
Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side

 , and
, and
Abstract
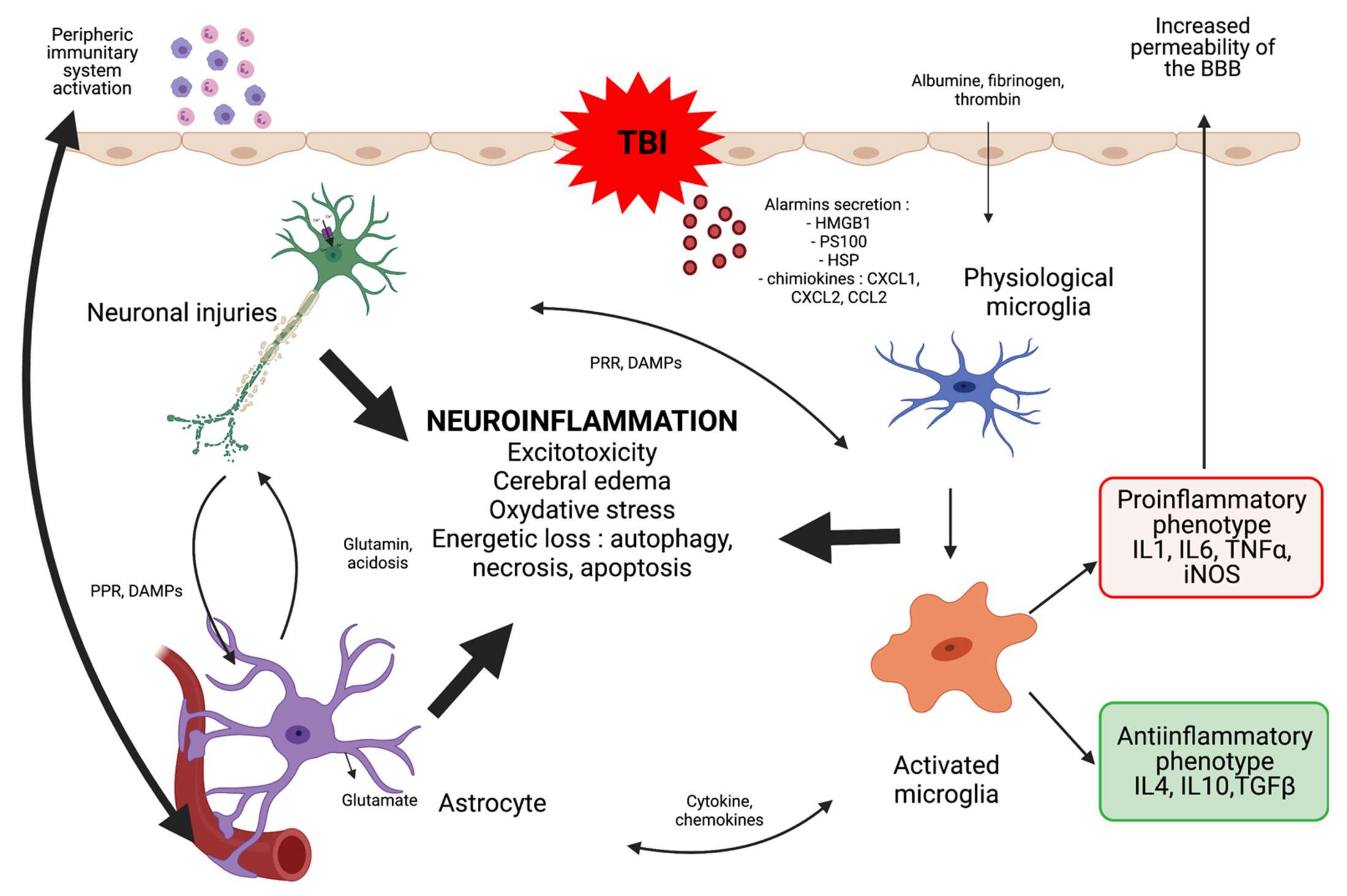
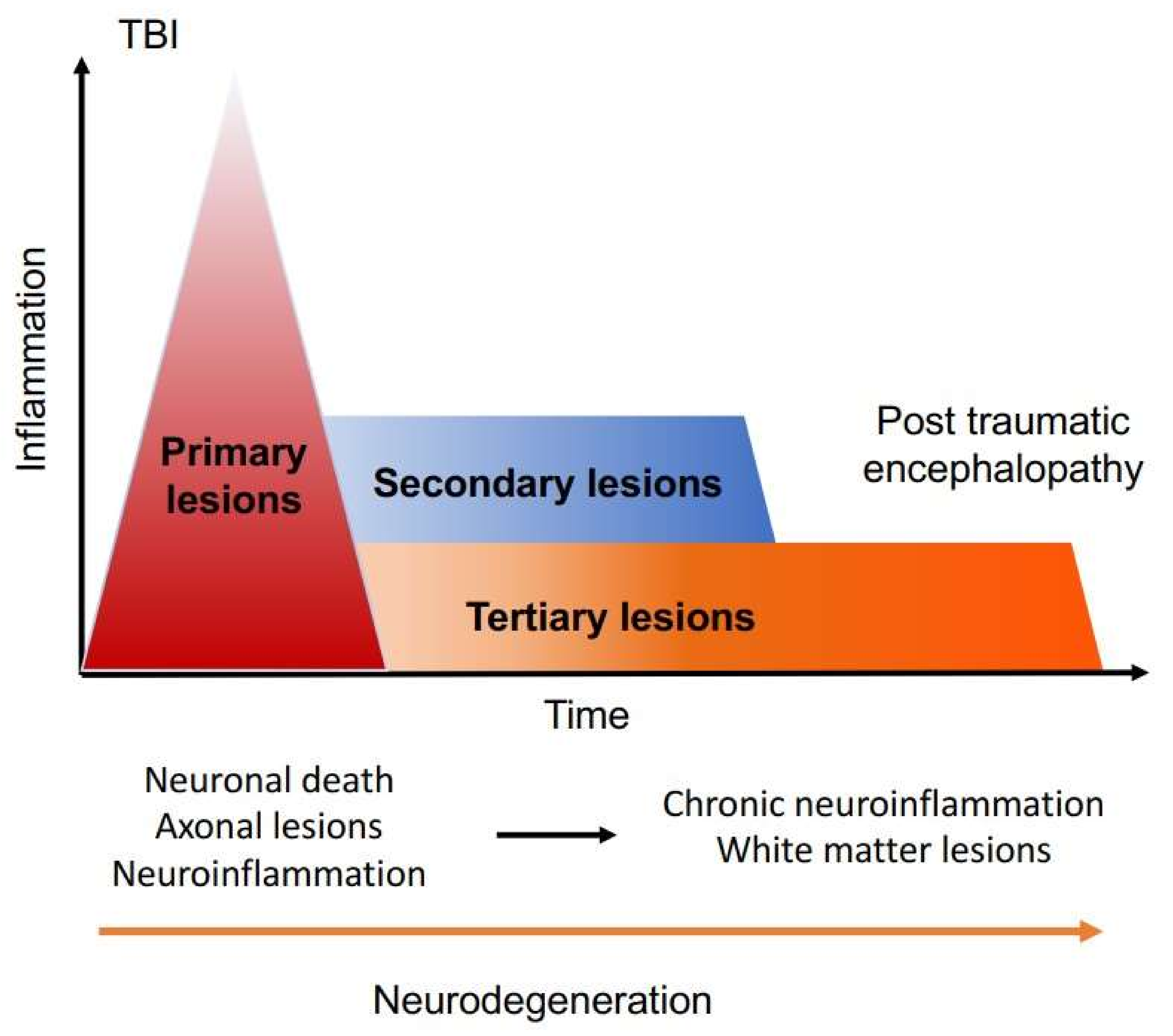
1. Introduction
2. Neuro-Anti-Inflammatory Therapeutics
2.1. Glucocorticoids
2.2. Nonsteroidal Anti-Inflammatory Drugs
2.3. Statins
2.4. Melatonin
2.5. Minocycline
2.6. Cyclosporin
2.7. Oxytocin
2.8. Erythropoietin
2.9. Others
3. Anesthetic Agents
3.1. Halogenated
3.2. Inert Gas
3.3. Propofol
3.4. Dexmedetomidine
3.5. Ketamine
4. Hormonal Treatments
4.1. Estrogens
4.2. Progesterone
5. Vitamin Supplementation
5.1. Vitamins B
5.2. Vitamin C
5.3. Vitamin D
5.4. Vitamin E
6. Ions
6.1. Magnésium
6.2. Zinc
7. Omega-3
8. Discussion

{kind=link}
{kind=link}
| Drugs | Mode of Action | Pediatric Studies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inflammation | Microglial/ Astrocytic Activation | Excitotoxicity | Anti-Oxydative | Apoptose | Œdema | Mitochondria | Neuronal Death/Neurogenesis | Cerebral Metabolism | BBB | |||
| Antiinflammatory drugs | GC | [387] | [44,50] | |||||||||
| NSAI | [56] | |||||||||||
| Statins | [70,71,72] | [64] | [388] | [59,60] | [389] | [61,62,66] | [389] | |||||
| Melatonine | [390] | [390] | [78] | [86,391,392,393] | [394] | [91] | ||||||
| Minocycline | [97,395] | [97,110] | [396] | [395,397,398] | ||||||||
| Ciclosporin | [113] | [399] | [112,115,116,117] | |||||||||
| Oxytocin | ||||||||||||
| Anti-TNF-α | [136,137,139] | [136] | [400] | [135] | ||||||||
| Anti-IL1 | [140] | [401] | ||||||||||
| Anti-IL-1β | [146] | [146] | [147] | |||||||||
| Anti IL-6 | [151] | |||||||||||
| Anti-HMGB1 | [156,402] | [155] | [156,157] | [402,403,404,405,406] | ||||||||
| Anesthesic agents | Hallogenous | |||||||||||
| Argon | [194] | |||||||||||
| Xenon | [186,407] | [183] | [186] | |||||||||
| Propofol | [199,203] | [202] | [408,409] | [198] | ||||||||
| Hormons | Œstrogenes | [231,232] | [228,229] | [227] | [227] | |||||||
| Progesterone | [262] | [242,262] | [244,245,246] | [403] | ||||||||
| Vitaminic supplementation | Vitamin B2 | [271] | [269] | [271] | ||||||||
| Vitamin B3 | [281,410] | [281,410] | [281] | [410] | [281] | [411] | ||||||
| Vitamin B6 | [284,286] | [284,286] | ||||||||||
| Vitamin B9 | [291] | |||||||||||
| Vitamin C | [294] | [293] | ||||||||||
| Vitamin D | [311] | [311] | [312] | [412] | ||||||||
| Vitamin E | [318] | [413] | ||||||||||
| Ions | Magnesium | [332] | [331,333] | [331] | ||||||||
| Zinc | [414] | |||||||||||
| Omega-3 | [365] | [366,367] | ||||||||||
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef]
- Asken, B.M.; Sullan, M.J.; DeKosky, S.T.; Jaffee, M.S.; Bauer, R.M. Research Gaps and Controversies in Chronic Traumatic Encephalopathy: A Review. JAMA Neurol. 2017, 74, 1255–1262. [Google Scholar] [CrossRef]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I.R. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 1–18. [Google Scholar] [CrossRef]
- Bayen, É.; Jourdan, C.; Azouvi, P.; Weiss, J.-J.; Pradat-Diehl, P. Prise en charge après lésion cérébrale acquise de type traumatisme crânien. Inf. Psychiatr. 2012, 88, 331–337. [Google Scholar] [CrossRef]
- Cambier, J.; Masson, M.; Masson, C.; Henri, D. (Eds.) Traumatismes crâniens. In Neurologie, 13th ed.; Elsevier Masson: Paris, France, 2012; pp. 408–417. ISBN 978-2-294-71451-1. [Google Scholar]
- Tazarourte, K.; Bensalah, N.; Rebillard, L.; Vigué, B. Epidémiologie des Traumatismes Crâniens; MAPAR: Paris, France, 2008. [Google Scholar]
- Masson, F.; Thicoipe, M.; Mokni, T.; Aye, P.; Erny, P.; Dabadie, P. Aquaitaine Group for Severe Brain Injury Study Epidemiology of traumatic comas: A prospective population-based study. Brain Inj. 2003, 17, 279–293. [Google Scholar] [CrossRef]
- Collaborators, M.C.T. Predicting outcome after traumatic brain injury: Practical prognostic models based on large cohort of international patients. BMJ 2008, 336, 425–429. [Google Scholar] [CrossRef]
- Davis, D.P.; Serrano, J.A.; Vilke, G.M.; Sise, M.J.; Kennedy, F.; Eastman, A.B.; Velky, T.; Hoyt, D.B. The predictive value of field versus arrival Glasgow Coma Scale score and TRISS calculations in moderate-to-severe traumatic brain injury. J. Trauma 2006, 60, 985–990. [Google Scholar] [CrossRef]
- Regel, G.; Lobenhoffer, P.; Grotz, M.; Pape, H.C.; Lehmann, U.; Tscherne, H. Treatment results of patients with multiple trauma: An analysis of 3406 cases treated between 1972 and 1991 at a German Level I Trauma Center. J. Trauma 1995, 38, 70–78. [Google Scholar] [CrossRef]
- Hills, M.W.; Deane, S.A. Head injury and facial injury: Is there an increased risk of cervical spine injury? J. Trauma 1993, 34, 549–553, discussion 553–554. [Google Scholar] [CrossRef]
- Sandiego, C.M.; Gallezot, J.-D.; Pittman, B.; Nabulsi, N.; Lim, K.; Lin, S.-F.; Matuskey, D.; Lee, J.-Y.; O’Connor, K.C.; Huang, Y.; et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl. Acad. Sci. USA 2015, 112, 12468–12473. [Google Scholar] [CrossRef]
- Tagliaferri, F.; Compagnone, C.; Korsic, M.; Servadei, F.; Kraus, J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 2006, 148, 255–268, discussion 268. [Google Scholar] [CrossRef]
- Adams, J.H.; Doyle, D.; Ford, I.; Gennarelli, T.A.; Graham, D.I.; McLellan, D.R. Diffuse axonal injury in head injury: Definition, diagnosis and grading. Histopathology 1989, 15, 49–59. [Google Scholar] [CrossRef]
- Li, X.-Y.; Feng, D.-F. Diffuse axonal injury: Novel insights into detection and treatment. J. Clin. Neurosci. 2009, 16, 614–619. [Google Scholar] [CrossRef]
- Carpentier, A.; Galanaud, D.; Puybasset, L.; Muller, J.-C.; Lescot, T.; Boch, A.-L.; Riedl, V.; Riedl, V.; Cornu, P.; Coriat, P.; et al. Early morphologic and spectroscopic magnetic resonance in severe traumatic brain injuries can detect “invisible brain stem damage” and predict “vegetative states”. J. Neurotrauma 2006, 23, 674–685. [Google Scholar] [CrossRef]
- Hammoud, D.A.; Wasserman, B.A. Diffuse axonal injuries: Pathophysiology and imaging. Neuroimaging Clin. N. Am. 2002, 12, 205–216. [Google Scholar] [CrossRef]
- Scheid, R.; Walther, K.; Guthke, T.; Preul, C.; von Cramon, D.Y. Cognitive sequelae of diffuse axonal injury. Arch. Neurol. 2006, 63, 418–424. [Google Scholar] [CrossRef]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef]
- Johannigman, J.A.; Zonies, D.; Dubose, J.; Blakeman, T.C.; Hanseman, D.; Branson, R.D. Reducing Secondary Insults in Traumatic Brain Injury. Mil. Med. 2015, 180, 50–55. [Google Scholar] [CrossRef]
- Groom, R.; Oakley, P.A. Secondary brain injury: Mechanisms and prevention. Curr. Anaesth. Crit. Care 1997, 8, 248–253. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 572. [Google Scholar] [CrossRef]
- Smith, C.; Gentleman, S.M.; Leclercq, P.D.; Murray, L.S.; Griffin, W.S.T.; Graham, D.I.; Nicoll, J.A.R. The neuroinflammatory response in humans after traumatic brain injury: Neuroinflammation after brain injury. Neuropathol. Appl. Neurobiol. 2013, 39, 654–666. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef]
- Martland, H.S. Punch Drunk. JAMA 1928, 91, 1103–1107. [Google Scholar] [CrossRef]
- Smith, D.H.; Johnson, V.E.; Trojanowski, J.Q.; Stewart, W. Chronic traumatic encephalopathy—Confusion and controversies. Nat. Rev. Neurol. 2019, 15, 179–183. [Google Scholar] [CrossRef]
- Omalu, B.I.; DeKosky, S.T.; Minster, R.L.; Kamboh, M.I.; Hamilton, R.L.; Wecht, C.H. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005, 57, 128–134, discussion 128–134. [Google Scholar] [CrossRef]
- Omalu, B.I.; DeKosky, S.T.; Hamilton, R.L.; Minster, R.L.; Kamboh, M.I.; Shakir, A.M.; Wecht, C.H. Chronic traumatic encephalopathy in a national football league player: Part II. Neurosurgery 2006, 59, 1086–1092, discussion 1092–1093. [Google Scholar] [CrossRef]
- Omalu, B.I.; Fitzsimmons, R.P.; Hammers, J.; Bailes, J. Chronic traumatic encephalopathy in a professional American wrestler. J. Forensic. Nurs. 2010, 6, 130–136. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Maroon, J.C.; Winkelman, R.; Bost, J.; Amos, A.; Mathyssek, C.; Miele, V. Chronic traumatic encephalopathy in contact sports: A systematic review of all reported pathological cases. PLoS ONE 2015, 10, e0117338. [Google Scholar] [CrossRef]
- Gardner, A.; Iverson, G.L.; McCrory, P. Chronic traumatic encephalopathy in sport: A systematic review. Br. J. Sports Med. 2014, 48, 84–90. [Google Scholar] [CrossRef]
- Broshek, D.K.; De Marco, A.P.; Freeman, J.R. A review of post-concussion syndrome and psychological factors associated with concussion. Brain Inj. 2015, 29, 228–237. [Google Scholar] [CrossRef]
- Azouvi, P.; Jourdan, C.; Bayen, E.; Darnoux, E.; Ghout, I.; Azerad, S.; Vallat-Azouvi, C.; Ruet, A.; Weiss, J.J.; Aegerter, P.; et al. L’étude PariS-TBI: Suivi longitudinal d’une cohorte de blessés après un traumatisme crânien (TC) sévère. Ann. Phys. Rehabil. Med. 2014, 57, e75. [Google Scholar] [CrossRef]
- Whitnall, L.; McMillan, T.M.; Murray, G.D.; Teasdale, G.M. Disability in young people and adults after head injury: 5–7 year follow up of a prospective cohort study. J. Neurol. Neurosurg. Psychiatry 2006, 77, 640–645. [Google Scholar] [CrossRef]
- Christensen, B.K.; Colella, B.; Inness, E.; Hebert, D.; Monette, G.; Bayley, M.; Green, R.E. Recovery of cognitive function after traumatic brain injury: A multilevel modeling analysis of Canadian outcomes. Arch. Phys. Med. Rehabil. 2008, 89, S3–S15. [Google Scholar] [CrossRef]
- Holbrook, T.L.; Anderson, J.P.; Sieber, W.J.; Browner, D.; Hoyt, D.B. Outcome after major trauma: Discharge and 6-month follow-up results from the Trauma Recovery Project. J. Trauma 1998, 45, 315–323, discussion 323–324. [Google Scholar] [CrossRef]
- Geeraerts, T.; Velly, L.; Abdennour, L.; Asehnoune, K.; Audibert, G.; Bouzat, P.; Bruder, N.; Carrillon, R.; Cottenceau, V.; Cotton, F.; et al. Management of severe traumatic brain injury (first 24hours). Anaesth. Crit. Care Pain Med. 2018, 37, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, G.W.J.; Rubiano, A.M.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; Shutter, L.; et al. Guidelines for the Management of Severe Traumatic Brain Injury: 2020 Update of the Decompressive Craniectomy Recommendations. Neurosurgery 2020, 87, 427–434. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef]
- Visser, J.; van Boxel-Dezaire, A.; Methorst, D.; Brunt, T.; de Kloet, E.R.; Nagelkerken, L. Differential regulation of interleukin-10 (IL-10) and IL-12 by glucocorticoids in vitro. Blood 1998, 91, 4255–4264. [Google Scholar] [CrossRef]
- Salmon, J.E.; Kapur, S.; Meryhew, N.L.; Runquist, O.A.; Kimberly, R.P. High-dose, pulse intravenous methylprednisolone enhances Fc gamma receptor-mediated mononuclear phagocyte function in systemic lupus erythematosus. Arthritis Rheum. 1989, 32, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Pipitone, N.; Bajocchi, G.; Hall, M.; Goulding, N.; Lee, A.; Kingsley, G.; Lanchbury, J.; Panayi, G. Corticosteroids inhibit lymphocyte binding to endothelium and intercellular adhesion: An additional mechanism for their anti-inflammatory and immunosuppressive effect. J. Immunol. 1997, 158, 5007–5016. [Google Scholar]
- Huizenga, N.A.T.M.; Koper, J.W.; de Lange, P.; Pols, H.A.P.; Stolk, R.P.; Burger, H.; Grobbee, D.E.; Brinkmann, A.O.; de Jong, F.H.; Lamberts, S.W.J. A Polymorphism in the Glucocorticoid Receptor Gene May Be Associated with an Increased Sensitivity to Glucocorticoids in vivo. J. Clin. Endocrinol. Metab. 1998, 83, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.D. High-dose glucocorticoid treatment improves neurological recovery in head-injured mice. J. Neurosurg. 1985, 62, 882–887. [Google Scholar] [CrossRef]
- Galicich, J.H.; French, L.A. Use of dexamethasone in the treatment of cerebral edema resulting from brain tumors and brain surgery. Am. Pract. Dig. Treat. 1961, 12, 169–174. [Google Scholar]
- Jeevaratnam, D.R.; Menon, D.K. Survey of intensive care of severely head injured patients in the United Kingdom. BMJ 1996, 312, 944–947. [Google Scholar] [CrossRef]
- Effect of intravenous corticosteroids on death within 14 days in 10 008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial. Lancet 2004, 364, 1321–1328. [CrossRef]
- Alderson, P.; Roberts, I. Corticosteroids for acute traumatic brain injury. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef] [PubMed]
- Saul, T.G.; Ducker, T.B.; Salcman, M.; Carro, E. Steroids in severe head injury: A prospective randomized clinical trial. J. Neurosurg. 1981, 54, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Grumme, T.; Baethmann, A.; Kolodziejczyk, D.; Krimmer, J.; Fischer, M.; Rothe, B.v.E.; Pelka, R.; Bennefeld, H.; Pöllauer, E.; Kostron, H.; et al. Treatment of patients with severe head injury by triamcinolone: A prospective, controlled multicenter clinical trial of 396 cases. Res. Exp. Med. 1995, 195, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Bergold, P.J. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp. Neurol. 2016, 275, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Wei, L.; Sun, Z.-d.; Zhao, S.-g.; Liu, X.-z. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC Neurosci. 2009, 10, 7. [Google Scholar] [CrossRef]
- Amin-Hanjani, S.; Stagliano, N.E.; Yamada, M.; Huang, P.L.; Liao, J.K.; Moskowitz, M.A. Mevastatin, an HMG-CoA Reductase Inhibitor, Reduces Stroke Damage and Upregulates Endothelial Nitric Oxide Synthase in Mice. Stroke 2001, 32, 980–986. [Google Scholar] [CrossRef]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Increase in phosphorylation of Akt and its downstream signaling targets and suppression of apoptosis by simvastatin after traumatic brain injury: Laboratory investigation. J. Neurosurg. 2008, 109, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Qu, C.; Goussev, A.; Jiang, H.; Lu, C.; Schallert, T.; Mahmood, A.; Chen, J.; Li, Y.; Chopp, M. Statins Increase Neurogenesis in the Dentate Gyrus, Reduce Delayed Neuronal Death in the Hippocampal CA3 Region, and Improve Spatial Learning in Rat after Traumatic Brain Injury. J. Neurotrauma 2007, 24, 1132–1146. [Google Scholar] [CrossRef]
- Lu, D.; Goussev, A.; Chen, J.; Pannu, P.; Li, Y.; Mahmood, A.; Chopp, M. Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J. Neurotrauma 2004, 21, 21–32. [Google Scholar] [CrossRef]
- Wang, H.; Lynch, J.R.; Song, P.; Yang, H.-J.; Yates, R.B.; Mace, B.; Warner, D.S.; Guyton, J.R.; Laskowitz, D.T. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp. Neurol. 2007, 206, 59–69. [Google Scholar] [CrossRef]
- Qu, C.; Lu, D.; Goussev, A.; Schallert, T.; Mahmood, A.; Chopp, M. Effect of atorvastatin on spatial memory, neuronal survival, and vascular density in female rats after traumatic brain injury. J. Neurosurg. 2005, 103, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Mahmood, A.; Lu, D.; Wu, H.; Xiong, Y.; Qu, C.; Chopp, M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery 2009, 65, 179–185, discussion 185–186. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gao, W.; Cheng, S.; Yin, D.; Li, F.; Wu, Y.; Sun, D.; Zhou, S.; Wang, D.; Zhang, Y.; et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J. Neuroinflamm. 2017, 14, 167. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, D.; Jiang, H.; Xiong, Y.; Qu, C.; Li, B.; Mahmood, A.; Zhou, D.; Chopp, M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma 2008, 25, 130–139. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Jiang, H.; Li, Y.; Zhang, L.; Robin, A.; Katakowski, M.; Lu, M.; Chopp, M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J. Cereb. Blood Flow Metab. 2005, 25, 281–290. [Google Scholar] [CrossRef]
- Lu, D.; Mahmood, A.; Goussev, A.; Schallert, T.; Qu, C.; Zhang, Z.G.; Li, Y.; Lu, M.; Chopp, M. Atorvastatin reduction of intravascular thrombosis, increase in cerebral microvascular patency and integrity, and enhancement of spatial learning in rats subjected to traumatic brain injury. J. Neurosurg. 2004, 101, 813–821. [Google Scholar] [CrossRef]
- Lynch, J.R.; Wang, H.; McGirt, M.J.; Floyd, J.; Friedman, A.H.; Coon, A.L.; Blessing, R.; Alexander, M.J.; Graffagnino, C.; Warner, D.S.; et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage: Results of a pilot randomized clinical trial. Stroke 2005, 36, 2024–2026. [Google Scholar] [CrossRef]
- Chen, S.-F.; Hung, T.-H.; Chen, C.-C.; Lin, K.-H.; Huang, Y.-N.; Tsai, H.-C.; Wang, J.-Y. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 2007, 81, 288–298. [Google Scholar] [CrossRef]
- Wang, K.-W.; Chen, H.-J.; Lu, K.; Liliang, P.-C.; Liang, C.-L.; Tsai, Y.-D.; Cho, C.-L. Simvastatin Attenuates the Cerebral Vascular Endothelial Inflammatory Response in a Rat Traumatic Brain Injury. Ann. Clin. Lab. Sci. 2014, 44, 145–150. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, S.; Shi, J.; Ai, J.; Qi, M.; Hang, C. Simvastatin reduces secondary brain injury caused by cortical contusion in rats: Possible involvement of TLR4/NF-κB pathway. Exp. Neurol. 2009, 216, 398–406. [Google Scholar] [CrossRef]
- Erdös, B.; Snipes, J.A.; Tulbert, C.D.; Katakam, P.; Miller, A.W.; Busija, D.W. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1264–H1270. [Google Scholar] [CrossRef] [PubMed]
- Turkoglu, O.F.; Eroglu, H.; Okutan, O.; Gurcan, O.; Bodur, E.; Sargon, M.F.; Öner, L.; Beskonaklı, E. Atorvastatin efficiency after traumatic brain injury in rats. Surg. Neurol. 2009, 72, 146–152, discussion 152. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Besson, V.C.; Beziaud, T.; Plotkine, M.; Marchand-Leroux, C. Combination therapy with fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, and simvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, on experimental traumatic brain injury. J Pharmacol. Exp. Ther. 2008, 326, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Sultan, W.; Sapkota, A.; Khurshid, H.; Qureshi, I.A.; Jahan, N.; Went, T.R.; Dominic, J.L.; Win, M.; Kannan, A.; Tara, A.; et al. Statins’ Effect on Cognitive Outcome After Traumatic Brain Injury: A Systematic Review. Cureus 2021, 13, e16953. [Google Scholar] [CrossRef] [PubMed]
- Osier, N.; McGreevy, E.; Pham, L.; Puccio, A.; Ren, D.; Conley, Y.P.; Alexander, S.; Dixon, C.E. Melatonin as a Therapy for Traumatic Brain Injury: A Review of Published Evidence. Int. J. Mol. Sci. 2018, 19, 1539. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar] [CrossRef]
- Lin, H.-W.; Lee, E.-J. Effects of melatonin in experimental stroke models in acute, sub-acute, and chronic stages. Neuropsychiatr. Dis. Treat. 2009, 5, 157–162. [Google Scholar] [CrossRef]
- Pei, Z.; Pang, S.F.; Cheung, R.T.F. Administration of Melatonin After Onset of Ischemia Reduces the Volume of Cerebral Infarction in a Rat Middle Cerebral Artery Occlusion Stroke Model. Stroke 2003, 34, 770–775. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Yamamoto, M.; Takei, N.; Kumazaki, M.; Ungsuparkorn, C.; Hida, H.; Sanberg, P.R.; Nishino, H. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. FASEB J. 2000, 14, 1307–1317. [Google Scholar]
- Shinozuka, K.; Staples, M.; Borlongan, C.V. Melatonin-based therapeutics for neuroprotection in stroke. Int. J. Mol. Sci. 2013, 14, 8924–8947. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Senol, N.; Nazıroğlu, M. Melatonin reduces traumatic brain injury-induced oxidative stress in the cerebral cortex and blood of rats. Neural Regen. Res. 2014, 9, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Ahmad, A.; Crupi, R.; Impellizzeri, D.; Morabito, R.; Esposito, E.; Cuzzocrea, S. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J. Endocrinol. 2013, 217, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Kaushik, P.; Tabassum, H.; Parvez, S. Melatonin Provides Neuroprotection Following Traumatic Brain Injury-Promoted Mitochondrial Perturbation in Wistar Rat. Cell. Mol. Neurobiol. 2021, 41, 765–781. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X.; et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic. Biol. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef]
- Ozdemir, D.; Uysal, N.; Gonenc, S.; Acikgoz, O.; Sonmez, A.; Topcu, A.; Ozdemir, N.; Duman, M.; Semin, I.; Ozkan, H. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol. Res. 2005, 54, 631–637. [Google Scholar] [CrossRef]
- Barlow, K.M.; Brooks, B.L.; MacMaster, F.P.; Kirton, A.; Seeger, T.; Esser, M.; Crawford, S.; Nettel-Aguirre, A.; Zemek, R.; Angelo, M.; et al. A double-blind, placebo-controlled intervention trial of 3 and 10 mg sublingual melatonin for post-concussion syndrome in youths (PLAYGAME): Study protocol for a randomized controlled trial. Trials 2014, 15, 271. [Google Scholar] [CrossRef]
- Barlow, K.M.; Esser, M.J.; Veidt, M.; Boyd, R. Melatonin as a Treatment after Traumatic Brain Injury: A Systematic Review and Meta-Analysis of the Pre-Clinical and Clinical Literature. J. Neurotrauma 2019, 36, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Grima, N.A.; Rajaratnam, S.M.W.; Mansfield, D.; Sletten, T.L.; Spitz, G.; Ponsford, J.L. Efficacy of melatonin for sleep disturbance following traumatic brain injury: A randomised controlled trial. BMC Med. 2018, 16, 8. [Google Scholar] [CrossRef] [PubMed]
- Osier, N.D.; Pham, L.; Pugh, B.J.; Puccio, A.; Ren, D.; Conley, Y.P.; Alexander, S.; Dixon, C.E. Brain injury results in lower levels of melatonin receptors subtypes MT1 and MT2. Neurosci. Lett. 2017, 650, 18–24. [Google Scholar] [CrossRef]
- Arvin, K.L.; Han, B.H.; Du, Y.; Lin, S.; Paul, S.M.; Holtzman, D.M. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann. Neurol. 2002, 52, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef]
- Yrjanheikki, J.; Keinanen, R.; Pellikka, M.; Hokfelt, T.; Koistinaho, J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 15769–15774. [Google Scholar] [CrossRef]
- Crack, P.J.; Gould, J.; Bye, N.; Ross, S.; Ali, U.; Habgood, M.D.; Morganti-Kossman, C.; Saunders, N.R.; Hertzog, P.J. Victorian Neurotrauma Research Group The genomic profile of the cerebral cortex after closed head injury in mice: Effects of minocycline. J. Neural. Transm. 2009, 116, 1–12. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Y.; Dodel, R.; Farlow, M.R.; Paul, S.M.; Du, Y. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci. Lett. 2001, 315, 61–64. [Google Scholar] [CrossRef]
- Tikka, T.M.; Koistinaho, J.E. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 2001, 166, 7527–7533. [Google Scholar] [CrossRef]
- Kovesdi, E.; Kamnaksh, A.; Wingo, D.; Ahmed, F.; Grunberg, N.E.; Long, J.B.; Kasper, C.E.; Agoston, D.V. Acute Minocycline Treatment Mitigates the Symptoms of Mild Blast-Induced Traumatic Brain Injury. Front. Neurol. 2012, 3. [Google Scholar] [CrossRef]
- Hanlon, L.A.; Huh, J.W.; Raghupathi, R. Minocycline Transiently Reduces Microglia/Macrophage Activation but Exacerbates Cognitive Deficits Following Repetitive Traumatic Brain Injury in the Neonatal Rat. J. Neuropathol. Exp. Neurol. 2016, 75, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Pechacek, K.M.; Reck, A.M.; Frankot, M.A.; Vonder Haar, C. Minocycline fails to treat chronic traumatic brain injury-induced impulsivity and attention deficits. Exp. Neurol. 2021, 348, 113924. [Google Scholar] [CrossRef] [PubMed]
- Sangobowale, M.; Nikulina, E.; Bergold, P.J. Minocycline plus N-acetylcysteine protect oligodendrocytes when first dosed 12 h after closed head injury in mice. Neurosci. Lett. 2018, 682, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Sangobowale, M.A.; Grin’kina, N.M.; Whitney, K.; Nikulina, E.; St. Laurent-Ariot, K.; Ho, J.S.; Bayzan, N.; Bergold, P.J. Minocycline plus N-Acetylcysteine Reduce Behavioral Deficits and Improve Histology with a Clinically Useful Time Window. J. Neurotrauma 2018, 35, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Abdel Baki, S.G.; Schwab, B.; Haber, M.; Fenton, A.A.; Bergold, P.J. Minocycline Synergizes with N-Acetylcysteine and Improves Cognition and Memory Following Traumatic Brain Injury in Rats. PLoS ONE 2010, 5, e12490. [Google Scholar] [CrossRef] [PubMed]
- Haber, M.; James, J.; Kim, J.; Sangobowale, M.; Irizarry, R.; Ho, J.; Nikulina, E.; Grin’kina, N.M.; Ramadani, A.; Hartman, I.; et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Koulaeinejad, N.; Haddadi, K.; Ehteshami, S.; Shafizad, M.; Salehifar, E.; Emadian, O.; Ali Mohammadpour, R.; Ala, S. Effects of Minocycline on Neurological Outcomes In Patients with Acute Traumatic Brain Injury: A Pilot Study. Iran. J. Pharm. Res. 2019, 18, 1086–1096. [Google Scholar] [CrossRef]
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 2018, 141, 459–471. [Google Scholar] [CrossRef]
- Russell, G.; Graveley, R.; Seid, J.; Al-Humidan, A.K.; Skjodt, H. Mechanisms of action of cyclosporine and effects on connective tissues. Semin. Arthritis Rheum. 1992, 21, 16–22. [Google Scholar] [CrossRef]
- Li, P.A.; Kristián, T.; He, Q.P.; Siesjö, B.K. Cyclosporin A Enhances Survival, Ameliorates Brain Damage, and Prevents Secondary Mitochondrial Dysfunction after a 30-Minute Period of Transient Cerebral Ischemia. Exp. Neurol. 2000, 165, 153–163. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Thompson, M.; Scheff, S.W. Continuous Infusion of Cyclosporin A Postinjury Significantly Ameliorates Cortical Damage Following Traumatic Brain Injury. Exp. Neurol. 2000, 161, 631–637. [Google Scholar] [CrossRef]
- Mbye, L.H.; Singh, I.N.; Sullivan, P.G.; Springer, J.E.; Hall, E.D. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp. Neurol. 2008, 209, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Thompson, M.B.; Scheff, S.W. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol. 1999, 160, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kilbaugh, T.J.; Bhandare, S.; Lorom, D.H.; Saraswati, M.; Robertson, C.L.; Margulies, S.S. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J. Neurotrauma 2011, 28, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Signoretti, S.; Marmarou, A.; Tavazzi, B.; Dunbar, J.; Amorini, A.M.; Lazzarino, G.; Vagnozzi, R. The Protective Effect of Cyclosporin A upon N-Acetylaspartate and Mitochondrial Dysfunction following Experimental Diffuse Traumatic Brain Injury. J. Neurotrauma 2004, 21, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Darvishzadeh, A.; Huang, K.W.; Malenka, R.C. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 2013, 501, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, S.; Bai, X.; Gao, Y.; Liu, G.; Wang, X.; Liu, D.; Li, T.; Hao, A.; Wang, Z. Oxytocin inhibits lipopolysaccharide-induced inflammation in microglial cells and attenuates microglial activation in lipopolysaccharide-treated mice. J. Neuroinflamm. 2016, 13, 77. [Google Scholar] [CrossRef]
- Wang, S.-C.; Lin, C.-C.; Chen, C.-C.; Tzeng, N.-S.; Liu, Y.-P. Effects of Oxytocin on Fear Memory and Neuroinflammation in a Rodent Model of Posttraumatic Stress Disorder. Int. J. Mol. Sci. 2018, 19, 3848. [Google Scholar] [CrossRef]
- Mairesse, J.; Zinni, M.; Pansiot, J.; Hassan-Abdi, R.; Demene, C.; Colella, M.; Charriaut-Marlangue, C.; Rideau Batista Novais, A.; Tanter, M.; Maccari, S.; et al. Oxytocin receptor agonist reduces perinatal brain damage by targeting microglia. Glia 2019, 67, 345–359. [Google Scholar] [CrossRef]
- Karelina, K.; Stuller, K.A.; Jarrett, B.; Zhang, N.; Wells, J.; Norman, G.J.; DeVries, A.C. Oxytocin Mediates Social Neuroprotection After Cerebral Ischemia. Stroke 2011, 42, 3606–3611. [Google Scholar] [CrossRef]
- Zinni, M.; Colella, M.; Batista Novais, A.R.; Baud, O.; Mairesse, J. Modulating the Oxytocin System During the Perinatal Period: A New Strategy for Neuroprotection of the Immature Brain? Front. Neurol. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Byts, N.; Samoylenko, A.; Fasshauer, T.; Ivanisevic, M.; Hennighausen, L.; Ehrenreich, H.; Sirén, A.-L. Essential role for Stat5 in the neurotrophic but not in the neuroprotective effect of erythropoietin. Cell Death Differ. 2008, 15, 783–792. [Google Scholar] [CrossRef]
- Lu, D.; Mahmood, A.; Qu, C.; Goussev, A.; Schallert, T.; Chopp, M. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J. Neurotrauma 2005, 22, 1011–1017. [Google Scholar] [CrossRef]
- Peng, W.; Xing, Z.; Yang, J.; Wang, Y.; Wang, W.; Huang, W. The efficacy of erythropoietin in treating experimental traumatic brain injury: A systematic review of controlled trials in animal models. J. Neurosurg. 2014, 121, 653–664. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Meng, Y.; Zhang, Y.; Qu, C.; Schallert, T.; Chopp, M. Delayed administration of erythropoietin reducing hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome following traumatic brain injury in rats: Comparison of treatment with single and triple dose. J. Neurosurg. 2010, 113, 598–608. [Google Scholar] [CrossRef]
- Nichol, A.; French, C.; Little, L.; Haddad, S.; Presneill, J.; Arabi, Y.; Bailey, M.; Cooper, D.J.; Duranteau, J.; Huet, O.; et al. Erythropoietin in traumatic brain injury (EPO-TBI): A double-blind randomised controlled trial. Lancet 2015, 386, 2499–2506. [Google Scholar] [CrossRef]
- Katiyar, V.; Chaturvedi, A.; Sharma, R.; Gurjar, H.K.; Goda, R.; Singla, R.; Ganeshkumar, A. Meta-Analysis with Trial Sequential Analysis on the Efficacy and Safety of Erythropoietin in Traumatic Brain Injury: A New Paradigm. World Neurosurg. 2020, 142, 465–475. [Google Scholar] [CrossRef]
- Jantzie, L.; El Demerdash, N.; Newville, J.C.; Robinson, S. Time to reconsider extended erythropoietin treatment for infantile traumatic brain injury? Exp. Neurol. 2019, 318, 205–215. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Dev. Ther. 2014, 8, 2221–2238. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.-C.; Lin, J.-W.; Chang, M.-W.; Wang, C.-C.; Kuo, J.-R.; Yang, C.-Z.; Chang, C.-P. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J. Neurochem. 2010, 115, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Cheong, C.-U.; Chang, C.-P.; Chao, C.-M.; Cheng, B.-C.; Yang, C.-Z.; Chio, C.-C. Etanercept attenuates traumatic brain injury in rats by reducing brain TNF- α contents and by stimulating newly formed neurogenesis. Mediat. Inflamm. 2013, 2013, 620837. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.-C.; Chang, C.-H.; Wang, C.-C.; Cheong, C.-U.; Chao, C.-M.; Cheng, B.-C.; Yang, C.-Z.; Chang, C.-P. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013, 14, 33. [Google Scholar] [CrossRef]
- Tobinick, E.; Rodriguez-Romanacce, H.; Levine, A.; Ignatowski, T.A.; Spengler, R.N. Immediate neurological recovery following perispinal etanercept years after brain injury. Clin. Drug Investig. 2014, 34, 361–366. [Google Scholar] [CrossRef]
- Ignatowski, T.A.; Spengler, R.N.; Dhandapani, K.M.; Folkersma, H.; Butterworth, R.F.; Tobinick, E. Perispinal etanercept for post-stroke neurological and cognitive dysfunction: Scientific rationale and current evidence. CNS Drugs 2014, 28, 679–697. [Google Scholar] [CrossRef]
- Tobinick, E.; Kim, N.M.; Reyzin, G.; Rodriguez-Romanacce, H.; DePuy, V. Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 2012, 26, 1051–1070. [Google Scholar] [CrossRef]
- Tehranian, R.; Andell-Jonsson, S.; Beni, S.M.; Yatsiv, I.; Shohami, E.; Bartfai, T.; Lundkvist, J.; Iverfeldt, K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. Neurotrauma 2002, 19, 939–951. [Google Scholar] [CrossRef]
- Jones, N.C.; Prior, M.J.W.; Burden-Teh, E.; Marsden, C.A.; Morris, P.G.; Murphy, S. Antagonism of the interleukin-1 receptor following traumatic brain injury in the mouse reduces the number of nitric oxide synthase-2-positive cells and improves anatomical and functional outcomes. Eur. J. Neurosci. 2005, 22, 72–78. [Google Scholar] [CrossRef]
- Flygt, J.; Ruscher, K.; Norberg, A.; Mir, A.; Gram, H.; Clausen, F.; Marklund, N. Neutralization of Interleukin-1β following Diffuse Traumatic Brain Injury in the Mouse Attenuates the Loss of Mature Oligodendrocytes. J. Neurotrauma 2018, 35, 2837–2849. [Google Scholar] [CrossRef]
- Newell, E.A.; Todd, B.P.; Mahoney, J.; Pieper, A.A.; Ferguson, P.J.; Bassuk, A.G. Combined Blockade of Interleukin-1α and -1β Signaling Protects Mice from Cognitive Dysfunction after Traumatic Brain Injury. eNeuro 2018, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, S.M.; Faden, A.I. Cortical interleukin-1 beta elevation after traumatic brain injury in the rat: No effect of two selective antagonists on motor recovery. Neurosci. Lett. 2000, 289, 5–8. [Google Scholar] [CrossRef]
- Evans, L.P.; Woll, A.W.; Wu, S.; Todd, B.P.; Hehr, N.; Hedberg-Buenz, A.; Anderson, M.G.; Newell, E.A.; Ferguson, P.J.; Mahajan, V.B.; et al. Modulation of Post-Traumatic Immune Response Using the IL-1 Receptor Antagonist Anakinra for Improved Visual Outcomes. J. Neurotrauma 2020, 37, 1463–1480. [Google Scholar] [CrossRef]
- Clausen, F.; Hånell, A.; Björk, M.; Hillered, L.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Clausen, F.; Hånell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.H.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase II randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef]
- Hergenroeder, G.W.; Moore, A.N.; McCoy, J.P.; Samsel, L.; Ward, N.H.; Clifton, G.L.; Dash, P.K. Serum IL-6: A candidate biomarker for intracranial pressure elevation following isolated traumatic brain injury. J. Neuroinflamm. 2010, 7, 19. [Google Scholar] [CrossRef]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 2013, 40, 471–475. [Google Scholar] [CrossRef]
- Müller, S.; Bianchi, M.E.; Knapp, S. Thermodynamics of HMGB1 interaction with duplex DNA. Biochemistry 2001, 40, 10254–10261. [Google Scholar] [CrossRef]
- Wang, H.; Vishnubhakat, J.M.; Bloom, O.; Zhang, M.; Ombrellino, M.; Sama, A.; Tracey, K.J. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery 1999, 126, 389–392. [Google Scholar] [CrossRef]
- Gao, H.-M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.-S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Chen, Z.; Chen, H.; Yuan, H.; Wang, Y.; Peng, X.; Wei, C.; Yang, J.; Xu, C. Inhibition of HMGB1 mediates neuroprotection of traumatic brain injury by modulating the microglia/macrophage polarization. Biochem. Biophys. Res. Commun. 2018, 497, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4: HMGB1-TLR4 Signaling Promotes Brain Edema after TBI. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef]
- Yang, L.; Wang, F.; Yang, L.; Yuan, Y.; Chen, Y.; Zhang, G.; Fan, Z. HMGB1 a-Box Reverses Brain Edema and Deterioration of Neurological Function in a Traumatic Brain Injury Mouse Model. Cell Physiol. Biochem. 2018, 46, 2532–2542. [Google Scholar] [CrossRef]
- Okuma, Y.; Wake, H.; Teshigawara, K.; Takahashi, Y.; Hishikawa, T.; Yasuhara, T.; Mori, S.; Takahashi, H.K.; Date, I.; Nishibori, M. Anti-High Mobility Group Box 1 Antibody Therapy May Prevent Cognitive Dysfunction After Traumatic Brain Injury. World Neurosurg. 2019, 122, e864–e871. [Google Scholar] [CrossRef]
- Cousin, M. Le cent cinquantenaire du chloroforme. Un agent anesthésique plus merveilleux et terrible encore que l’éther. Ann. Françaises Anesth. Reanim. 1997, 16, 1037–1044. [Google Scholar] [CrossRef]
- Alkire, M.T.; Haier, R.J.; Fallon, J.H. Toward a Unified Theory of Narcosis: Brain Imaging Evidence for a Thalamocortical Switch as the Neurophysiologic Basis of Anesthetic-Induced Unconsciousness. Conscious. Cogn. 2000, 9, 370–386. [Google Scholar] [CrossRef]
- Eilers, H.; Kindler, C.H.; Bickler, P.E. Different Effects of Volatile Anesthetics and Polyhalogenated Alkanes on Depolarization-Evoked Glutamate Release in Rat Cortical Brain Slices. Anesth. Analg. 1999, 88, 1168–1174. [Google Scholar] [CrossRef]
- Larsen, M.; Grøndahl, T.Ø.; Haugstad, T.S.; Langmoen, I.A. The effect of the volatile anesthetic isoflurane on Ca2+-dependent glutamate release from rat cerebral cortex. Brain Res. 1994, 663, 335–337. [Google Scholar] [CrossRef]
- Jones, M.V.; Harrison, N.L. Effects of volatile anesthetics on the kinetics of inhibitory postsynaptic currents in cultured rat hippocampal neurons. J. Neurophysiol. 1993, 70, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Lenz, C.; Frietsch, T.; Fütterer, C.; Rebel, A.; van Ackern, K.; Kuschinsky, W.; Waschke, K.F. Local Coupling of Cerebral Blood Flow to Cerebral Glucose Metabolism during Inhalational Anesthesia in Rats: Desflurane versus Isoflurane. Anesthesiology 1999, 91, 1720. [Google Scholar] [CrossRef] [PubMed]
- Mielck, F.; Stephan, H.; Buhre, W.; Weyland, A.; Sonntag, H. Effects of 1 MAC desflurane on cerebral metabolism, blood flow and carbon dioxide reactivity in humans. Br. J. Anaesth. 1998, 81, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Li, H.; Feng, C.; Zuo, Z. Inhibition of brain ischemia-caused notch activation in microglia may contribute to isoflurane postconditioning-induced neuroprotection in male rats. CNS Neurol. Disord. Drug Targets 2014, 13, 718–732. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ge, M.; Yin, J.; Dai, Z.; Xie, L.; Li, Y.; Liu, X.; Peng, L.; Zhang, G.; Si, J.; et al. Isoflurane post-conditioning down-regulates expression of aquaporin 4 in rats with cerebral ischemia/reperfusion injury and is possibly related to bone morphogenetic protein 4/Smad1/5/8 signaling pathway. Biomed. Pharmacother. 2018, 97, 429–438. [Google Scholar] [CrossRef]
- Wang, S.; Yin, J.; Ge, M.; Dai, Z.; Li, Y.; Si, J.; Ma, K.; Li, L.; Yao, S. Transforming growth-beta 1 contributes to isoflurane postconditioning against cerebral ischemia-reperfusion injury by regulating the c-Jun N-terminal kinase signaling pathway. Biomed. Pharmacother. 2016, 78, 280–290. [Google Scholar] [CrossRef]
- Statler, K.D.; Alexander, H.; Vagni, V.; Holubkov, R.; Dixon, C.E.; Clark, R.S.B.; Jenkins, L.; Kochanek, P.M. Isoflurane exerts neuroprotective actions at or near the time of severe traumatic brain injury. Brain Res. 2006, 1076, 216–224. [Google Scholar] [CrossRef]
- Statler, K.D.; Kochanek, P.M.; Dixon, C.E.; Alexander, H.L.; Warner, D.S.; Clark, R.S.; Wisniewski, S.R.; Graham, S.H.; Jenkins, L.W.; Marion, D.W.; et al. Isoflurane improves long-term neurologic outcome versus fentanyl after traumatic brain injury in rats. J. Neurotrauma 2000, 17, 1179–1189. [Google Scholar] [CrossRef]
- Hertle, D.; Beynon, C.; Zweckberger, K.; Vienenkötter, B.; Jung, C.S.; Kiening, K.; Unterberg, A.; Sakowitz, O.W. Influence of isoflurane on neuronal death and outcome in a rat model of traumatic brain injury. Acta Neurochir. Suppl. 2012, 114, 383–386. [Google Scholar] [CrossRef]
- Beck-Schimmer, B.; Baumann, L.; Restin, T.; Eugster, P.; Hasler, M.; Booy, C.; Schläpfer, M. Sevoflurane attenuates systemic inflammation compared with propofol, but does not modulate neuro-inflammation: A laboratory rat study. Eur. J. Anaesthesiol. 2017, 34, 764–775. [Google Scholar] [CrossRef]
- Hwang, J.-W.; Jeon, Y.-T.; Lim, Y.-J.; Park, H.-P. Sevoflurane Postconditioning-Induced Anti-Inflammation via Inhibition of the Toll-Like Receptor-4/Nuclear Factor Kappa B Pathway Contributes to Neuroprotection against Transient Global Cerebral Ischemia in Rats. Int. J. Mol. Sci. 2017, 18, 2347. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.-D.; Saiyin, H.; Yu, Q.; Liang, W.-M. Effects of sevoflurane preconditioning on microglia/macrophage dynamics and phagocytosis profile against cerebral ischemia in rats. CNS Neurosci. Ther. 2018, 24, 564–571. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, X.; Yang, L.; Miao, X.; Lu, D.-H.; Yang, X.-Y.; Zhou, Z.-B.; Kang, W.-B.; Chen, K.-Y.; Zhou, L.-H.; et al. Neonatal Exposure to Low-Dose (1.2%) Sevoflurane Increases Rats’ Hippocampal Neurogenesis and Synaptic Plasticity in Later Life. Neurotox. Res. 2018, 34, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Zhang, L.; Su, J.; Cai, D.; Xu, Q. Sevoflurane postconditioning improves long-term learning and memory of neonatal hypoxia-ischemia brain damage rats via the PI3K/Akt-mPTP pathway. Brain Res. 2016, 1630, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xiong, X.; Liu, J.; Gu, L.; Li, F.; Wan, Y.; Xu, S. Emulsified Isoflurane Protects Against Transient Focal Cerebral Ischemia Injury in Rats via the PI3K/Akt Signaling Pathway. Anesth. Analg. 2016, 122, 1377–1384. [Google Scholar] [CrossRef]
- Pi, Z.; Lin, H.; Yang, J. Isoflurane reduces pain and inhibits apoptosis of myocardial cells through the phosphoinositide 3-kinase/protein kinase B signaling pathway in mice during cardiac surgery. Mol. Med. Rep. 2018, 17, 6497–6505. [Google Scholar] [CrossRef]
- Thal, S.C.; Luh, C.; Schaible, E.-V.; Timaru-Kast, R.; Hedrich, J.; Luhmann, H.J.; Engelhard, K.; Zehendner, C.M. Volatile anesthetics influence blood-brain barrier integrity by modulation of tight junction protein expression in traumatic brain injury. PLoS ONE 2012, 7, e50752. [Google Scholar] [CrossRef]
- Statler, K.D.; Alexander, H.; Vagni, V.; Dixon, C.E.; Clark, R.S.B.; Jenkins, L.; Kochanek, P.M. Comparison of Seven Anesthetic Agents on Outcome after Experimental Traumatic Brain Injury in Adult, Male Rats. J. Neurotrauma 2006, 23, 97–108. [Google Scholar] [CrossRef]
- Neag, M.-A.; Mitre, A.-O.; Catinean, A.; Mitre, C.-I. An Overview on the Mechanisms of Neuroprotection and Neurotoxicity of Isoflurane and Sevoflurane in Experimental Studies. Brain Res. Bull. 2020, 165, 281–289. [Google Scholar] [CrossRef]
- Maze, M.; Laitio, T. Neuroprotective Properties of Xenon. Mol. Neurobiol. 2020, 57, 118–124. [Google Scholar] [CrossRef]
- Ma, D.; Wilhelm, S.; Maze, M.; Franks, N.P. Neuroprotective and neurotoxic properties of the “inert” gas, xenon. Br. J. Anaesth. 2002, 89, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.; Franks, N.P.; Dickinson, R. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor mediates xenon neuroprotection against hypoxia-ischemia. Anesthesiology 2010, 112, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Campos-Pires, R.; Armstrong, S.P.; Sebastiani, A.; Luh, C.; Gruss, M.; Radyushkin, K.; Hirnet, T.; Werner, C.; Engelhard, K.; Franks, N.P.; et al. Xenon improves neurologic outcome and reduces secondary injury following trauma in an in vivo model of traumatic brain injury. Crit. Care Med. 2015, 43, 149–158. [Google Scholar] [CrossRef]
- Campos-Pires, R.; Hirnet, T.; Valeo, F.; Ong, B.E.; Radyushkin, K.; Aldhoun, J.; Saville, J.; Edge, C.J.; Franks, N.P.; Thal, S.C.; et al. Xenon improves long-term cognitive function, reduces neuronal loss and chronic neuroinflammation, and improves survival after traumatic brain injury in mice. Br. J. Anaesth. 2019, 123, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Campos-Pires, R.; Onggradito, H.; Ujvari, E.; Karimi, S.; Valeo, F.; Aldhoun, J.; Edge, C.J.; Franks, N.P.; Dickinson, R. Xenon treatment after severe traumatic brain injury improves locomotor outcome, reduces acute neuronal loss and enhances early beneficial neuroinflammation: A randomized, blinded, controlled animal study. Crit. Care 2020, 24, 667. [Google Scholar] [CrossRef] [PubMed]
- Filev, A.D.; Silachev, D.N.; Ryzhkov, I.A.; Lapin, K.N.; Babkina, A.S.; Grebenchikov, O.A.; Pisarev, V.M. Effect of Xenon Treatment on Gene Expression in Brain Tissue after Traumatic Brain Injury in Rats. Brain Sci. 2021, 11, 889. [Google Scholar] [CrossRef] [PubMed]
- Abraini, J.H.; Kriem, B.; Balon, N.; Rostain, J.-C.; Risso, J.-J. Gamma-aminobutyric acid neuropharmacological investigations on narcosis produced by nitrogen, argon, or nitrous oxide. Anesth. Analg. 2003, 96, 746–749. [Google Scholar] [CrossRef]
- Sanders, R.D.; Ma, D.; Maze, M. Argon neuroprotection. Crit. Care 2010, 14, 117. [Google Scholar] [CrossRef]
- David, H.N.; Haelewyn, B.; Degoulet, M.; Colomb, D.G.; Risso, J.-J.; Abraini, J.H. Ex Vivo and In Vivo Neuroprotection Induced by Argon When Given after an Excitotoxic or Ischemic Insult. PLoS ONE 2012, 7, e30934. [Google Scholar] [CrossRef]
- Zhuang, L.; Yang, T.; Zhao, H.; Fidalgo, A.R.; Vizcaychipi, M.P.; Sanders, R.D.; Yu, B.; Takata, M.; Johnson, M.R.; Ma, D. The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit. Care Med. 2012, 40, 1724–1730. [Google Scholar] [CrossRef]
- Creed, J.; Cantillana-Riquelme, V.; Yan, B.H.; Ma, S.; Chu, D.; Wang, H.; Turner, D.A.; Laskowitz, D.T.; Hoffmann, U. Argon Inhalation for 24 h After Closed-Head Injury Does not Improve Recovery, Neuroinflammation, or Neurologic Outcome in Mice. Neurocrit. Care 2021, 34, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Fossi, F.; Magliocca, A.; Pascente, R.; Sammali, E.; Baldini, F.; Tolomeo, D.; Micotti, E.; Citerio, G.; Stocchetti, N.; et al. Efficacy of acute administration of inhaled argon on traumatic brain injury in mice. Br. J. Anaesth. 2021, 126, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Furuya, H.; Patel, P.M. Neuroprotective effects of anesthetic agents. J. Anesth. 2005, 19, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Flores Gutiérrez, J.; Nistri, A. Neuroprotective effect of propofol against excitotoxic injury to locomotor networks of the rat spinal cord in vitro. Eur. J. Neurosci. 2016, 44, 2418–2430. [Google Scholar] [CrossRef]
- Koerner, I.P.; Brambrink, A.M. Brain protection by anesthetic agents. Curr. Opin. Anaesthesiol. 2006, 19, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Eberspcher, E.; Heimann, K.; Hollweck, R.; Werner, C.; Schneider, G.; Engelhard, K. The Effect of Electroencephalogram-Targeted High- and Low-Dose Propofol Infusion on Histopathological Damage After Traumatic Brain Injury in the Rat. Anesth. Analg. 2006, 103, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xiao, W.; Wang, J.; Wu, J.; Ren, J.; Hou, J.; Gu, J.; Fan, K.; Yu, B. Propofol Inhibits NLRP3 Inflammasome and Attenuates Blast-Induced Traumatic Brain Injury in Rats. Inflammation 2016, 39, 2094–2103. [Google Scholar] [CrossRef]
- Peters, C.E.; Korcok, J.; Gelb, A.W.; Wilson, J.X. Anesthetic Concentrations of Propofol Protect against Oxidative Stress in Primary Astrocyte Cultures. Anesthesiology 2001, 94, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Zhang, J.; Xu, J.; Sheng, G.; Huang, G. Propofol administration modulates AQP-4 expression and brain edema after traumatic brain injury. Cell Biochem. Biophys. 2013, 67, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Wu, J.; Kabadi, S.V.; Sabirzhanov, B.; Guanciale, K.; Hanscom, M.; Faden, J.; Cardiff, K.; Bengson, C.J.; Faden, A.I. Propofol limits microglial activation after experimental brain trauma through inhibition of nicotinamide adenine dinucleotide phosphate oxidase. Anesthesiology 2013, 119, 1370–1388. [Google Scholar] [CrossRef]
- Liu, F.; Chen, M.-R.; Liu, J.; Zou, Y.; Wang, T.-Y.; Zuo, Y.-X.; Wang, T.-H. Propofol administration improves neurological function associated with inhibition of pro-inflammatory cytokines in adult rats after traumatic brain injury. Neuropeptides 2016, 58, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.C.; Timaru-Kast, R.; Wilde, F.; Merk, P.; Johnson, F.; Frauenknecht, K.; Sebastiani, A.; Sommer, C.; Staib-Lasarzik, I.; Werner, C.; et al. Propofol impairs neurogenesis and neurologic recovery and increases mortality rate in adult rats after traumatic brain injury. Crit. Care Med. 2014, 42, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, L.; Shen, J.; Wang, Z.; Qian, Y. Effect of propofol on mucous permeability and inflammatory mediators expression in the intestine following traumatic brain injury in rats. Cytokine 2007, 40, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, X.; Wu, Y.-G.; Lyu, L.; Zhou, Z.-W.; Zhang, J.-N. Dexmedetomidine attenuates traumatic brain injury: Action pathway and mechanisms. Neural. Regen. Res. 2018, 13, 819–826. [Google Scholar] [CrossRef]
- Zhang, M.-H.; Zhou, X.-M.; Cui, J.-Z.; Wang, K.-J.; Feng, Y.; Zhang, H.-A. Neuroprotective effects of dexmedetomidine on traumatic brain injury: Involvement of neuronal apoptosis and HSP70 expression. Mol. Med. Rep. 2018, 17, 8079–8086. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Vogel, T.; Gao, X.; Lin, B.; Kulwin, C.; Chen, J. Neuroprotective effect of dexmedetomidine in a murine model of traumatic brain injury. Sci. Rep. 2018, 8, 4935. [Google Scholar] [CrossRef]
- Shen, M.; Wang, S.; Wen, X.; Han, X.-R.; Wang, Y.-J.; Zhou, X.-M.; Zhang, M.-H.; Wu, D.-M.; Lu, J.; Zheng, Y.-L. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharmacother. 2017, 95, 885–893. [Google Scholar] [CrossRef]
- Qiu, Z.; Lu, P.; Wang, K.; Zhao, X.; Li, Q.; Wen, J.; Zhang, H.; Li, R.; Wei, H.; Lv, Y.; et al. Dexmedetomidine Inhibits Neuroinflammation by Altering Microglial M1/M2 Polarization Through MAPK/ERK Pathway. Neurochem. Res. 2020, 45, 345–353. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, P.; Zheng, H.; Ren, Y.-Q.; Yan, H. Dexmedetomidine reduces hippocampal microglia inflammatory response induced by surgical injury through inhibiting NLRP3. Chin. J. Traumatol. 2019, 22, 161–165. [Google Scholar] [CrossRef]
- Zheng, B.; Zhang, S.; Ying, Y.; Guo, X.; Li, H.; Xu, L.; Ruan, X. Administration of Dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci. Rep. 2018, 38, BSR20180892. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Chen, Y.; Luan, H.; Zhang, X.; Zhao, Z.; Wu, Y. Dexmedetomidine reduces inflammation in traumatic brain injury by regulating the inflammatory responses of macrophages and splenocytes. Exp. Ther. Med. 2019, 18, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.Z.; Matias, B.I.; Maciel, A.L.; Abelaira, H.M.; Ignácio, Z.M.; de Moura, A.B.; Matos, D.; Danielski, L.G.; Petronilho, F.; Carvalho, A.F.; et al. Mechanism of synergistic action on behavior, oxidative stress and inflammation following co-treatment with ketamine and different antidepressant classes. Pharmacol. Rep. 2017, 69, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Luggya, T.S.; Roche, T.; Ssemogerere, L.; Kintu, A.; Kasumba, J.M.; Kwizera, A.; Tindimwebwa, J.V. Effect of low-dose ketamine on post-operative serum IL-6 production among elective surgical patients: A randomized clinical trial. Afr. Health Sci. 2017, 17, 500–507. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marcoux, F.W.; Goodrich, J.E.; Dominick, M.A. Ketamine prevents ischemic neuronal injury. Brain Res. 1988, 452, 329–335. [Google Scholar] [CrossRef]
- Chang, L.C.; Raty, S.R.; Ortiz, J.; Bailard, N.S.; Mathew, S.J. The emerging use of ketamine for anesthesia and sedation in traumatic brain injuries. CNS Neurosci. Ther. 2013, 19, 390–395. [Google Scholar] [CrossRef]
- Liang, J.; Wu, S.; Xie, W.; He, H. Ketamine ameliorates oxidative stress-induced apoptosis in experimental traumatic brain injury via the Nrf2 pathway. DDDT 2018, 12, 845–853. [Google Scholar] [CrossRef]
- Wagner, A.K.; McCullough, E.H.; Niyonkuru, C.; Ozawa, H.; Loucks, T.L.; Dobos, J.A.; Brett, C.A.; Santarsieri, M.; Dixon, C.E.; Berga, S.L.; et al. Acute serum hormone levels: Characterization and prognosis after severe traumatic brain injury. J. Neurotrauma 2011, 28, 871–888. [Google Scholar] [CrossRef]
- Bazarian, J.J.; Blyth, B.; Mookerjee, S.; He, H.; McDermott, M.P. Sex differences in outcome after mild traumatic brain injury. J. Neurotrauma 2010, 27, 527–539. [Google Scholar] [CrossRef]
- Groswasser, Z.; Cohen, M.; Keren, O. Female TBI patients recover better than males. Brain Inj. 1998, 12, 805–808. [Google Scholar] [CrossRef]
- Garringer, J.A.; Niyonkuru, C.; McCullough, E.H.; Loucks, T.; Dixon, C.E.; Conley, Y.P.; Berga, S.; Wagner, A.K. Impact of Aromatase Genetic Variation on Hormone Levels and Global Outcome after Severe TBI. J. Neurotrauma 2013, 30, 1415–1425. [Google Scholar] [CrossRef]
- Roof, R.L.; Duvdevani, R.; Braswell, L.; Stein, D.G. Progesterone Facilitates Cognitive Recovery and Reduces Secondary Neuronal Loss Caused by Cortical Contusion Injury in Male Rats. Exp. Neurol. 1994, 129, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Gatson, J.W.; Warren, V.; Abdelfattah, K.; Wolf, S.; Hynan, L.S.; Moore, C.; Diaz-Arrastia, R.; Minei, J.P.; Madden, C.; Wigginton, J.G. Detection of β-amyloid oligomers as a predictor of neurological outcome after brain injury: Laboratory investigation. J. Neurosurg. 2013, 118, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Pepe, P.; Wigginton, J.; Gatson, J.; Simpkins, J.; Maass, D.; AbdelFattah, K.; Idris, A.; Warren, V.; Minei, J. Single-dose estrogen infusion can amplify brain levels of Sonic hedgehog, a signal protein for neuro stem cells and repair following the indirect brain injury resulting after severe torso burns. Crit. Care 2013, 17. [Google Scholar] [CrossRef]
- McClean, J.; Nuñez, J.L. 17α-Estradiol is neuroprotective in male and female rats in a model of early brain injury. Exp. Neurol. 2008, 210, 41–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neese, S.L.; Clough, R.W.; Banz, W.J.; Smith, D.C. Z-Bisdehydrodoisynolic acid (Z-BDDA): An estrogenic seco-steroid that enhances behavioral recovery following moderate fluid percussion brain injury in male rats. Brain Res. 2010, 1362, 93–101. [Google Scholar] [CrossRef]
- Behl, C.; Manthey, D. Neuroprotective activites of estrogen: An update. J. Neurocytol. 2000, 29, 351–358. [Google Scholar] [CrossRef]
- Green, P.S.; Yang, S.-H.; Simpkins, J.W. Neuroprotective Effects of Phenolic A Ring Oestrogens. In Novartis Foundation Symposia; Chadwick, D.J., Goode, J.A., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2008; pp. 202–220. ISBN 978-0-471-49203-0. [Google Scholar]
- Brann, D.W.; Dhandapani, K.; Wakade, C.; Mahesh, V.B.; Khan, M.M. Neurotrophic and neuroprotective actions of estrogen: Basic mechanisms and clinical implications. Steroids 2007, 72, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, M.-A.; Santos-Galindo, M.; Bellini, M.-J.; Azcoitia, I.; Garcia-Segura, L.M. Actions of estrogens on glial cells: Implications for neuroprotection. Biochim. Biophys. Acta 2010, 1800, 1106–1112. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory Effects of Estrogen on Microglial Activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef]
- Emerson, C.S.; Headrick, J.P.; Vink, R. Estrogen improves biochemical and neurologic outcome following traumatic brain injury in male rats, but not in females. Brain Res. 1993, 608, 95–100. [Google Scholar] [CrossRef]
- Kim, H.; Cam-Etoz, B.; Zhai, G.; Hubbard, W.J.; Zinn, K.R.; Chaudry, I.H. Salutary Effects of Estrogen Sulfate for Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, Y.; Zhang, L.; Liu, M.; Zhao, J.; Zhang, Z.; Ma, Y.; Hou, W. Estrogen Attenuates Traumatic Brain Injury by Inhibiting the Activation of Microglia and Astrocyte-Mediated Neuroinflammatory Responses. Mol. Neurobiol. 2021, 58, 1052–1061. [Google Scholar] [CrossRef]
- Li, L.-Z.; Bao, Y.-J.; Zhao, M. 17beta-estradiol attenuates programmed cell death in cortical pericontusional zone following traumatic brain injury via upregulation of ERalpha and inhibition of caspase-3 activation. Neurochem. Int. 2011, 58, 126–133. [Google Scholar] [CrossRef]
- Kövesdi, E.; Szabó-Meleg, E.; Abrahám, I.M. The Role of Estradiol in Traumatic Brain Injury: Mechanism and Treatment Potential. Int. J. Mol. Sci. 2020, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.-L.; Wang, X.; Zou, Y.-J.; Xing, J.-S.; Lou, J.-C.; Zou, S.; Ma, B.-B.; Ding, Y.; Zhang, B. Bazedoxifene protects cerebral autoregulation after traumatic brain injury and attenuates impairments in blood-brain barrier damage: Involvement of anti-inflammatory pathways by blocking MAPK signaling. Inflamm. Res. 2019, 68, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Asl, S.Z.; Khaksari, M.; Khachki, A.S.; Shahrokhi, N.; Nourizade, S. Contribution of estrogen receptors alpha and beta in the brain response to traumatic brain injury. J. Neurosurg. 2013, 119, 353–361. [Google Scholar] [CrossRef]
- Khaksari, M.; Abbasloo, E.; Dehghan, F.; Soltani, Z.; Asadikaram, G. The brain cytokine levels are modulated by estrogen following traumatic brain injury: Which estrogen receptor serves as modulator? Int. Immunopharmacol. 2015, 28, 279–287. [Google Scholar] [CrossRef]
- Roof, R.L.; Hall, E.D. Gender Differences in Acute CNS Trauma and Stroke: Neuroprotective Effects of Estrogen and Progesterone. J. Neurotrauma 2000, 17, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Su, C. Progesterone and neuroprotection. Horm. Behav. 2013, 63, 284–290. [Google Scholar] [CrossRef]
- Stein, D.G. A clinical/translational perspective: Can a developmental hormone play a role in the treatment of traumatic brain injury? Horm. Behav. 2013, 63, 291–300. [Google Scholar] [CrossRef]
- Roof, R.L.; Duvdevani, R.; Heyburn, J.W.; Stein, D.G. Progesterone Rapidly Decreases Brain Edema: Treatment Delayed up to 24 Hours Is Still Effective. Exp. Neurol. 1996, 138, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.W.; Bauer, M.E.; Hoffman, S.W.; Stein, D.G. Serum Progesterone Levels Correlate with Decreased Cerebral Edema after Traumatic Brain Injury in Male Rats. J. Neurotrauma 2001, 18, 901–909. [Google Scholar] [CrossRef]
- Guo, Q.; Sayeed, I.; Baronne, L.M.; Hoffman, S.W.; Guennoun, R.; Stein, D.G. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp. Neurol. 2006, 198, 469–478. [Google Scholar] [CrossRef]
- Vink, R.; Van Den Heuvel, C. Recent advances in the development of multifactorial therapies for the treatment of traumatic brain injury. Expert Opin. Investig. Drugs 2004, 13, 1263–1274. [Google Scholar] [CrossRef]
- Chen, G.; Shi, J.; Jin, W.; Wang, L.; Xie, W.; Sun, J.; Hang, C. Progesterone administration modulates TLRs/NF-kappaB signaling pathway in rat brain after cortical contusion. Ann. Clin. Lab. Sci. 2008, 38, 65–74. [Google Scholar] [PubMed]
- Pan, D.-S.; Liu, W.-G.; Yang, X.-F.; Cao, F. Inhibitory effect of progesterone on inflammatory factors after experimental traumatic brain injury. Biomed. Environ. Sci. 2007, 20, 432–438. [Google Scholar] [PubMed]
- Roof, R.L.; Hoffman, S.W.; Stein, D.G. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol. Chem. Neuropathol. 1997, 31, 1–11. [Google Scholar] [CrossRef]
- Ghoumari, A.M.; Ibanez, C.; El-Etr, M.; Leclerc, P.; Eychenne, B.; O’Malley, B.W.; Baulieu, E.E.; Schumacher, M. Progesterone and its metabolites increase myelin basic protein expression in organotypic slice cultures of rat cerebellum. J. Neurochem. 2003, 86, 848–859. [Google Scholar] [CrossRef]
- Koenig, H.L.; Schumacher, M.; Ferzaz, B.; Thi, A.N.; Ressouches, A.; Guennoun, R.; Jung-Testas, I.; Robel, P.; Akwa, Y.; Baulieu, E.E. Progesterone synthesis and myelin formation by Schwann cells. Science 1995, 268, 1500–1503. [Google Scholar] [CrossRef]
- Ibanez, C.; Shields, S.A.; El-Etr, M.; Baulieu, E.-E.; Schumacher, M.; Franklin, R.J.M. Systemic progesterone administration results in a partial reversal of the age-associated decline in CNS remyelination following toxin-induced demyelination in male rats: Progesterone and CNS remyelination. Neuropathol. Appl. Neurobiol. 2004, 30, 80–89. [Google Scholar] [CrossRef]
- Wagner, C.K. Progesterone receptors and neural development: A gap between bench and bedside? Endocrinology 2008, 149, 2743–2749. [Google Scholar] [CrossRef]
- Wagner, C.K. The many faces of progesterone: A role in adult and developing male brain. Front. Neuroendocrinol. 2006, 27, 340–359. [Google Scholar] [CrossRef]
- Quadros, P.S.; Wagner, C.K. Regulation of Progesterone Receptor Expression by Estradiol Is Dependent on Age, Sex and Region in the Rat Brain. Endocrinology 2008, 149, 3054–3061. [Google Scholar] [CrossRef]
- Quadros, P.S.; Pfau, J.L.; Wagner, C.K. Distribution of progesterone receptor immunoreactivity in the fetal and neonatal rat forebrain. J. Comp. Neurol. 2007, 504, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Uysal, N.; Baykara, B.; Kiray, M.; Cetin, F.; Aksu, I.; Dayi, A.; Gurpinar, T.; Ozdemir, D.; Arda, M.N. Combined treatment with progesterone and magnesium sulfate positively affects traumatic brain injury in immature rats. Turk. Neurosurg. 2013, 23, 129–137. [Google Scholar] [CrossRef]
- Baykara, B.; Aksu, I.; Buyuk, E.; Kiray, M.; Sisman, A.; Baykara, B.; Dayi, A.; Tas, A.; Ozdemir, D.; Arda, M.; et al. Progesterone treatment decreases traumatic brain injury induced anxiety and is correlated with increased serum IGF-1 levels; prefrontal cortex, amygdala, hippocampus neuron density; and reduced serum corticosterone levels in immature rats. Biotech. Histochem. 2013, 88, 250–257. [Google Scholar] [CrossRef]
- Trotter, A.; Bokelmann, B.; Sorgo, W.; Bechinger-Kornhuber, D.; Heinemann, H.; Schmücker, G.; Oesterle, M.; Köhntop, B.; Brisch, K.H.; Pohlandt, F. Follow-up examination at the age of 15 months of extremely preterm infants after postnatal estradiol and progesterone replacement. J. Clin. Endocrinol. Metab. 2001, 86, 601–603. [Google Scholar] [CrossRef]
- Trotter, A.; Steinmacher, J.; Kron, M.; Pohlandt, F. Neurodevelopmental follow-up at five years corrected age of extremely low birth weight infants after postnatal replacement of 17β-estradiol and progesterone. J. Clin. Endocrinol. Metab. 2012, 97, 1041–1047. [Google Scholar] [CrossRef]
- Stein, D.G. Progesterone exerts neuroprotective effects after brain injury. Brain Res. Rev. 2008, 57, 386–397. [Google Scholar] [CrossRef]
- Wright, D.W.; Kellermann, A.L.; Hertzberg, V.S.; Clark, P.L.; Frankel, M.; Goldstein, F.C.; Salomone, J.P.; Dent, L.L.; Harris, O.A.; Ander, D.S.; et al. ProTECT: A Randomized Clinical Trial of Progesterone for Acute Traumatic Brain Injury. Ann. Emerg. Med. 2007, 49, 391–402.e2. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Wei, J.; Yan, W.; Wang, W.; Lu, Z. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: A randomized controlled trial. Crit. Care 2008, 12, R61. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Aminmansour, B.; Ghorbani, A.; Rahmani, P.; Nourian, M.; Rezvani, M.; Torkashvand, M.; Moradi, M. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: A randomized clinical trial with placebo group. Adv. Biomed. Res. 2012, 1, 58. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, M.; Boustani, M.R.; Pak, N.; Panahi, F.; Salehpour, F.; Lotfinia, I.; Meshkini, A.; Daghighi, S.; vahedi, P.; Khani, M.; et al. Effect of progesterone administration on prognosis of patients with diffuse axonal injury due to severe head trauma. Clin. Neurol. Neurosurg. 2013, 115, 2019–2022. [Google Scholar] [CrossRef]
- Wright, D.W.; Yeatts, S.D.; Silbergleit, R.; Palesch, Y.Y.; Hertzberg, V.S.; Frankel, M.; Goldstein, F.C.; Caveney, A.F.; Howlett-Smith, H.; Bengelink, E.M.; et al. Very Early Administration of Progesterone for Acute Traumatic Brain Injury. N. Engl. J. Med. 2014, 371, 2457–2466. [Google Scholar] [CrossRef]
- Skolnick, B.E.; Maas, A.I.; Narayan, R.K.; van der Hoop, R.G.; MacAllister, T.; Ward, J.D.; Nelson, N.R.; Stocchetti, N. A Clinical Trial of Progesterone for Severe Traumatic Brain Injury. N. Engl. J. Med. 2014, 371, 2467–2476. [Google Scholar] [CrossRef]
- Hultquist, D.E.; Xu, F.; Quandt, K.S.; Shlafer, M.; Mack, C.P.; Till, G.O.; Seekamp, A.; Betz, A.L.; Ennis, S.R. Evidence that NADPH-dependent methemoglobin reductase and administered riboflavin protect tissues from oxidative injury. Am. J. Hematol. 1993, 42, 13–18. [Google Scholar] [CrossRef]
- Lin, Y.; Desbois, A.; Jiang, S.; Hou, S.T. Group B vitamins protect murine cerebellar granule cells from glutamate/NMDA toxicity. Neuroreport 2004, 15, 2241–2244. [Google Scholar] [CrossRef]
- Hoane, M.R.; Wolyniak, J.G.; Akstulewicz, S.L. Administration of riboflavin improves behavioral outcome and reduces edema formation and glial fibrillary acidic protein expression after traumatic brain injury. J. Neurotrauma 2005, 22, 1112–1122. [Google Scholar] [CrossRef]
- Barbre, A.B.; Hoane, M.R. Magnesium and riboflavin combination therapy following cortical contusion injury in the rat. Brain Res. Bull. 2006, 69, 639–646. [Google Scholar] [CrossRef]
- Vonder Haar, C. The Use of Nicotinamide as a Treatment for Experimental Traumatic Brain Injury and Stroke: A Review and Evaluation. Clin. Pharm. Biopharm. 2013, S1. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. The vitamin nicotinamide: Translating nutrition into clinical care. Molecules 2009, 14, 3446–3485. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Pierce, J.L.; Holland, M.A.; Anderson, G.D. Nicotinamide treatment induces behavioral recovery when administered up to 4 h following cortical contusion injury in the rat. Neuroscience 2008, 154, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Vonder Haar, C.; Maass, W.R.; Jacobs, E.A.; Hoane, M.R. Deficits in discrimination after experimental frontal brain injury are mediated by motivation and can be improved by nicotinamide administration. J. Neurotrauma 2014, 31, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Akstulewicz, S.L.; Toppen, J. Treatment with vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. J. Neurotrauma 2003, 20, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Goffus, A.M.; Anderson, G.D.; Hoane, M. Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid. Med. Cell Longev. 2010, 3, 145–152. [Google Scholar] [CrossRef]
- Hoane, M.R.; Pierce, J.L.; Kaufman, N.A.; Beare, J.E. Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid. Med. Cell Longev. 2008, 1, 46–53. [Google Scholar] [CrossRef]
- Hoane, M.R.; Tan, A.A.; Pierce, J.L.; Anderson, G.D.; Smith, D.C. Nicotinamide treatment reduces behavioral impairments and provides cortical protection after fluid percussion injury in the rat. J. Neurotrauma 2006, 23, 1535–1548. [Google Scholar] [CrossRef]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef]
- Vonder Haar, C.; Anderson, G.D.; Hoane, M.R. Continuous nicotinamide administration improves behavioral recovery and reduces lesion size following bilateral frontal controlled cortical impact injury. Behav. Brain Res. 2011, 224, 311–317. [Google Scholar] [CrossRef]
- Bender, D.A. Non-nutritional uses of vitamin B6. Br. J. Nutr. 1999, 81, 7–20. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.-Y.; Kim, D.H.; Lee, B.-H.; Kwon, Y.-G.; Won, M.H. Time course of changes in pyridoxal 5’-phosphate (vitamin B6 active form) and its neuroprotection in experimental ischemic damage. Exp. Neurol. 2007, 206, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Oka, T. Modulation of gene expression by vitamin B6. Nutr. Res. Rev. 2001, 14, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, L.; Bergami, R.; Fiorentini, D.; Marchetti, M.; Landi, L.; Tolomelli, B. Vitamin B6 deficiency affects antioxidant defences in rat liver and heart. Biochem. Mol. Biol. Int. 1998, 46, 689–697. [Google Scholar] [CrossRef]
- Kuypers, N.J.; Hoane, M.R. Pyridoxine administration improves behavioral and anatomical outcome after unilateral contusion injury in the rat. J. Neurotrauma 2010, 27, 1275–1282. [Google Scholar] [CrossRef]
- Xu, Y.; Sladky, J.T.; Brown, M.J. Dose-dependent expression of neuronopathy after experimental pyridoxine intoxication. Neurology 1989, 39, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Krinke, G.; Schaumburg, H.H.; Spencer, P.S.; Suter, J.; Thomann, P.; Hess, R. Pyridoxine megavitaminosis produces degeneration of peripheral sensory neurons (sensory neuronopathy) in the dog. Neurotoxicology 1981, 2, 13–24. [Google Scholar]
- Fenech, M. The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutat. Res. 2001, 475, 57–67. [Google Scholar] [CrossRef]
- Naim, M.Y.; Friess, S.; Smith, C.; Ralston, J.; Ryall, K.; Helfaer, M.A.; Margulies, S.S. Folic acid enhances early functional recovery in a piglet model of pediatric head injury. Dev. Neurosci. 2010, 32, 466–479. [Google Scholar] [CrossRef]
- Vonder Haar, C.; Emery, M.A.; Hoane, M.R. Chronic folic acid administration confers no treatment effects in either a high or low dose following unilateral controlled cortical impact injury in the rat. Restor. Neurol. Neurosci. 2012, 30, 291–302. [Google Scholar] [CrossRef]
- Grünewald, R.A. Ascorbic acid in the brain. Brain Res. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Awasthi, D.; Church, D.F.; Torbati, D.; Carey, M.E.; Pryor, W.A. Oxidative stress following traumatic brain injury in rats. Surg. Neurol. 1997, 47, 575–581, discussion 581–582. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; Sokolova, T.V.; Ritov, V.B.; Quinn, P.J.; Rose, M.; Kochanek, P.; Graham, S.H.; Kagan, V.E. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000, 75, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mecocci, P.; Frei, B. Plasma vitamin C levels are decreased and correlated with brain damage in patients with intracranial hemorrhage or head trauma. Stroke 2001, 32, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Moor, E.; Shohami, E.; Kanevsky, E.; Grigoriadis, N.; Symeonidou, C.; Kohen, R. Impairment of the ability of the injured aged brain in elevating urate and ascorbate. Exp. Gerontol. 2006, 41, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-W.; Wang, H.-K.; Chen, H.-J.; Liliang, P.-C.; Liang, C.-L.; Tsai, Y.-D.; Cho, C.-L.; Lu, K. Simvastatin Combined with Antioxidant Attenuates the Cerebral Vascular Endothelial Inflammatory Response in a Rat Traumatic Brain Injury. BioMed Res. Int. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Ishaq, G.M.; Saidu, Y.; Bilbis, L.S.; Muhammad, S.A.; Jinjir, N.; Shehu, B.B. Effects of α-tocopherol and ascorbic acid in the severity and management of traumatic brain injury in albino rats. J. Neurosci. Rural Pract. 2013, 4, 292–297. [Google Scholar] [CrossRef]
- Lin, J.-L.; Huang, Y.-H.; Shen, Y.-C.; Huang, H.-C.; Liu, P.-H. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef]
- Maekawa, T.; Uchida, T.; Nakata-Horiuchi, Y.; Kobayashi, H.; Kawauchi, S.; Kinoshita, M.; Saitoh, D.; Sato, S. Oral ascorbic acid 2-glucoside prevents coordination disorder induced via laser-induced shock waves in rat brain. PLoS ONE 2020, 15, e0230774. [Google Scholar] [CrossRef]
- Putzu, A.; Daems, A.-M.; Lopez-Delgado, J.C.; Giordano, V.F.; Landoni, G. The Effect of Vitamin C on Clinical Outcome in Critically Ill Patients: A Systematic Review With Meta-Analysis of Randomized Controlled Trials. Crit. Care Med. 2019, 47, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Razmkon, A.; Sadidi, A.; Sherafat-Kazemzadeh, E.; Mehrafshan, A.; Jamali, M.; Malekpour, B.; Saghafinia, M. Administration of vitamin C and vitamin E in severe head injury: A randomized double-blind controlled trial. Clin. Neurosurg. 2011, 58, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Leichtle, S.W.; Sarma, A.K.; Strein, M.; Yajnik, V.; Rivet, D.; Sima, A.; Brophy, G.M. High-Dose Intravenous Ascorbic Acid: Ready for Prime Time in Traumatic Brain Injury? Neurocrit. Care 2020, 32, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.W.; Sharma, B. A review of the neuroprotective role of vitamin D in traumatic brain injury with implications for supplementation post-concussion. Brain Inj. 2016, 30, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Cekic, M.; Stein, D.G. Traumatic brain injury and aging: Is a combination of progesterone and vitamin D hormone a simple solution to a complex problem? Neurotherapeutics 2010, 7, 81–90. [Google Scholar] [CrossRef]
- Hua, F.; Reiss, J.I.; Tang, H.; Wang, J.; Fowler, X.; Sayeed, I.; Stein, D.G. Progesterone and low-dose vitamin D hormone treatment enhances sparing of memory following traumatic brain injury. Horm. Behav. 2012, 61, 642–651. [Google Scholar] [CrossRef]
- Tang, H.; Hua, F.; Wang, J.; Yousuf, S.; Atif, F.; Sayeed, I.; Stein, D.G. Progesterone and vitamin D combination therapy modulates inflammatory response after traumatic brain injury. Brain Inj. 2015, 29, 1165–1174. [Google Scholar] [CrossRef]
- Tang, H.; Hua, F.; Wang, J.; Sayeed, I.; Wang, X.; Chen, Z.; Yousuf, S.; Atif, F.; Stein, D.G. Progesterone and vitamin D: Improvement after traumatic brain injury in middle-aged rats. Horm. Behav. 2013, 64, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, K.; Hu, T.; Wang, G.; Wang, W.; Zhang, J. Vitamin D3 Supplement Attenuates Blood-Brain Barrier Disruption and Cognitive Impairments in a Rat Model of Traumatic Brain Injury. Neuromol. Med. 2021, 23, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Jeong, S.W.; Kim, M.Y.; Park, J.B.; Kim, M.S. The Effect of Vitamin D Supplementation in Patients with Acute Traumatic Brain Injury. World Neurosurg. 2019, 126, e1421–e1426. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kumar, A.; Choudhary, A.; Sharma, S.; Khurana, L.; Sharma, N.; Kumar, V.; Bisht, A. Neuroprotective Role of Oral Vitamin D Supplementation on Consciousness and Inflammatory Biomarkers in Determining Severity Outcome in Acute Traumatic Brain Injury Patients: A Double-Blind Randomized Clinical Trial. Clin. Drug Investig. 2020, 40, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.L.; Lyeth, B.G.; Jenkins, L.W.; Taft, W.C.; DeLorenzo, R.J.; Hayes, R.L. Effect of D, alpha-tocopheryl succinate and polyethylene glycol on performance tests after fluid percussion brain injury. J. Neurotrauma 1989, 6, 71–81. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef]
- Stein, D.G.; Halks-Miller, M.; Hoffman, S.W. Intracerebral administration of alpha-tocopherol-containing liposomes facilitates behavioral recovery in rats with bilateral lesions of the frontal cortex. J. Neurotrauma 1991, 8, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Han, Y.; Ye, W.; Liu, F.; Zhuang, K.; Wu, G. Alpha tocopherol treatment reduces the expression of Nogo-A and NgR in rat brain after traumatic brain injury. J. Surg. Res. 2013, 182, e69–e77. [Google Scholar] [CrossRef] [PubMed]
- Koç, R.K.; Kurtsoy, A.; Paşaoğlu, H.; Karaküçük, E.I.; Oktem, I.S.; Meral, M. Lipid peroxidation and oedema in experimental brain injury: Comparison of treatment with methylprednisolone, tirilazad mesylate and vitamin E. Res. Exp. Med. 1999, 199, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, J.; Smrcka, M.; Bienertova-Vasku, J. Therapeutic potential of vitamin E and its derivatives in traumatic brain injury-associated dementia. Neurol. Sci. 2018, 39, 989–998. [Google Scholar] [CrossRef]
- Hall, E.D.; Yonkers, P.A.; Andrus, P.K.; Cox, J.W.; Anderson, D.K. Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. J. Neurotrauma 1992, 9, S425–S442. [Google Scholar]
- Huskisson, E.; Maggini, S.; Ruf, M. The influence of micronutrients on cognitive function and performance. J. Int. Med. Res. 2007, 35, 1–19. [Google Scholar] [CrossRef]
- van den Heuvel, C.; Vink, R. The role of magnesium in traumatic brain injury. Clin. Calcium 2004, 14, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.P.; Gulati, A. Use of magnesium in traumatic brain injury. Neurotherapeutics 2010, 7, 91–99. [Google Scholar] [CrossRef]
- Vink, R.; Cook, N.L.; van den Heuvel, C. Magnesium in acute and chronic brain injury: An update. Magnes. Res. 2009, 22, 158S–162S. [Google Scholar] [CrossRef]
- Saatman, K.E.; Bareyre, F.M.; Grady, M.S.; McIntosh, T.K. Acute cytoskeletal alterations and cell death induced by experimental brain injury are attenuated by magnesium treatment and exacerbated by magnesium deficiency. J. Neuropathol. Exp. Neurol. 2001, 60, 183–194. [Google Scholar] [CrossRef]
- Muir, J.K.; Raghupathi, R.; Emery, D.L.; Bareyre, F.M.; McIntosh, T.K. Postinjury magnesium treatment attenuates traumatic brain injury-induced cortical induction of p53 mRNA in rats. Exp. Neurol. 1999, 159, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, T.; Osugi, T.; Satoh, H.; McIntosh, T.K.; Nabeshima, T. Pre-Injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J. Neurotrauma 2005, 22, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Esen, F.; Erdem, T.; Aktan, D.; Kalayci, R.; Cakar, N.; Kaya, M.; Telci, L. Effects of magnesium administration on brain edema and blood-brain barrier breakdown after experimental traumatic brain injury in rats. J. Neurosurg. Anesthesiol. 2003, 15, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Park, C.O.; Hyun, D.K. Apoptotic change in response to magnesium therapy after moderate diffuse axonal injury in rats. Yonsei Med. J. 2004, 45, 908–916. [Google Scholar] [CrossRef]
- Ghabriel, M.N.; Thomas, A.; Vink, R. Magnesium restores altered aquaporin-4 immunoreactivity following traumatic brain injury to a pre-injury state. Acta Neurochir. Suppl. 2006, 96, 402–406. [Google Scholar] [CrossRef]
- Bareyre, F.M.; Saatman, K.E.; Raghupathi, R.; McIntosh, T.K. Postinjury treatment with magnesium chloride attenuates cortical damage after traumatic brain injury in rats. J. Neurotrauma 2000, 17, 1029–1039. [Google Scholar] [CrossRef]
- Fromm, L.; Heath, D.L.; Vink, R.; Nimmo, A.J. Magnesium attenuates post-traumatic depression/anxiety following diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 529S–533S. [Google Scholar] [CrossRef]
- Vink, R.; O’Connor, C.A.; Nimmo, A.J.; Heath, D.L. Magnesium attenuates persistent functional deficits following diffuse traumatic brain injury in rats. Neurosci. Lett. 2003, 336, 41–44. [Google Scholar] [CrossRef]
- Turner, R.J.; Dasilva, K.W.; O’Connor, C.; van den Heuvel, C.; Vink, R. Magnesium gluconate offers no more protection than magnesium sulphate following diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 541S–544S. [Google Scholar] [CrossRef]
- Temkin, N.R.; Anderson, G.D.; Winn, H.R.; Ellenbogen, R.G.; Britz, G.W.; Schuster, J.; Lucas, T.; Newell, D.W.; Mansfield, P.N.; Machamer, J.E.; et al. Magnesium sulfate for neuroprotection after traumatic brain injury: A randomised controlled trial. Lancet Neurol. 2007, 6, 29–38. [Google Scholar] [CrossRef]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef]
- Natale, J.E.; Guerguerian, A.-M.; Joseph, J.G.; McCarter, R.; Shao, C.; Slomine, B.; Christensen, J.; Johnston, M.V.; Shaffner, D.H. Pilot study to determine the hemodynamic safety and feasibility of magnesium sulfate infusion in children with severe traumatic brain injury. Pediatr. Crit. Care Med. 2007, 8, 1–9. [Google Scholar] [CrossRef]
- Levenson, C.W. Zinc and Traumatic Brain Injury: From Chelation to Supplementation. Med. Sci. 2020, 8, 36. [Google Scholar] [CrossRef]
- Portbury, S.D.; Hare, D.J.; Sgambelloni, C.; Finkelstein, D.I.; Adlard, P.A. A time-course analysis of changes in cerebral metal levels following a controlled cortical impact. Metallomics 2016, 8, 193–200. [Google Scholar] [CrossRef]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Eidson, K.A.; Capra, B.A.; Garcia, J.M.; Boone, D.R.; Hawkins, B.E.; Uchida, T.; Dewitt, D.S.; Prough, D.S. Injured Fluoro-Jade-positive hippocampal neurons contain high levels of zinc after traumatic brain injury. Brain Res. 2007, 1127, 119–126. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Elevated Cytoplasmic Free Zinc and Increased Reactive Oxygen Species Generation in the Context of Brain Injury. Acta Neurochir. Suppl. 2016, 121, 347–353. [Google Scholar] [CrossRef]
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E. Role of zinc and copper ions in the pathogenetic mechanisms of traumatic brain injury and Alzheimer’s disease. Rev. Neurosci. 2020, 31, 233–243. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Weiss, J.H. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur. J. Neurosci. 2000, 12, 3813–3818. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Koh, J.-Y. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp. Neurol. 2002, 177, 407–418. [Google Scholar] [CrossRef]
- Doering, P.; Stoltenberg, M.; Penkowa, M.; Rungby, J.; Larsen, A.; Danscher, G. Chemical blocking of zinc ions in CNS increases neuronal damage following traumatic brain injury (TBI) in mice. PLoS ONE 2010, 5, e10131. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Frederickson, C.J.; DeWitt, D.S.; Saban, R.; Parsley, M.O.; Stephenson, R.; Velasco, M.; Uchida, T.; Shimamura, M.; Prough, D.S. Protective effects of zinc chelation in traumatic brain injury correlate with upregulation of neuroprotective genes in rat brain. Neurosci. Lett. 2004, 355, 221–225. [Google Scholar] [CrossRef]
- McClain, C.J.; Twyman, D.L.; Ott, L.G.; Rapp, R.P.; Tibbs, P.A.; Norton, J.A.; Kasarskis, E.J.; Dempsey, R.J.; Young, B. Serum and urine zinc response in head-injured patients. J. Neurosurg. 1986, 64, 224–230. [Google Scholar] [CrossRef]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; VanLandingham, J.W.; Levenson, C.W. Zinc supplementation provides behavioral resiliency in a rat model of traumatic brain injury. Physiol. Behav. 2011, 104, 942–947. [Google Scholar] [CrossRef]
- Young, B.; Ott, L.; Kasarskis, E.; Rapp, R.; Moles, K.; Dempsey, R.J.; Tibbs, P.A.; Kryscio, R.; McClain, C. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J. Neurotrauma 1996, 13, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, M.I.; Blasco-Ibáñez, J.M.; Crespo, C.; Marqués-Marí, A.I.; Martínez-Guijarro, F.J. Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience 2003, 116, 791–806. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Eidson, K.; Cowart, J.; Crookshanks, J.; Boone, D.K.; Shah, S.; Uchida, T.; DeWitt, D.S.; Prough, D.S. Chelation of neurotoxic zinc levels does not improve neurobehavioral outcome after traumatic brain injury. Neurosci. Lett. 2008, 440, 155–159. [Google Scholar] [CrossRef]
- Scrimgeour, A.G.; Carrigan, C.T.; Condlin, M.L.; Urso, M.L.; van den Berg, R.M.; van Helden, H.P.M.; Montain, S.J.; Joosen, M.J.A. Dietary Zinc Modulates Matrix Metalloproteinases in Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2495–2506. [Google Scholar] [CrossRef]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; Levenson, C.W. Use of zinc as a treatment for traumatic brain injury in the rat: Effects on cognitive and behavioral outcomes. Neurorehabil. Neural Repair 2012, 26, 907–913. [Google Scholar] [CrossRef]
- Khazdouz, M.; Mazidi, M.; Ehsaei, M.-R.; Ferns, G.; Kengne, A.P.; Norouzy, A.-R. Impact of Zinc Supplementation on the Clinical Outcomes of Patients with Severe Head Trauma: A Double-Blind Randomized Clinical Trial. J. Diet Suppl. 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Niemoller, T.D.; Stark, D.T.; Bazan, N.G. Omega-3 fatty acid docosahexaenoic acid is the precursor of neuroprotectin D1 in the nervous system. World Rev. Nutr. Diet 2009, 99, 46–54. [Google Scholar] [CrossRef]
- Hasadsri, L.; Wang, B.H.; Lee, J.V.; Erdman, J.W.; Llano, D.A.; Barbey, A.K.; Wszalek, T.; Sharrock, M.F.; Wang, H.J. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J. Neurotrauma 2013, 30, 897–906. [Google Scholar] [CrossRef]
- Michael-Titus, A.T.; Priestley, J.V. Omega-3 fatty acids and traumatic neurological injury: From neuroprotection to neuroplasticity? Trends Neurosci. 2014, 37, 30–38. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J. Neurotrauma 2004, 21, 1457–1467. [Google Scholar] [CrossRef]
- Kumar, P.R.; Essa, M.M.; Al-Adawi, S.; Dradekh, G.; Memon, M.A.; Akbar, M.; Manivasagam, T. Omega-3 Fatty acids could alleviate the risks of traumatic brain injury—A mini review. J. Tradit. Complement Med. 2014, 4, 89–92. [Google Scholar] [CrossRef]
- Ferrucci, L.; Cherubini, A.; Bandinelli, S.; Bartali, B.; Corsi, A.; Lauretani, F.; Martin, A.; Andres-Lacueva, C.; Senin, U.; Guralnik, J.M. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J. Clin. Endocrinol. Metab. 2006, 91, 439–446. [Google Scholar] [CrossRef]
- Vreugdenhil, M.; Bruehl, C.; Voskuyl, R.A.; Kang, J.X.; Leaf, A.; Wadman, W.J. Polyunsaturated fatty acids modulate sodium and calcium currents in CA1 neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 12559–12563. [Google Scholar] [CrossRef]
- Ménard, C.; Patenaude, C.; Gagné, A.-M.; Massicotte, G. AMPA receptor-mediated cell death is reduced by docosahexaenoic acid but not by eicosapentaenoic acid in area CA1 of hippocampal slice cultures. J. Neurosci. Res. 2009, 87, 876–886. [Google Scholar] [CrossRef]
- Desai, A.; Kevala, K.; Kim, H.-Y. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS ONE 2014, 9, e86472. [Google Scholar] [CrossRef]
- Wang, T.; Van, K.C.; Gavitt, B.J.; Grayson, J.K.; Lu, Y.-C.; Lyeth, B.G.; Pichakron, K.O. Effect of fish oil supplementation in a rat model of multiple mild traumatic brain injuries. Restor. Neurol. Neurosci. 2013, 31, 647–659. [Google Scholar] [CrossRef]
- Pu, H.; Guo, Y.; Zhang, W.; Huang, L.; Wang, G.; Liou, A.K.; Zhang, J.; Zhang, P.; Leak, R.K.; Wang, Y.; et al. Omega-3 polyunsaturated fatty acid supplementation improves neurologic recovery and attenuates white matter injury after experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1474–1484. [Google Scholar] [CrossRef]
- Mills, J.D.; Hadley, K.; Bailes, J.E. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 2011, 68, 474–481, discussion 481. [Google Scholar] [CrossRef]
- Chen, X.; Chen, C.; Fan, S.; Wu, S.; Yang, F.; Fang, Z.; Fu, H.; Li, Y. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2018, 15, 116. [Google Scholar] [CrossRef]
- Zhang, E.; Wan, X.; Yang, L.; Wang, D.; Chen, Z.; Chen, Y.; Liu, M.; Zhang, G.; Wu, J.; Han, H.; et al. Omega-3 Polyunsaturated Fatty Acids Alleviate Traumatic Brain Injury by Regulating the Glymphatic Pathway in Mice. Front. Neurol. 2020, 11, 707. [Google Scholar] [CrossRef]
- Barrett, E.C.; McBurney, M.I.; Ciappio, E.D. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv. Nutr. 2014, 5, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Van Steenwinckel, J.; Schang, A.-L.; Krishnan, M.L.; Degos, V.; Delahaye-Duriez, A.; Bokobza, C.; Csaba, Z.; Verdonk, F.; Montané, A.; Sigaut, S.; et al. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain 2019, 142, 3806–3833. [Google Scholar] [CrossRef] [PubMed]
- Dang, B.; Chen, W.; He, W.; Chen, G. Rehabilitation Treatment and Progress of Traumatic Brain Injury Dysfunction. Neural Plast. 2017, 2017, 1582182. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, P.M.; Landgrebe, M.; Frank, E.; Langguth, B. Repetitive transcranial magnetic stimulation for the treatment of chronic tinnitus after traumatic brain injury: A case study. J. Head Trauma Rehabil. 2013, 28, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Neville, I.S.; Hayashi, C.Y.; El Hajj, S.A.; Zaninotto, A.L.C.; Sabino, J.P.; Sousa, L.M.; Nagumo, M.M.; Brunoni, A.R.; Shieh, B.D.F.S.; Amorim, R.L.O.; et al. Repetitive Transcranial Magnetic Stimulation (rTMS) for the cognitive rehabilitation of traumatic brain injury (TBI) victims: Study protocol for a randomized controlled trial. Trials 2015, 16, 440. [Google Scholar] [CrossRef]
- Fregni, F.; Li, S.; Zaninotto, A.; Santana Neville, I.; Paiva, W.; Nunn, D. Clinical utility of brain stimulation modalities following traumatic brain injury: Current evidence. NDT 2015, 1573. [Google Scholar] [CrossRef]
- Dhaliwal, S.K.; Meek, B.P.; Modirrousta, M.M. Non-Invasive Brain Stimulation for the Treatment of Symptoms Following Traumatic Brain Injury. Front. Psychiatry 2015, 6, 119. [Google Scholar] [CrossRef]
- Tam, S.-F.; Man, W.-K. Evaluating computer-assisted memory retraining programmes for people with post-head injury amnesia. Brain Inj. 2004, 18, 461–470. [Google Scholar] [CrossRef]
- Bergquist, T.; Gehl, C.; Mandrekar, J.; Lepore, S.; Hanna, S.; Osten, A.; Beaulieu, W. The effect of internet-based cognitive rehabilitation in persons with memory impairments after severe traumatic brain injury. Brain Inj. 2009, 23, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Florez-Perdomo, W.A.; Shrivatava, A.; Chouksey, P.; Raj, S.; Moscote-Salazar, L.R.; Rahman, M.M.; Sutar, R.; Agrawal, A. Role of Music Therapy in Traumatic Brain Injury: A Systematic Review and Meta-analysis. World Neurosurg. 2021, 146, 197–204. [Google Scholar] [CrossRef]
- Baker, F.; Wigram, T.; Gold, C. The effects of a song-singing programme on the affective speaking intonation of people with traumatic brain injury. Brain Inj. 2005, 19, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Siponkoski, S.-T.; Martínez-Molina, N.; Kuusela, L.; Laitinen, S.; Holma, M.; Ahlfors, M.; Jordan-Kilkki, P.; Ala-Kauhaluoma, K.; Melkas, S.; Pekkola, J.; et al. Music Therapy Enhances Executive Functions and Prefrontal Structural Neuroplasticity after Traumatic Brain Injury: Evidence from a Randomized Controlled Trial. J. Neurotrauma 2020, 37, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Sinnakaruppan, I.; Downey, B.; Morrison, S. Head injury and family carers: A pilot study to investigate an innovative community-based educational programme for family carers and patients. Brain Inj. 2005, 19, 283–308. [Google Scholar] [CrossRef]
- Brunetti, M.; Martelli, N.; Colasante, A.; Piantelli, M.; Musiani, P.; Aiello, F.B. Spontaneous and glucocorticoid-induced apoptosis in human mature T lymphocytes. Blood 1995, 86, 4199–4205. [Google Scholar] [CrossRef]
- Zacco, A.; Togo, J.; Spence, K.; Ellis, A.; Lloyd, D.; Furlong, S.; Piser, T. 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors Protect Cortical Neurons from Excitotoxicity. J. Neurosci. 2003, 23, 11104–11111. [Google Scholar] [CrossRef] [PubMed]
- Béziaud, T.; Ru Chen, X.; El Shafey, N.; Fréchou, M.; Teng, F.; Palmier, B.; Beray-Berthat, V.; Soustrat, M.; Margaill, I.; Plotkine, M.; et al. Simvastatin in traumatic brain injury: Effect on brain edema mechanisms. Crit. Care Med. 2011, 39, 2300–2307. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Lu, X.; Zhang, L.; Zhu, L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem. Int. 2014, 76, 23–31. [Google Scholar] [CrossRef]
- Feng, Y.-M.; Jia, Y.-F.; Su, L.-Y.; Wang, D.; Lv, L.; Xu, L.; Yao, Y.-G. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy 2013, 9, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Huang, H.-J.; Lee, H.-C.; Hung, K.-C.; Wu, R.-T.; Lin, A.M.-Y. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation: Kainic acid and α-synuclein aggregation. J. Pineal Res. 2012, 52, 312–321. [Google Scholar] [CrossRef]
- Reiter, R.; Paredes, S.; Korkmaz, A.; Jou, M.-J.; Tan, D.-X. Melatonin combats molecular terrorism at the mitochondrial level. Interdiscip. Toxicol. 2008, 1, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Moretti, R.; Le Charpentier, T.; Sigaut, S.; Lebon, S.; Schwendimann, L.; Oré, M.-V.; Zuiani, C.; Milan, V.; Josserand, J.; et al. Role of microglia in a mouse model of paediatric traumatic brain injury. Brain Behav. Immun. 2017, 63, 197–209. [Google Scholar] [CrossRef]
- Homsi, S.; Federico, F.; Croci, N.; Palmier, B.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Minocycline effects on cerebral edema: Relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 2009, 1291, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, L.A.; Raghupathi, R.; Huh, J.W. Differential effects of minocycline on microglial activation and neurodegeneration following closed head injury in the neonate rat. Exp. Neurol. 2017, 290, 1–14. [Google Scholar] [CrossRef]
- Simon, D.W.; Aneja, R.K.; Alexander, H.; Bell, M.J.; Bayır, H.; Kochanek, P.M.; Clark, R.S.B. Minocycline Attenuates High Mobility Group Box 1 Translocation, Microglial Activation, and Thalamic Neurodegeneration after Traumatic Brain Injury in Post-Natal Day 17 Rats. J. Neurotrauma 2018, 35, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Signoretti, S.; Dunbar, J.G.; Marmarou, A. The effect of Cyclosporin A on brain edema formation following experimental cortical contusion. In Brain Edema XII; Kuroiwa, T., Baethmann, A., Czernicki, Z., Hoff, J.T., Ito, U., Katayama, Y., Marmarou, A., Mendelow, B.A.D., Reulen, H.-J., Eds.; Springer: Vienna, Austria, 2003; pp. 301–303. ISBN 978-3-7091-7220-9. [Google Scholar]
- Aykanat, Ö.; Karakoyun, D.O.; Türkoğlu, M.E.; Dinç, C. Anti-edematous, anti-inflammatory and neuroprotective effect of etanercept in acute stage in experimental head injury. Ulus. Travma. Acil. Cerrahi. Derg. 2017, 23, 173–180. [Google Scholar] [CrossRef]
- Ozen, I.; Ruscher, K.; Nilsson, R.; Flygt, J.; Clausen, F.; Marklund, N. Interleukin-1 Beta Neutralization Attenuates Traumatic Brain Injury-Induced Microglia Activation and Neuronal Changes in the Globus Pallidus. Int. J. Mol. Sci. 2020, 21, 387. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.M.; Shultz, S.R.; Ozturk, E.; Dill, L.K.; Sun, M.; Casillas-Espinosa, P.; Jones, N.C.; Crack, P.J.; O’Brien, T.J.; Semple, B.D. Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: Chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp. Neurol. 2019, 320, 112979. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.L.; Saraswati, M. Progesterone protects mitochondrial function in a rat model of pediatric traumatic brain injury. J Bioenerg Biomembr 2015, 47, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lengel, D.; Huh, J.W.; Barson, J.R.; Raghupathi, R. Progesterone treatment following traumatic brain injury in the 11-day-old rat attenuates cognitive deficits and neuronal hyperexcitability in adolescence. Exp. Neurol. 2020, 330, 113329. [Google Scholar] [CrossRef] [PubMed]
- Geddes, R.I.; Sribnick, E.A.; Sayeed, I.; Stein, D.G. Progesterone treatment shows benefit in a pediatric model of moderate to severe bilateral brain injury. PLoS ONE 2014, 9, e87252. [Google Scholar] [CrossRef]
- Robertson, C.L.; Fidan, E.; Stanley, R.M.; Noje, C.; Bayir, H. Progesterone for neuroprotection in pediatric traumatic brain injury. Pediatr. Crit. Care Med. 2015, 16, 236–244. [Google Scholar] [CrossRef]
- Harris, K.; Armstrong, S.P.; Campos-Pires, R.; Kiru, L.; Franks, N.P.; Dickinson, R. Neuroprotection against traumatic brain injury by xenon, but not argon, is mediated by inhibition at the N-methyl-D-aspartate receptor glycine site. Anesthesiology 2013, 119, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, E.; Demirbilek, S.; Kadir But, A.; Saricicek, V.; Gulec, M.; Akyol, O.; Ozcan Ersoy, M. Antioxidant properties of propofol and erythropoietin after closed head injury in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 922–927. [Google Scholar] [CrossRef]
- Kaptanoglu, E.; Sen, S.; Beskonakli, E.; Surucu, H.S.; Tuncel, M.; Kilinc, K.; Taskin, Y. Antioxidant Actions and Early Ultrastructural Findings of Thiopental and Propofol in Experimental Spinal Cord Injury. J. Neurosurg. Anesthesiol. 2002, 14, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Gilbert, D.R.; Holland, M.A.; Pierce, J.L. Nicotinamide reduces acute cortical neuronal death and edema in the traumatically injured brain. Neurosci. Lett. 2006, 408, 35–39. [Google Scholar] [CrossRef]
- Smith, A.C.; Holden, R.C.; Rasmussen, S.M.; Hoane, M.R.; Hylin, M.J. Effects of nicotinamide on spatial memory and inflammation after juvenile traumatic brain injury. Behav. Brain Res. 2019, 364, 123–132. [Google Scholar] [CrossRef]
- Cui, C.; Song, S.; Cui, J.; Feng, Y.; Gao, J.; Jiang, P. Vitamin D Receptor Activation Influences NADPH Oxidase (NOX2) Activity and Protects against Neurological Deficits and Apoptosis in a Rat Model of Traumatic Brain Injury. Oxid. Med. Cell Longev. 2017, 2017, 9245702. [Google Scholar] [CrossRef]
- Ikeda, Y.; Mochizuki, Y.; Nakamura, Y.; Dohi, K.; Matsumoto, H.; Jimbo, H.; Hayashi, M.; Matsumoto, K.; Yoshikawa, T.; Murase, H.; et al. Protective effect of a novel vitamin E derivative on experimental traumatic brain edema in rats—Preliminary study. Acta Neurochir. Suppl. 2000, 76, 343–345. [Google Scholar] [CrossRef]
- Li, Y.; Hawkins, B.E.; DeWitt, D.S.; Prough, D.S.; Maret, W. The relationship between transient zinc ion fluctuations and redox signaling in the pathways of secondary cellular injury: Relevance to traumatic brain injury. Brain Res. 2010, 1330, 131–141. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquens, A.; Needham, E.J.; Zanier, E.R.; Degos, V.; Gressens, P.; Menon, D. Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side. Int. J. Mol. Sci. 2022, 23, 11193. https://doi.org/10.3390/ijms231911193
Jacquens A, Needham EJ, Zanier ER, Degos V, Gressens P, Menon D. Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side. International Journal of Molecular Sciences. 2022; 23(19):11193. https://doi.org/10.3390/ijms231911193
Chicago/Turabian StyleJacquens, Alice, Edward J. Needham, Elisa R. Zanier, Vincent Degos, Pierre Gressens, and David Menon. 2022. "Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side" International Journal of Molecular Sciences 23, no. 19: 11193. https://doi.org/10.3390/ijms231911193
APA StyleJacquens, A., Needham, E. J., Zanier, E. R., Degos, V., Gressens, P., & Menon, D. (2022). Neuro-Inflammation Modulation and Post-Traumatic Brain Injury Lesions: From Bench to Bed-Side. International Journal of Molecular Sciences, 23(19), 11193. https://doi.org/10.3390/ijms231911193