
A Comparative DFT Study on Process Control Agents in the Mechanochemical Synthesis of PbTe

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
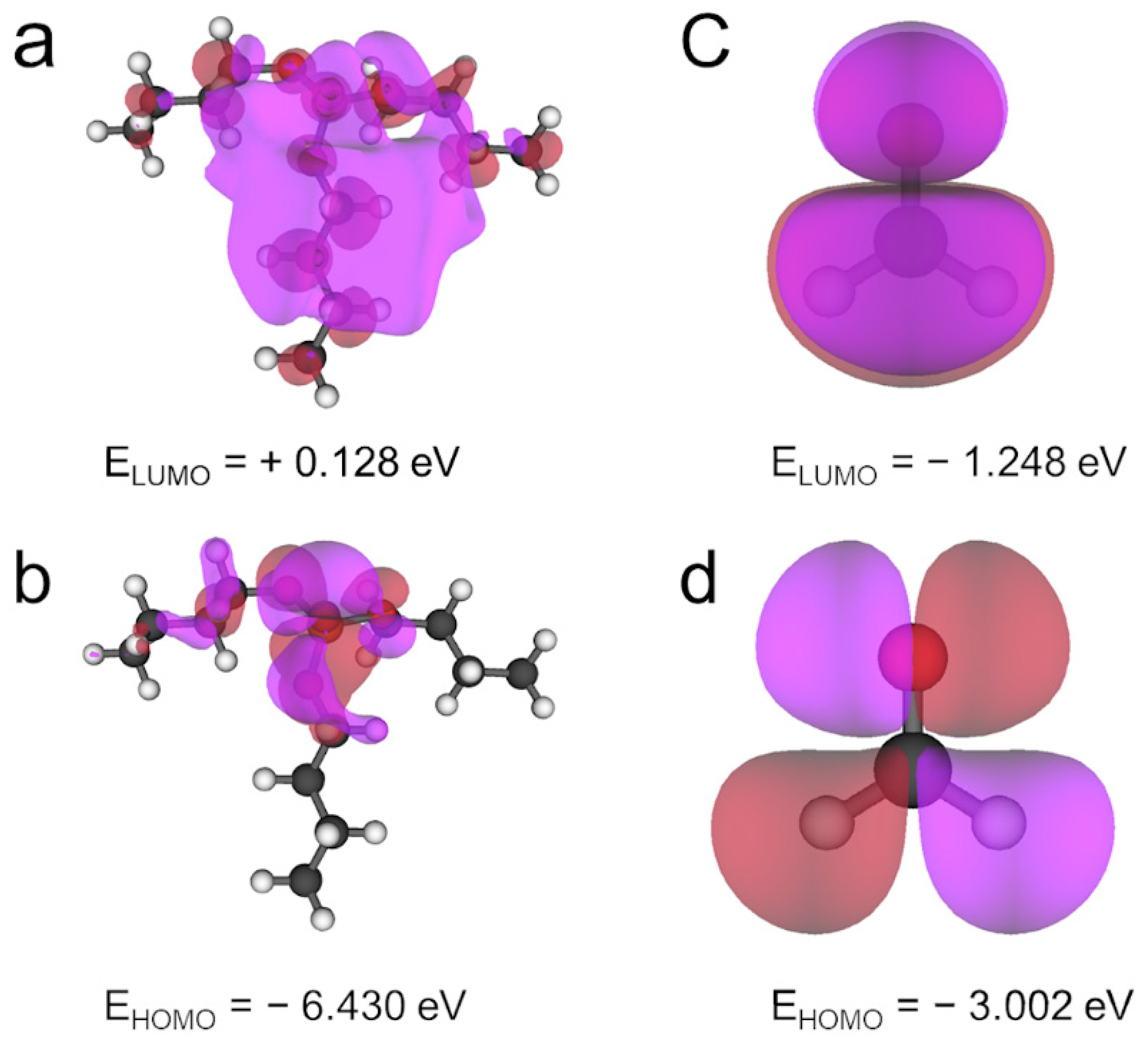
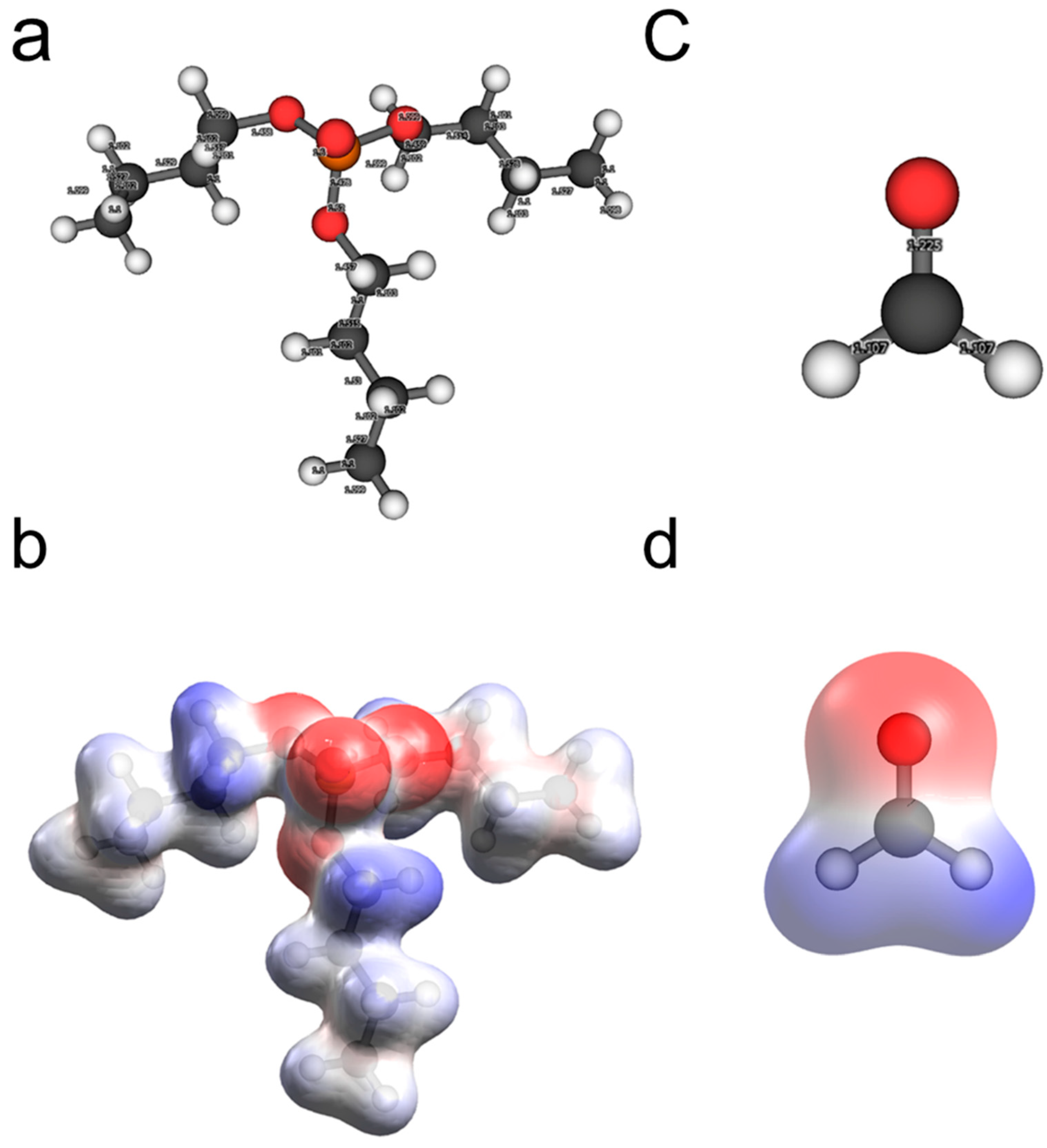
2.1. Surface Modifier Molecules
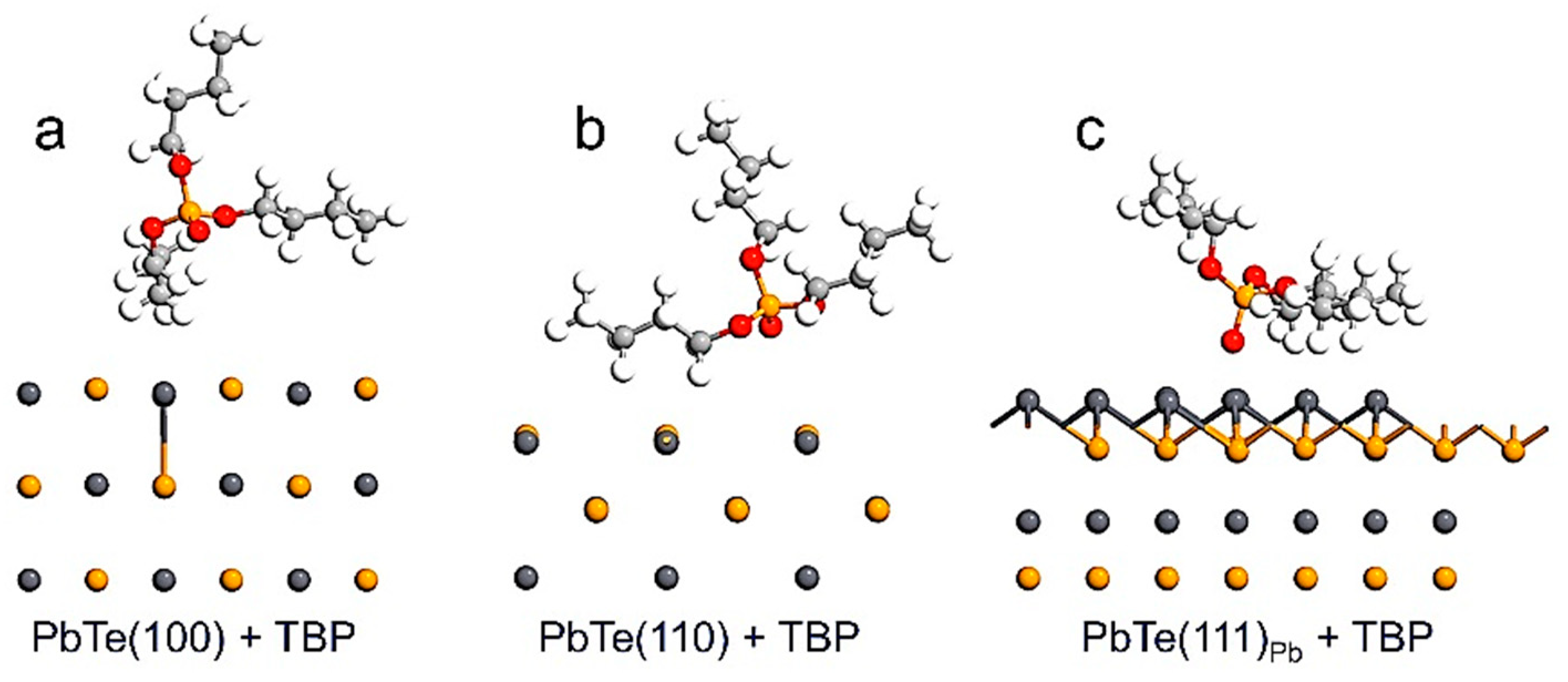
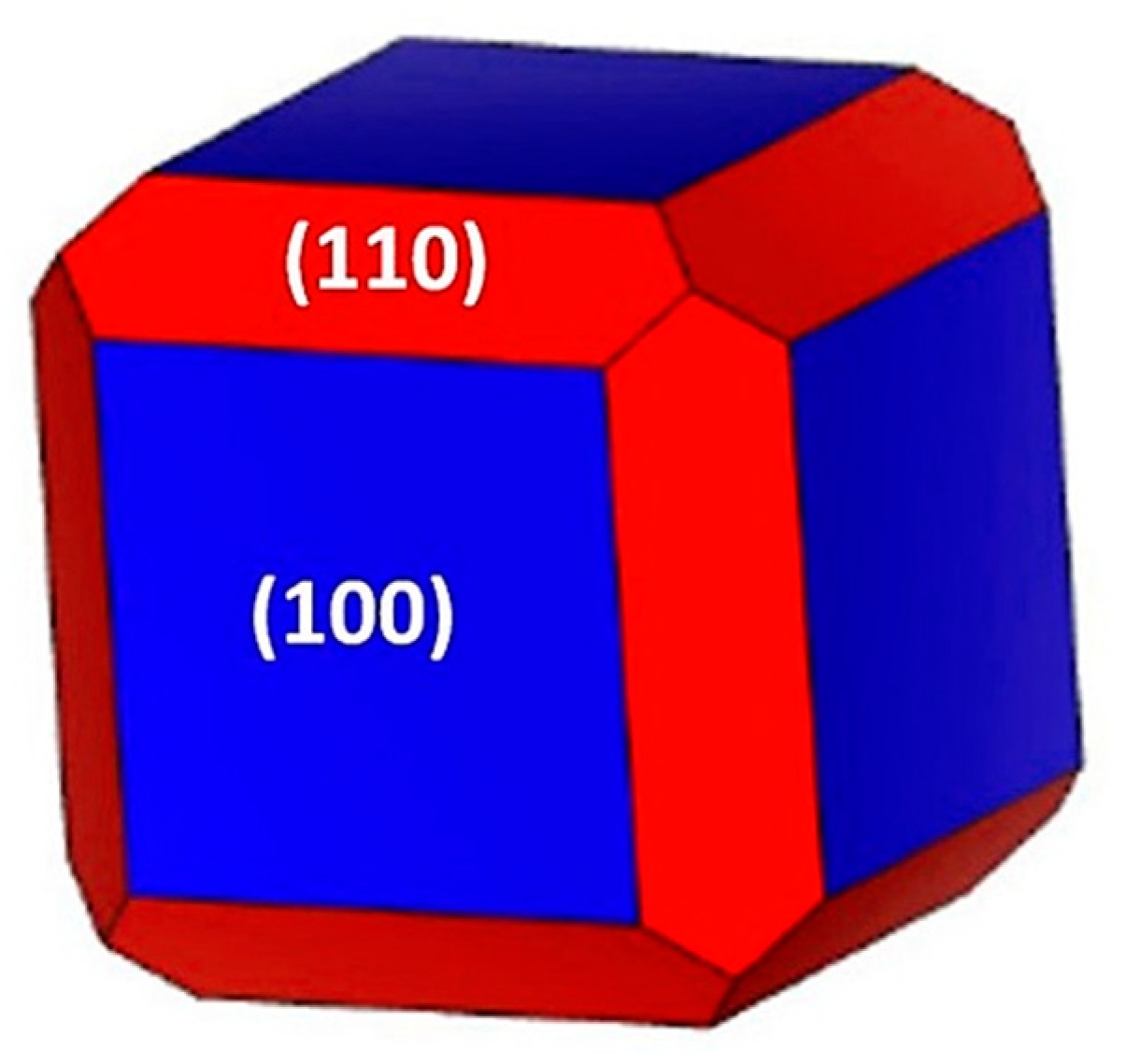
2.2. Slab Model Analysis for TBP as Surface Modifier
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rojas-Chávez, H.; Miralrio, A.; Cruz-Martínez, H.; Carbajal-Franco, G.; Valdés-Madrigal, M.A. Oriented-Attachment- And Defect-Dependent PbTe Quantum Dots Growth: Shape Transformations Supported by Experimental Insights and DFT Calculations. Inorg. Chem. 2021, 60, 7196. [Google Scholar] [CrossRef] [PubMed]
- Edalati, K.; Bachmaier, A.; Beloshenko, V.A.; Beygelzimer, Y.; Blank, V.D.; Botta, W.J.; Bryła, K.; Čížek, J.; Divinski, S.; Enikeev, N.A.; et al. Nanomaterials by severe plastic deformation: Review of historical developments and recent advances. Mater. Res. Lett. 2022, 10, 163. [Google Scholar] [CrossRef]
- Révész, Á.; Takacs, L. Coating a Cu plate with a Zr–Ti powder mixture using surface mechanical attrition treatment. Surf. Coat. Technol. 2009, 203, 3026. [Google Scholar] [CrossRef]
- Rojas-Chávez, H.; Miralrio, A.; Cruz-Martínez, H.; Martínez-Espinosa, J.A.; Carbajal-Franco, G.; Juárez-García, J.M. Identifying the mechanistic insights into PbSeO3 decomposition, during milling, to give way to PbSe: An experimental and theoretical approach. Comput. Mater. Sci. 2022, 206, 111291. [Google Scholar] [CrossRef]
- Huang, J.Y.; Wu, Y.K.; Ye, H.Q. Ball milling of ductile metals. Mater. Sci. Eng. A 1995, 199, 165. [Google Scholar] [CrossRef]
- Mingliang, M.; Xinkuan, L.; Shengqi, X.; Donglang, C.; Jing’en, Z. Effect of material characteristics on the ignition of the combustion reactions induced by ball milling. J. Mater. Process. Technol. 2001, 116, 124. [Google Scholar] [CrossRef]
- Wu, Z.F.; Zeng, M.Q.; Ouyang, L.Z.; Zhang, X.P.; Zhu, M. Ostwald ripening of Pb nanocrystalline phase in mechanically milled Al–Pb alloys and the influence of Cu additive. Scr. Mater. 2005, 53, 529. [Google Scholar] [CrossRef]
- Rojas-Chávez, H.; Cruz-Martínez, H.; Flores-Rojas, E.; Juárez-García, J.M.; González-Domínguez, J.L.; Daneu, N.; Santoyo-Salazar, J. The mechanochemical synthesis of PbTe nanostructures: Following the Ostwald ripening effect during milling. Phys. Chem. Chem. Phys. 2018, 20, 27082. [Google Scholar] [CrossRef]
- Gandhi, P.M.; Valluri, S.K.; Schoenitz, M.; Dreizin, E. Effect of organic liquid process control agents on properties of ball-milled powders. Adv. Powder Technol. 2022, 33, 103332. [Google Scholar] [CrossRef]
- Motozuka, S.; Sato, H.; Kuwata, H.; Bito, M.; Okazaki, Y. Effects of interfacial interactions between metal and process control agents during ball milling on the microstructure of the milled Fe-based nanocrystalline alloy powder. Heliyon 2022, 8, 10325. [Google Scholar] [CrossRef]
- Borgman, J.M.; Conway, P.P.; Torres-Sanchez, C. The use of inorganic process control agents to mill titanium-niobium powders suitable for the selective laser melting process. Powder Technol. 2022, 407, 117546. [Google Scholar] [CrossRef]
- Huber, F.; Berwanger, J.; Polesya, S.; Mankovsky, S.; Ebert, H.; Giessibl, F.J. Chemical bond formation showing a transition from physisorption to chemisorption. Science 2019, 366, 235. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, H.; Ahn, B. Microstructure and reactivity of cryomolds Al-Ni energetic material with nanoscale lamellar structure. J. Mater. Sci. 2022. [Google Scholar] [CrossRef]
- Kleiner, S.; Bertocco, F.; Khalid, F.A.; Beffort, O. Decomposition of process control agent during mechanical milling and its influence on displacement reactions in the Al–TiO2 system. Mater. Chem. Phys. 2005, 89, 362. [Google Scholar] [CrossRef]
- Canakci, A.; Varol, T.; Ozsahin, S. Analysis of the effect of a new process control agent technique on the mechanical milling process using a neural network model: Measurement and modeling. Measurement 2013, 46, 1818. [Google Scholar] [CrossRef]
- Ammar, H.Y.; Eid, K.M.; Badran, H.M. Interaction and detection of formaldehyde on pristine and doped boron nitride nano-cage: DFT calculations. Mater. Today Commun. 2020, 25, 101408. [Google Scholar] [CrossRef]
- Fan, H.; Song, X.; Xu, Y.; Yu, J. Insights into the modification for improving the surface property of calcium sulfate whisker: Experimental and DFT simulation study. Appl. Surf. Sci. 2019, 478, 594. [Google Scholar] [CrossRef]
- Liu, L.; Min, F.; Chen, J.; Lu, F.; Shen, L. The adsorption of dodecylamine and oleic acid on kaolinite surfaces: Insights from DFT calculation and experimental investigation. Appl. Surf. Sci. 2019, 470, 27. [Google Scholar] [CrossRef]
- Noda, Y.; Masumoto, K.; Ohba, S.; Saito, Y.; Toriumi, K.; Iwata, Y.; Shibuya, I. IUCr, Temperature dependence of atomic thermal parameters of lead chalcogenides, PbS, PbSe and PbTe. Acta Cryst. C 1987, 43, 1443. [Google Scholar] [CrossRef]
- Vainio, M.J.; Johnson, M.S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model. 2007, 47, 2462. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | VIE (eV) | VEA (eV) | Egap (eV) |
|---|---|---|---|
| TBP | 8.272 | –0.739 | 6.302 |
| CH2O | 9.936 | –1.283 | 1.756 |
| Surface | Structure | ΔE (kcal·mol−1) | Eads (kcal·mol−1) |
|---|---|---|---|
| (100) | #1 | 0.00 | −45.29 |
| (110) | #1 | 0.00 | −44.36 |
| (110) | #2 | 1.75 | −42.61 |
| (110) | #3 | 0.96 | −43.39 |
| (111)Pb | #1 | 9.09 | 8.25 |
| (111)Pb | #2 | 17.14 | 16.30 |
| (111)Pb | #3 | 0.00 | −0.84 |
| Surface | n | Energy [Ry] | Area [Å2] | γ [meV·Å−2] |
|---|---|---|---|---|
| (100) | 27 | −4429.33 | 185.97 | 26.54 |
| (110) | 27 | −4428.52 | 263.00 | 39.82 |
| (111)Pb | 18 | −2952.41 | 139.36 | 46.91 |
| Surface | Str | Eint [kcal·mol−1] | Edis [kcal·mol−1] | QTBP [e] | γ [meV·Å−2] | Δγ [meV·Å−2] | Ratio to γ100 |
|---|---|---|---|---|---|---|---|
| (100) | #1 | −12.69 | −32.60 | −0.06 | 15.97 | −10.57 | 1.00 |
| (110) | #3 | −13.11 | −31.25 | −0.07 | 32.50 | −7.32 | 2.03 |
| (111)Pb | #3 | −32.29 | −31.45 | −0.19 | 46.64 | −0.27 | 2.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas-Chávez, H.; Miralrio, A.; Juárez-García, J.M.; Carbajal-Franco, G.; Cruz-Martínez, H.; Montejo-Alvaro, F.; Valdés-Madrigal, M.A. A Comparative DFT Study on Process Control Agents in the Mechanochemical Synthesis of PbTe. Int. J. Mol. Sci. 2022, 23, 11194. https://doi.org/10.3390/ijms231911194
Rojas-Chávez H, Miralrio A, Juárez-García JM, Carbajal-Franco G, Cruz-Martínez H, Montejo-Alvaro F, Valdés-Madrigal MA. A Comparative DFT Study on Process Control Agents in the Mechanochemical Synthesis of PbTe. International Journal of Molecular Sciences. 2022; 23(19):11194. https://doi.org/10.3390/ijms231911194
Chicago/Turabian StyleRojas-Chávez, Hugo, Alan Miralrio, José M. Juárez-García, Guillermo Carbajal-Franco, Heriberto Cruz-Martínez, Fernando Montejo-Alvaro, and Manuel A. Valdés-Madrigal. 2022. "A Comparative DFT Study on Process Control Agents in the Mechanochemical Synthesis of PbTe" International Journal of Molecular Sciences 23, no. 19: 11194. https://doi.org/10.3390/ijms231911194
APA StyleRojas-Chávez, H., Miralrio, A., Juárez-García, J. M., Carbajal-Franco, G., Cruz-Martínez, H., Montejo-Alvaro, F., & Valdés-Madrigal, M. A. (2022). A Comparative DFT Study on Process Control Agents in the Mechanochemical Synthesis of PbTe. International Journal of Molecular Sciences, 23(19), 11194. https://doi.org/10.3390/ijms231911194