossRef]
- Guinez, C.; Mir, A.M.; Dehennaut, V.; Cacan, R.; Harduin-Lepers, A.; Michalski, J.C.; Lefebvre, T. Protein ubiquitination is modulated by O-GlcNAc glycosylation. FASEB J. 2008, 22, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The human O-GlcNAcome database and meta-analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Malard, F.; Berendt, R.; Wulff-Fuentes, E.; Danner, L. Find out if your protein is O-GlcNAc modified: The O-GlcNAc database. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Nagel, A.K.; Ball, L.E. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef]
- März, P.; Stetefeld, J.; Bendfeldt, K.; Nitsch, C.; Reinstein, J.; Shoeman, R.L.; Dimitriades-Schmutz, B.; Schwager, M.; Leiser, D.; Ozcan, S.; et al. Ataxin-10 interacts with O-linked beta-N-acetylglucosamine transferase in the brain. J. Biol. Chem. 2006, 281, 20263–20270. [Google Scholar] [CrossRef]
- Cheung, W.D.; Sakabe, K.; Housley, M.P.; Dias, W.B.; Hart, G.W. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J. Biol. Chem. 2008, 283, 33935–33941. [Google Scholar] [CrossRef]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Issad, T.; Masson, E.; Pagesy, P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010, 36, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Hart, G.W. New insights: A role for O-GlcNAcylation in diabetic complications. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 150–161. [Google Scholar] [CrossRef]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 12, 99. [Google Scholar] [CrossRef]
- De Queiroz, R.M.; Carvalho, E.; Dias, W.B. O-GlcNAcylation: The Sweet Side of the Cancer. Front. Oncol. 2014, 4, 132. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Sodi, V.L.; Reginato, M.J. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J. Mol. Biol. 2016, 428, 3282–3294. [Google Scholar] [CrossRef]
- Banerjee, P.S.; Ma, J.; Hart, G.W. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 6050–6055. [Google Scholar] [CrossRef]
- Lunde, I.G.; Aronsen, J.M.; Kvaløy, H.; Qvigstad, E.; Sjaastad, I.; Tønnessen, T.; Christensen, G.; Grønning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef]
- Mesubi, O.O.; Rokita, A.G.; Abrol, N.; Wu, Y.; Chen, B.; Wang, Q.; Granger, J.M.; Tucker-Bartley, A.; Luczak, E.D.; Murphy, K.R.; et al. Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. J. Clin. Investig. 2021, 131, e95747. [Google Scholar] [CrossRef]
- Gu, J.H.; Shi, J.; Dai, C.L.; Ge, J.B.; Zhao, Y.; Chen, Y.; Yu, Q.; Qin, Z.H.; Iqbal, K.; Liu, F.; et al. O-GlcNAcylation Reduces Ischemia-Reperfusion-Induced Brain Injury. Sci. Rep. 2017, 7, 10686. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ha, H.J.; Chung, E.S.; Baek, S.H.; Cho, Y.; Kim, H.K.; Han, J.; Sul, J.H.; Lee, J.; Kim, E.; et al. O-GlcNAcylation ameliorates the pathological manifestations of Alzheimer’s disease by inhibiting necroptosis. Sci. Adv. 2021, 7, eabd3207. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, E.G.; Albarran, E.; White, C.W., 3rd; Bieri, G.; Sanchez-Diaz, C.; Pratt, K.; Snethlage, C.E.; Ding, J.B.; Villeda, S.A. Neuronal O-GlcNAcylation Improves Cognitive Function in the Aged Mouse Brain. Curr. Biol. 2019, 29, 3359–3369.e4. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef]
- Rajamani, U.; Essop, M.F. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am. J. Physiol. Cell Physiol. 2010, 299, C139–C147. [Google Scholar] [CrossRef]
- Yue, X.; Wang, J.; Chang, C.Y.; Liu, J.; Yang, X.; Zhou, F.; Qiu, X.; Bhatt, V.; Guo, J.Y.; Su, X.; et al. Leukemia inhibitory factor drives glucose metabolic reprogramming to promote breast tumorigenesis. Cell Death Dis. 2022, 13, 370. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef] [PubMed]
- Umapathi, P.; Mesubi, O.O.; Banerjee, P.S.; Abrol, N.; Wang, Q.; Luczak, E.D.; Wu, Y.; Granger, J.M.; Wei, A.C.; Reyes Gaido, O.E.; et al. Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death. Circulation 2021, 143, 1687–1703. [Google Scholar] [CrossRef] [PubMed]
- Limbutara, K.; Chou, C.L.; Knepper, M.A. Quantitative Proteomics of All 14 Renal Tubule Segments in Rat. J. Am. Soc. Nephrol. 2020, 31, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chou, C.L.; Knepper, M.A. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. J. Am. Soc. Nephrol. 2015, 26, 2669–2677. [Google Scholar] [CrossRef]
- Lagerlöf, O.; Slocomb, J.E.; Hong, I.; Aponte, Y.; Blackshaw, S.; Hart, G.W.; Huganir, R.L. The nutrient sensor OGT in PVN neurons regulates feeding. Science 2016, 351, 1293–1296. [Google Scholar] [CrossRef]
- Lagerlöf, O.; Hart, G.W.; Huganir, R.L. O-GlcNAc transferase regulates excitatory synapse maturity. Proc. Natl. Acad. Sci. USA 2017, 114, 1684–1689. [Google Scholar] [CrossRef]
- Li, M.D.; Vera, N.B.; Yang, Y.; Zhang, B.; Ni, W.; Ziso-Qejvanaj, E.; Ding, S.; Zhang, K.; Yin, R.; Wang, S.; et al. Adipocyte OGT governs diet-induced hyperphagia and obesity. Nat. Commun. 2018, 9, 5103. [Google Scholar] [CrossRef]
- Watson, L.J.; Facundo, H.T.; Ngoh, G.A.; Ameen, M.; Brainard, R.E.; Lemma, K.M.; Long, B.W.; Prabhu, S.D.; Xuan, Y.T.; Jones, S.P. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 17797–17802. [Google Scholar] [CrossRef]
- Dassanayaka, S.; Brittian, K.R.; Long, B.W.; Higgins, L.A.; Bradley, J.A.; Audam, T.N.; Jurkovic, A.; Gumpert, A.M.; Harrison, L.T.; Hartyánszky, I.; et al. Cardiomyocyte Oga haploinsufficiency increases O-GlcNAcylation but hastens ventricular dysfunction following myocardial infarction. PLoS ONE 2020, 15, e0242250. [Google Scholar] [CrossRef]
- Abramowitz, L.K.; Harly, C.; Das, A.; Bhandoola, A.; Hanover, J.A. Blocked O-GlcNAc cycling disrupts mouse hematopoeitic stem cell maintenance and early T cell development. Sci. Rep. 2019, 9, 12569. [Google Scholar] [CrossRef]
- Ono, S.; Kume, S.; Yasuda-Yamahara, M.; Yamahara, K.; Takeda, N.; Chin-Kanasaki, M.; Araki, H.; Sekine, O.; Yokoi, H.; Mukoyama, M.; et al. O-linked β-N-acetylglucosamine modification of proteins is essential for foot process maturation and survival in podocytes. Nephrol. Dial. Transplant. 2017, 32, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47 (Suppl. 1), 3–13. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, S.; Kume, S.; Chin-Kanasaki, M.; Tomita, I.; Yasuda-Yamahara, M.; Yamahara, K.; Takeda, N.; Osawa, N.; Yanagita, M.; Araki, S.I.; et al. Protein O-GlcNAcylation Is Essential for the Maintenance of Renal Energy Homeostasis and Function via Lipolysis during Fasting and Diabetes. J. Am. Soc. Nephrol. 2019, 30, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, S.; Li, X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell. Mol. Life Sci. 2021, 78, 5731–5741. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron. Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Kellum, J.A.; Ronco, C. Subclinical AKI—An emerging syndrome with important consequences. Nat. Rev. Nephrol. 2012, 8, 735–739. [Google Scholar] [CrossRef]
- Haase, M.; Devarajan, P.; Haase-Fielitz, A.; Bellomo, R.; Cruz, D.N.; Wagener, G.; Krawczeski, C.D.; Koyner, J.L.; Murray, P.; Zappitelli, M.; et al. The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: A multicenter pooled analysis of prospective studies. J. Am. Coll. Cardiol. 2011, 57, 1752–1761. [Google Scholar] [CrossRef]
- Fang, F.; Hu, X.; Dai, X.; Wang, S.; Bai, Z.; Chen, J.; Pan, J.; Li, X.; Wang, J.; Li, Y. Subclinical acute kidney injury is associated with adverse outcomes in critically ill neonates and children. Crit. Care 2018, 22, 256. [Google Scholar] [CrossRef]
- Dépret, F.; Hollinger, A.; Cariou, A.; Deye, N.; Vieillard-Baron, A.; Fournier, M.C.; Jaber, S.; Damoisel, C.; Lu, Q.; Monnet, X.; et al. Incidence and Outcome of Subclinical Acute Kidney Injury Using penKid in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2020, 202, 822–829. [Google Scholar] [CrossRef]
- Sun, D.Q.; Wang, T.Y.; Zheng, K.I.; Targher, G.; Byrne, C.D.; Chen, Y.P.; Zheng, M.H. Subclinical Acute Kidney Injury in COVID-19 Patients: A Retrospective Cohort Study. Nephron. Clin. Pract. 2020, 144, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Selby, N.M.; Shaw, S.; Woodier, N.; Fluck, R.J.; Kolhe, N.V. Gentamicin-associated acute kidney injury. QJM 2009, 102, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Mattson, D.L. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat. Rev. Nephrol. 2019, 15, 290–300. [Google Scholar] [CrossRef]
- Ravera, M.; Re, M.; Deferrari, L.; Vettoretti, S.; Deferrari, G. Importance of blood pressure control in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17 (Suppl. 2), S98–S103. [Google Scholar] [CrossRef]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Mann, J.F.; Yi, Q.; Zinman, B.; Dinneen, S.F.; Hoogwerf, B.; Hallé, J.P.; Young, J.; Rashkow, A.; Joyce, C.; et al. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001, 286, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Warnock, D.G.; Muntner, P.; McCullough, P.A.; Zhang, X.; McClure, L.A.; Zakai, N.; Cushman, M.; Newsome, B.B.; Kewalramani, R.; Steffes, M.W.; et al. Kidney function, albuminuria, and all-cause mortality in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study. Am. J. Kidney Dis. 2010, 56, 861–871. [Google Scholar] [CrossRef]
- Ninomiya, T.; Perkovic, V.; de Galan, B.E.; Zoungas, S.; Pillai, A.; Jardine, M.; Patel, A.; Cass, A.; Neal, B.; Poulter, N.; et al. Albuminuria and kidney function independently predict cardiovascular and renal outcomes in diabetes. J. Am. Soc. Nephrol. 2009, 20, 1813–1821. [Google Scholar] [CrossRef]
- Lea, J.; Greene, T.; Hebert, L.; Lipkowitz, M.; Massry, S.; Middleton, J.; Rostand, S.G.; Miller, E.; Smith, W.; Bakris, G.L. The relationship between magnitude of proteinuria reduction and risk of end-stage renal disease: Results of the African American study of kidney disease and hypertension. Arch. Intern. Med. 2005, 165, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.N.; Troyanov, S.; Scholey, J.W.; Cattran, D.C.; Toronto Glomerulonephritis Registry. Remission of proteinuria improves prognosis in IgA nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Troost, J.P.; Trachtman, H.; Spino, C.; Kaskel, F.J.; Friedman, A.; Moxey-Mims, M.M.; Fine, R.N.; Gassman, J.J.; Kopp, J.B.; Walsh, L.; et al. Proteinuria Reduction and Kidney Survival in Focal Segmental Glomerulosclerosis. Am. J. Kidney Dis. 2021, 77, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Rilla, K.; Oikari, S.; Jokela, T.A.; Hyttinen, J.M.; Kärnä, R.; Tammi, R.H.; Tammi, M.I. Hyaluronan synthase 1 (HAS1) requires higher cellular UDP-GlcNAc concentration than HAS2 and HAS3. J. Biol. Chem. 2013, 288, 5973–5983. [Google Scholar] [CrossRef]
- Yang, S.; Zou, L.Y.; Bounelis, P.; Chaudry, I.; Chatham, J.C.; Marchase, R.B. Glucosamine administration during resuscitation improves organ function after trauma hemorrhage. Shock 2006, 25, 600–607. [Google Scholar] [CrossRef]
- Ryczko, M.C.; Pawling, J.; Chen, R.; Abdel Rahman, A.M.; Yau, K.; Copeland, J.K.; Zhang, C.; Surendra, A.; Guttman, D.S.; Figeys, D.; et al. Metabolic Reprogramming by Hexosamine Biosynthetic and Golgi N-Glycan Branching Pathways. Sci. Rep. 2016, 6, 23043. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. The protective effects of PUGNAc on cardiac function after trauma-hemorrhage are mediated via increased protein O-GlcNAc levels. Shock 2007, 27, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.N.; Lee, Y.J.; Kim, M.O.; Ryu, J.M.; Han, H.J. Glucosamine-induced Sp1 O-GlcNAcylation ameliorates hypoxia-induced SGLT dysfunction in primary cultured renal proximal tubule cells. J. Cell. Physiol. 2014, 229, 1557–1568. [Google Scholar] [CrossRef]
- Hu, J.; Chen, R.; Jia, P.; Fang, Y.; Liu, T.; Song, N.; Xu, X.; Ji, J.; Ding, X. Augmented O-GlcNAc signaling via glucosamine attenuates oxidative stress and apoptosis following contrast-induced acute kidney injury in rats. Free Radic. Biol. Med. 2017, 103, 121–132. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Y.; Zhao, S.; Chen, J.; Jin, S.; Jia, P.; Ding, X. Remote Ischemic Preconditioning Ameliorates Acute Kidney Injury due to Contrast Exposure in Rats through Augmented O-GlcNAcylation. Oxid. Med. Cell. Longev. 2018, 2018, 4895913. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.R.; Dias, T.B.; Natov, P.S.; Zachara, N.E. Stress-induced O-GlcNAcylation: An adaptive process of injured cells. Biochem. Soc. Trans. 2017, 45, 237–249. [Google Scholar] [CrossRef]
- Sohn, K.C.; Lee, K.Y.; Park, J.E.; Do, S.I. OGT functions as a catalytic chaperone under heat stress response: A unique defense role of OGT in hyperthermia. Biochem. Biophys. Res. Commun. 2004, 322, 1045–1051. [Google Scholar] [CrossRef]
- Matsumoto, T.; Urushido, M.; Ide, H.; Ishihara, M.; Hamada-Ode, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Taguchi, T.; et al. Small Heat Shock Protein Beta-1 (HSPB1) Is Upregulated and Regulates Autophagy and Apoptosis of Renal Tubular Cells in Acute Kidney Injury. PLoS ONE 2015, 10, e0126229. [Google Scholar] [CrossRef]
- Kim, M.G.; Jung Cho, E.; Won Lee, J.; Sook Ko, Y.; Young Lee, H.; Jo, S.K.; Cho, W.Y.; Kim, H.K. The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+ CD25+ Foxp3 + regulatory T cells in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2014, 85, 62–71. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell. 2014, 54, 820–831. [Google Scholar] [CrossRef]
- Conde, E.; Giménez-Moyano, S.; Martín-Gómez, L.; Rodríguez, M.; Ramos, M.E.; Aguado-Fraile, E.; Blanco-Sanchez, I.; Saiz, A.; García-Bermejo, M.L. HIF-1α induction during reperfusion avoids maladaptive repair after renal ischemia/reperfusion involving miR127-3p. Sci. Rep. 2017, 7, 41099. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Ohn, T.; Kedersha, N.; Hickman, T.; Tisdale, S.; Anderson, P. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 2008, 10, 1224–1231. [Google Scholar] [CrossRef]
- Wang, S.; Kwon, S.H.; Su, Y.; Dong, Z. Stress granules are formed in renal proximal tubular cells during metabolic stress and ischemic injury for cell survival. Am. J. Physiol. Renal Physiol. 2019, 317, F116–F123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, M.; Li, M.D.; Zhang, K.; Zhang, B.; Wang, S.; Liu, Y.; Ni, W.; Ong, Q.; Mi, J.; et al. O-GlcNAc transferase inhibits visceral fat lipolysis and promotes diet-induced obesity. Nat. Commun. 2020, 11, 181. [Google Scholar] [CrossRef]
- Shin, S.J.; Chen, C.H.; Kuo, W.C.; Chan, H.C.; Chan, H.C.; Lin, K.D.; Ke, L.Y. Disruption of retinoid homeostasis induces RBP4 overproduction in diabetes: O-GlcNAcylation involved. Metabolism 2020, 113, 154403. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, K.D.; Ke, L.Y.; Liang, C.J.; Kuo, W.C.; Lee, M.Y.; Lee, Y.L.; Hsiao, P.J.; Hsu, C.C.; Shin, S.J. O-GlcNAcylation disrupts STRA6-retinol signals in kidneys of diabetes. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1059–1069. [Google Scholar] [CrossRef]
- Degrell, P.; Cseh, J.; Mohás, M.; Molnár, G.A.; Pajor, L.; Chatham, J.C.; Fülöp, N.; Wittmann, I. Evidence of O-linked N-acetylglucosamine in diabetic nephropathy. Life Sci. 2009, 84, 389–393. [Google Scholar] [CrossRef]
- Goldberg, H.; Whiteside, C.; Fantus, I.G. O-linked β-N-acetylglucosamine supports p38 MAPK activation by high glucose in glomerular mesangial cells. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E713–E726. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Kim, D.I.; Lim, S.K.; Choi, J.H.; Han, H.J.; Yoon, K.C.; Park, S.H. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: The possible role in the development of diabetic nephropathy. J. Biol. Chem. 2014, 289, 13519–13530. [Google Scholar] [CrossRef]
- Palmer, M.B.; Abedini, A.; Jackson, C.; Blady, S.; Chatterjee, S.; Sullivan, K.M.; Townsend, R.R.; Brodbeck, J.; Almaani, S.; Srivastava, A.; et al. The Role of Glomerular Epithelial Injury in Kidney Function Decline in Patients with Diabetic Kidney Disease in the TRIDENT Cohort. Kidney Int. Rep. 2021, 6, 1066–1080. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef]
- Gyarmati, G.; Shroff, U.N.; Izuhara, A.; Hou, X.; Da Sacco, S.; Sedrakyan, S.; Lemley, K.V.; Amann, K.; Perin, L.; Peti-Peterdi, J. Intravital imaging reveals glomerular capillary distension and endothelial and immune cell activation early in Alport syndrome. JCI Insight 2022, 7, e152676. [Google Scholar] [CrossRef]
- Eddy, A.A. Proteinuria and interstitial injury. Nephrol. Dial. Transplant. 2004, 19, 277–281. [Google Scholar] [CrossRef]
- Liu, W.J.; Xu, B.H.; Ye, L.; Liang, D.; Wu, H.L.; Zheng, Y.Y.; Deng, J.K.; Li, B.; Liu, H.F. Urinary proteins induce lysosomal membrane permeabilization and lysosomal dysfunction in renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2015, 308, F639–F649. [Google Scholar] [CrossRef]
- Caruso-Neves, C.; Pinheiro, A.A.; Cai, H.; Souza-Menezes, J.; Guggino, W.B. PKB and megalin determine the survival or death of renal proximal tubule cells. Proc. Natl. Acad. Sci. USA 2006, 103, 18810–18815. [Google Scholar] [CrossRef]
- Ohse, T.; Inagi, R.; Tanaka, T.; Ota, T.; Miyata, T.; Kojima, I.; Ingelfinger, J.R.; Ogawa, S.; Fujita, T.; Nangaku, M. Albumin induces endoplasmic reticulum stress and apoptosis in renal proximal tubular cells. Kidney Int. 2006, 70, 1447–1455. [Google Scholar] [CrossRef]
- Peruchetti, D.B.; Silva-Aguiar, R.P.; Siqueira, G.M.; Dias, W.B.; Caruso-Neves, C. High glucose reduces megalin-mediated albumin endocytosis in renal proximal tubule cells through protein kinase B O-GlcNAcylation. J. Biol. Chem. 2018, 293, 11388–11400. [Google Scholar] [CrossRef]
- Russo, L.M.; Sandoval, R.M.; Campos, S.B.; Molitoris, B.A.; Comper, W.D.; Brown, D. Impaired tubular uptake explains albuminuria in early diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 489–494. [Google Scholar] [CrossRef]
- Coffey, S.; Costacou, T.; Orchard, T.; Erkan, E. Akt Links Insulin Signaling to Albumin Endocytosis in Proximal Tubule Epithelial Cells. PLoS ONE 2015, 10, e0140417. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 2012, 7, e37427. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wu, S.; Dai, C.L.; Li, Y.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.X. Diverse regulation of AKT and GSK-3β by O-GlcNAcylation in various types of cells. FEBS Lett. 2012, 586, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gu, J.H.; Dai, C.L.; Gu, J.; Jin, X.; Sun, J.; Iqbal, K.; Liu, F.; Gong, C.X. O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci. Rep. 2015, 5, 14500. [Google Scholar] [CrossRef]
- Hodrea, J.; Balogh, D.B.; Hosszu, A.; Lenart, L.; Besztercei, B.; Koszegi, S.; Sparding, N.; Genovese, F.; Wagner, L.J.; Szabo, A.J.; et al. Reduced O-GlcNAcylation and tubular hypoxia contribute to the antifibrotic effect of SGLT2 inhibitor dapagliflozin in the diabetic kidney. Am. J. Physiol. Renal Physiol. 2020, 318, F1017–F1029. [Google Scholar] [CrossRef] [PubMed]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; Charbonnel, B.; Cherney, D.Z.I.; Dagogo-Jack, S.; Pratley, R.; Greenberg, M.; Wang, S.; Huyck, S.; et al. Association of SGLT2 Inhibitors with Cardiovascular and Kidney Outcomes in Patients With Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lima, V.V.; Giachini, F.R.; Choi, H.; Carneiro, F.S.; Carneiro, Z.N.; Fortes, Z.B.; Carvalho, M.H.; Webb, R.C.; Tostes, R.C. Impaired vasodilator activity in deoxycorticosterone acetate-salt hypertension is associated with increased protein O-GlcNAcylation. Hypertension 2009, 53, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Silva-Aguiar, R.P.; Bezerra, N.C.F.; Lucena, M.C.; Sirtoli, G.M.; Sudo, R.T.; Zapata-Sudo, G.; Takiya, C.M.; Pinheiro, A.A.S.; Dias, W.B.; Caruso-Neves, C. O-GlcNAcylation reduces proximal tubule protein reabsorption and promotes proteinuria in spontaneously hypertensive rats. J. Biol. Chem. 2018, 293, 12749–12758. [Google Scholar] [CrossRef]
- Wang, W.; Silva, L.M.; Wang, H.H.; Kavanaugh, M.A.; Pottorf, T.S.; Allard, B.A.; Jacobs, D.T.; Dong, R.; Cornelius, J.T.; Chaturvedi, A.; et al. Ttc21b deficiency attenuates autosomal dominant polycystic kidney disease in a kidney tubular- and maturation-dependent manner. Kidney Int. 2022, 102, 577–591. [Google Scholar] [CrossRef]
- Burt, R.A.; Alghusen, I.M.; John Ephrame, S.; Villar, M.T.; Artigues, A.; Slawson, C. Mapping the O-GlcNAc Modified Proteome: Applications for Health and Disease. Front. Mol. Biosci. 2022, 9, 920727. [Google Scholar] [CrossRef]
- Fehl, C.; Hanover, J.A. Tools, tactics and objectives to interrogate cellular roles of O-GlcNAc in disease. Nat. Chem. Biol. 2022, 18, 8–17. [Google Scholar] [CrossRef]
- Ma, J.; Hou, C.; Wu, C. Demystifying the O-GlcNAc Code: A Systems View. Chem. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J. Chemical Reporters and Their Bioorthogonal Reactions for Labeling Protein O-GlcNAcylation. Molecules 2018, 23, 2411. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Rookmaaker, M.B.; Joles, J.A.; Kramann, R.; Nguyen, T.Q.; van Griensven, M.; LaPointe, V.L.S. Defining the variety of cell types in developing and adult human kidneys by single-cell RNA sequencing. NPJ Regen. Med. 2021, 6, 45. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Aguiar, R.P.; Peruchetti, D.B.; Pinheiro, A.A.S.; Caruso-Neves, C.; Dias, W.B. O-GlcNAcylation in Renal (Patho)Physiology. Int. J. Mol. Sci. 2022, 23, 11260. https://doi.org/10.3390/ijms231911260
Silva-Aguiar RP, Peruchetti DB, Pinheiro AAS, Caruso-Neves C, Dias WB. O-GlcNAcylation in Renal (Patho)Physiology. International Journal of Molecular Sciences. 2022; 23(19):11260. https://doi.org/10.3390/ijms231911260
Chicago/Turabian StyleSilva-Aguiar, Rodrigo P., Diogo B. Peruchetti, Ana Acacia S. Pinheiro, Celso Caruso-Neves, and Wagner B. Dias. 2022. "O-GlcNAcylation in Renal (Patho)Physiology" International Journal of Molecular Sciences 23, no. 19: 11260. https://doi.org/10.3390/ijms231911260
APA StyleSilva-Aguiar, R. P., Peruchetti, D. B., Pinheiro, A. A. S., Caruso-Neves, C., & Dias, W. B. (2022). O-GlcNAcylation in Renal (Patho)Physiology. International Journal of Molecular Sciences, 23(19), 11260. https://doi.org/10.3390/ijms231911260