
Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis


Abstract

1. Introduction
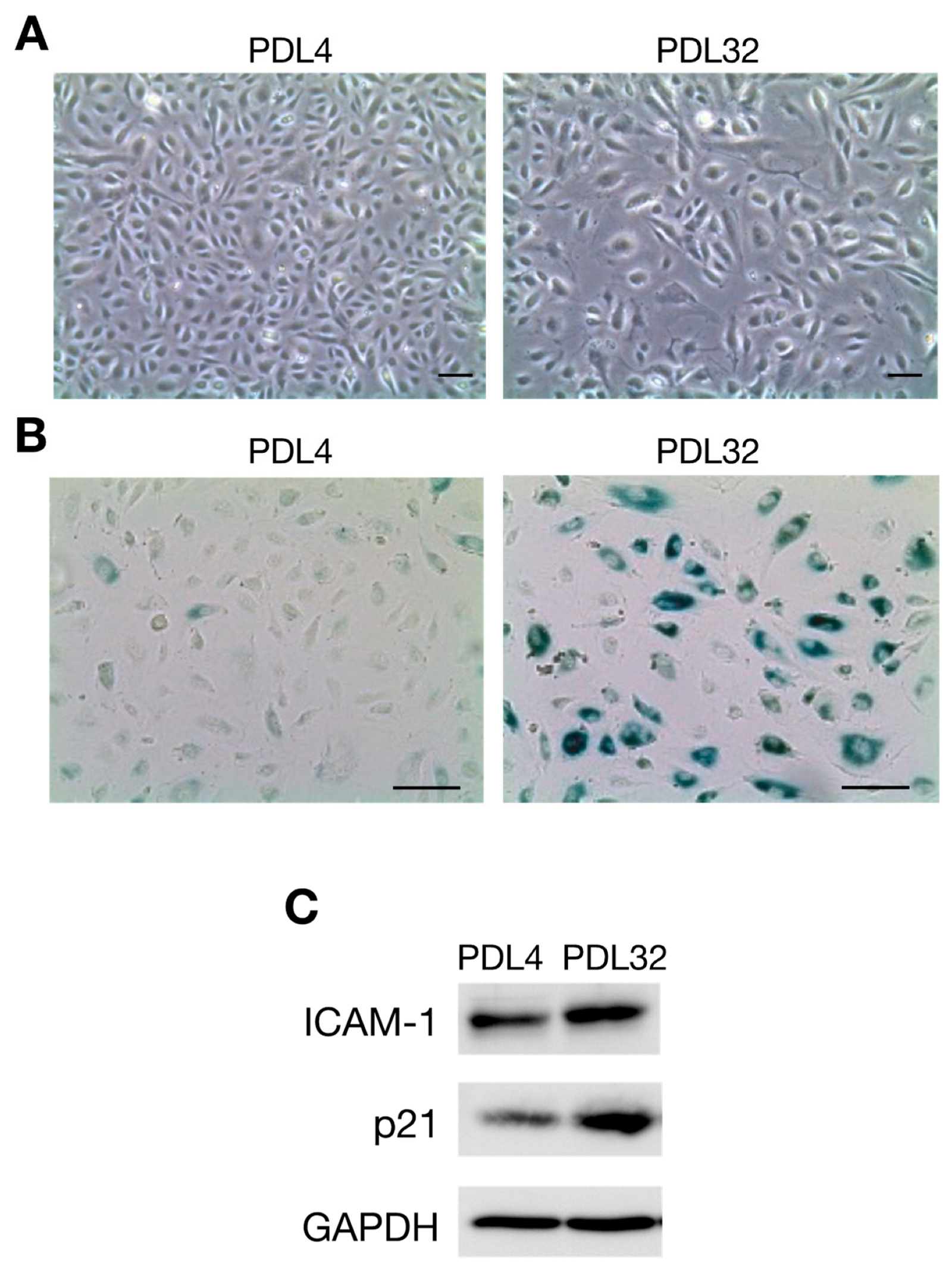
2. Atherosclerosis and Cellular Senescence
3. Atherosclerosis and LPS
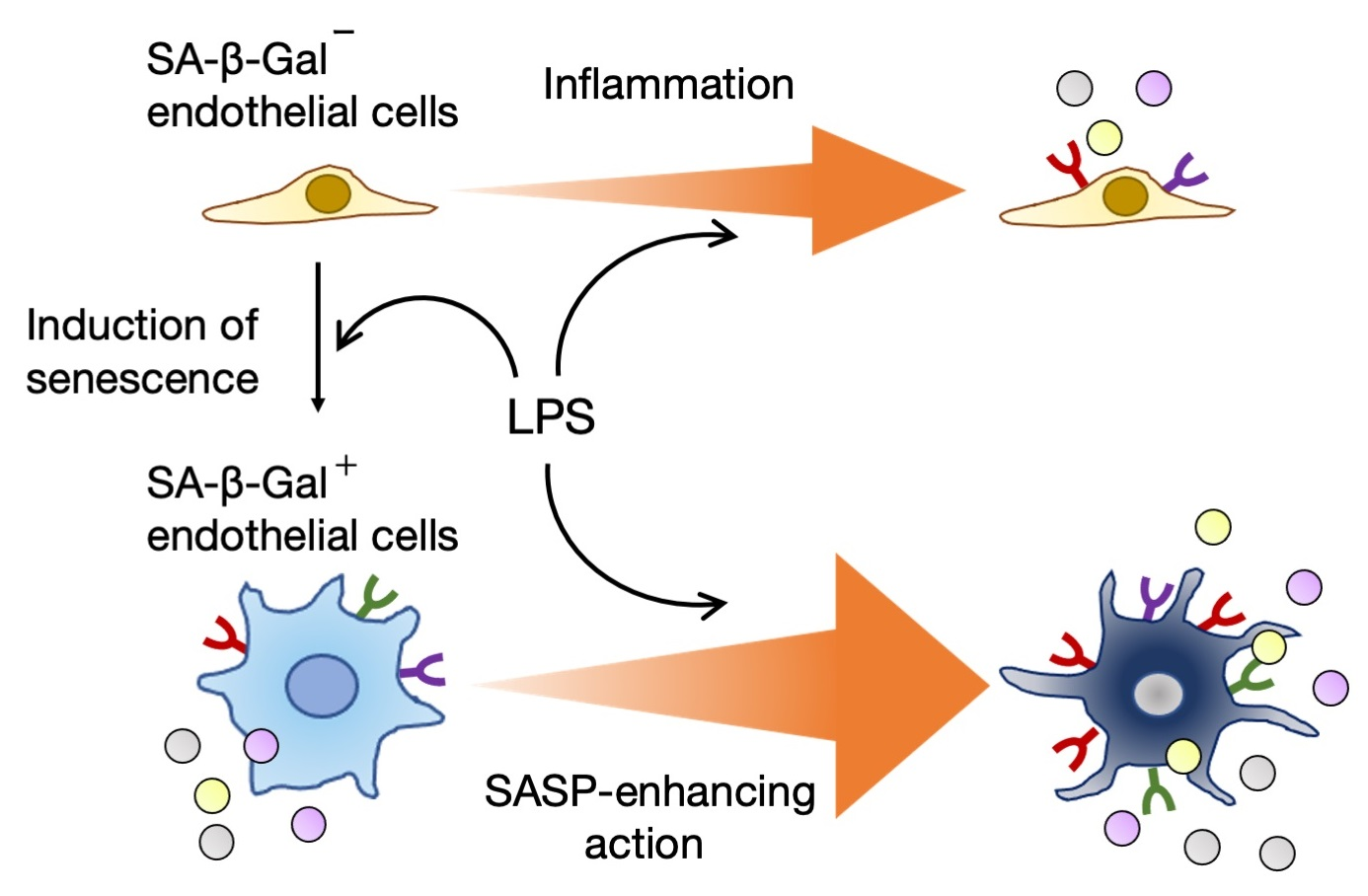
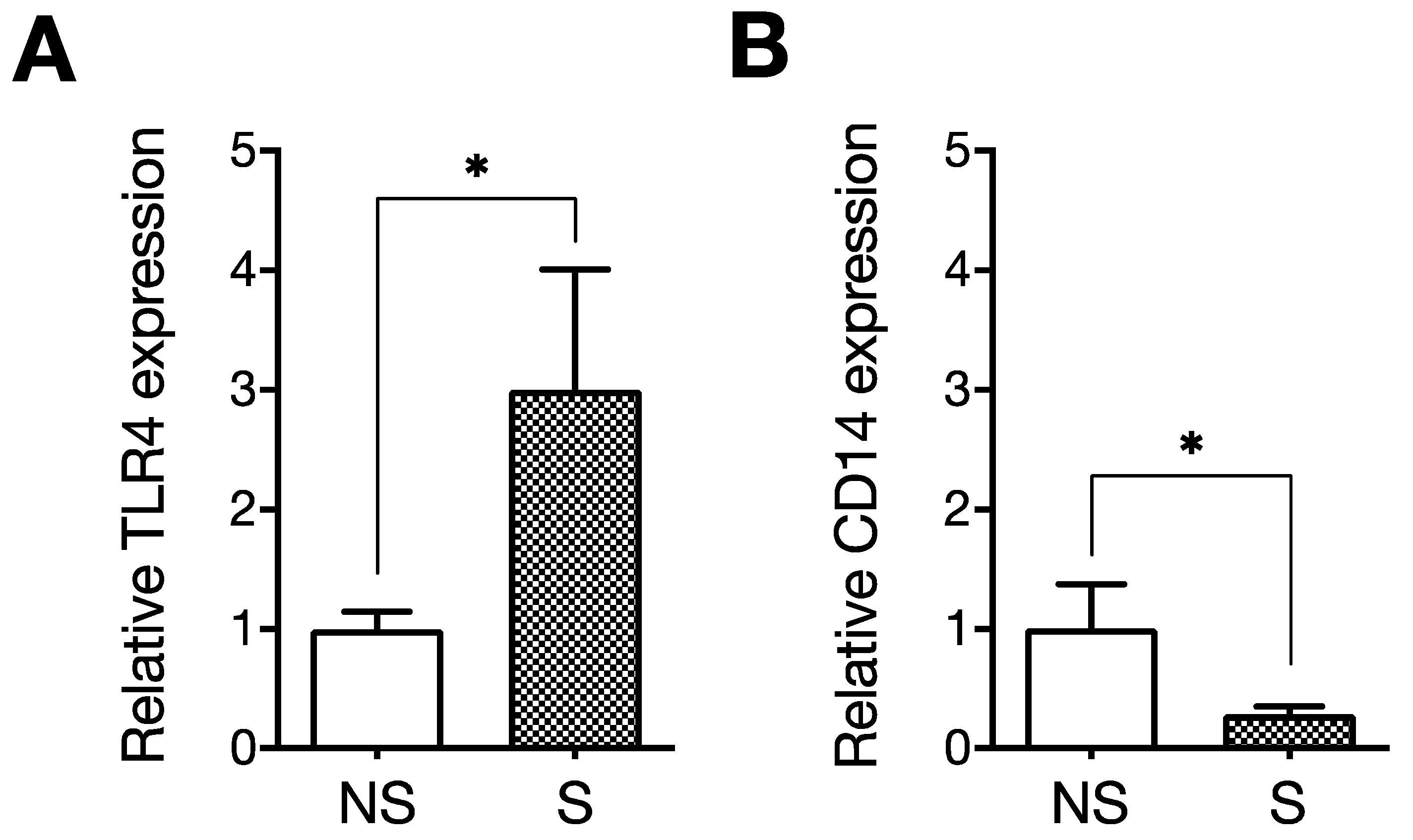
4. LPS Induces Cellular Senescence
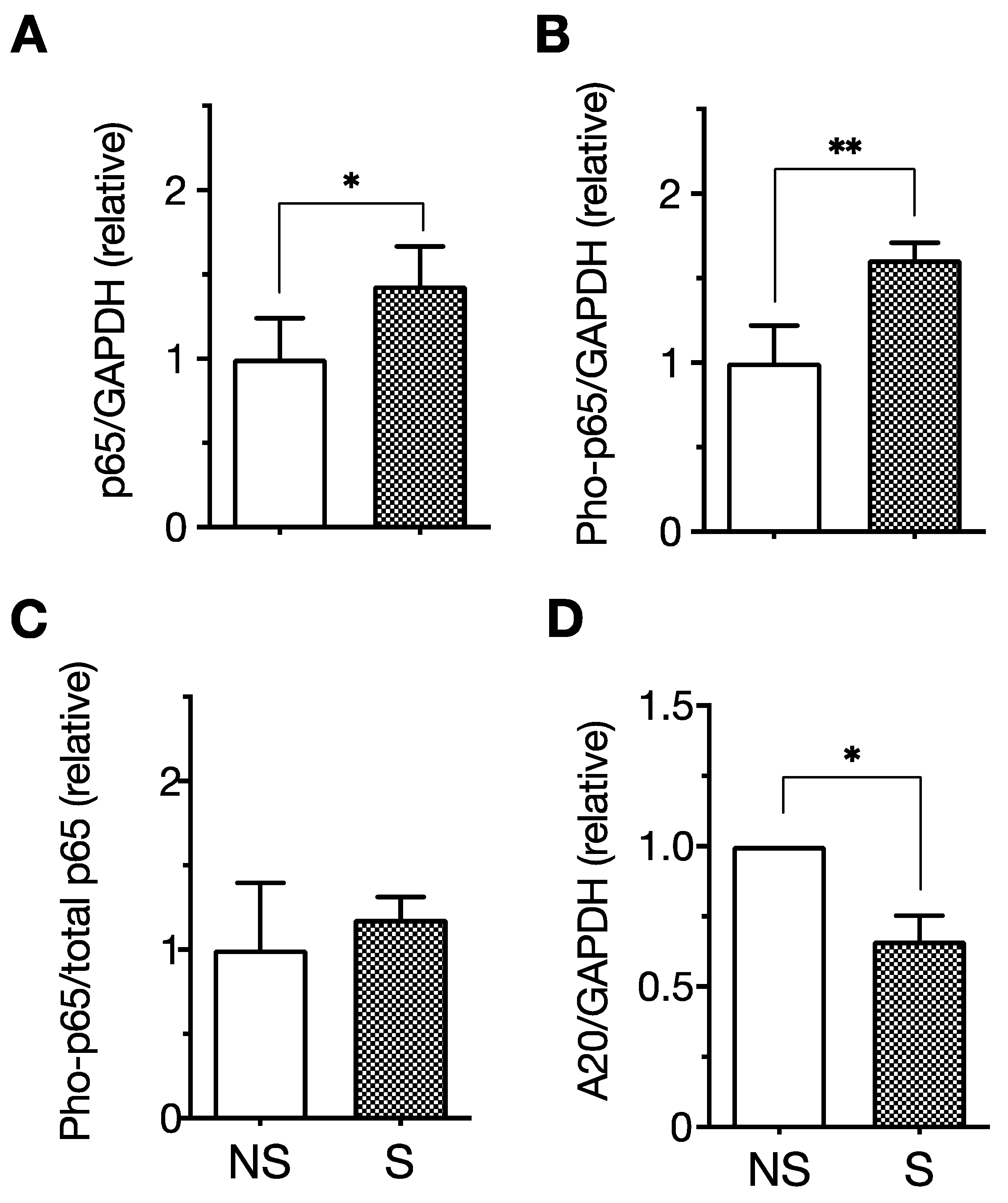
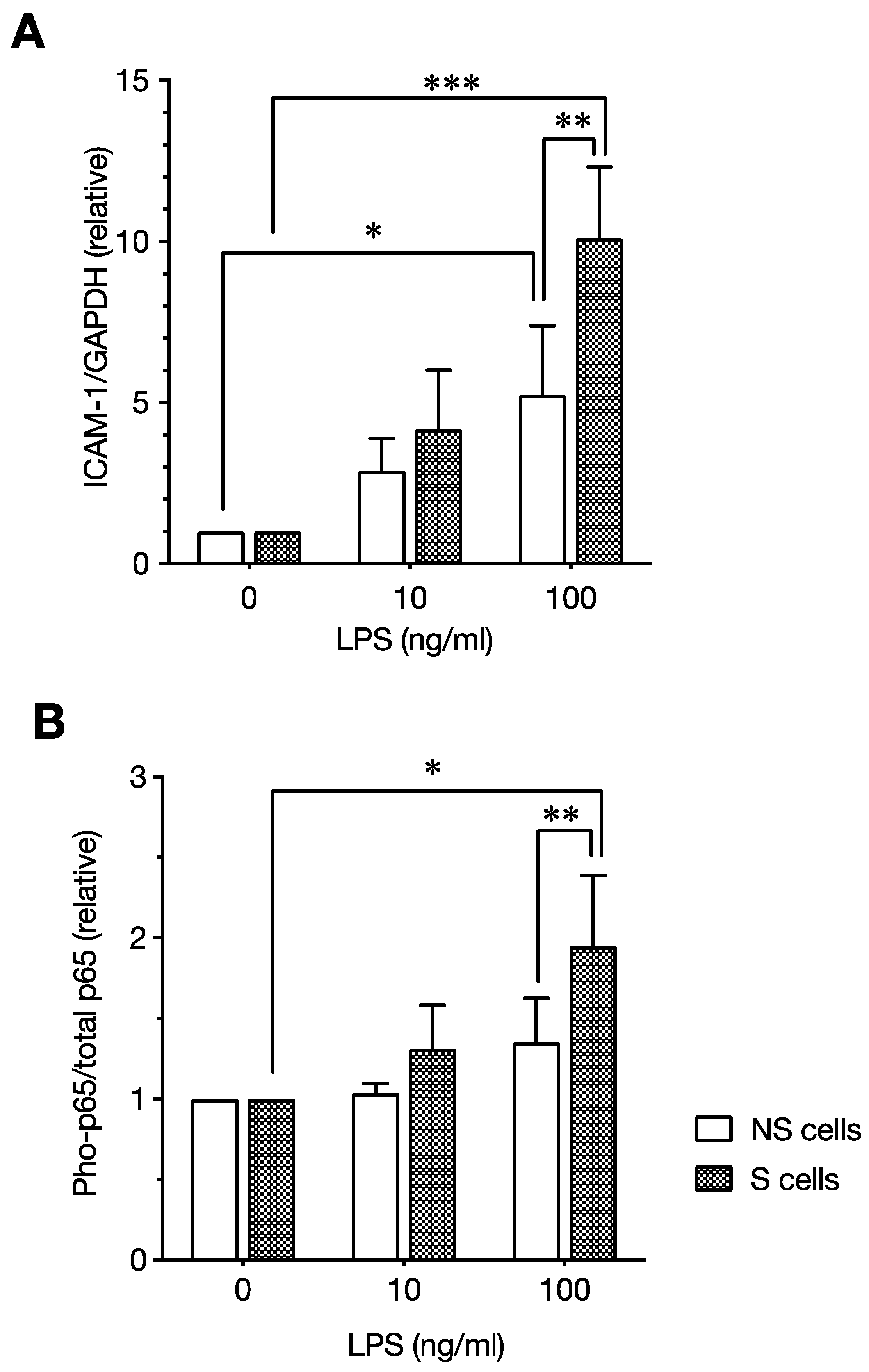
5. LPS Enhances SASP-Associated Proinflammatory Responses of Senescent Cells
6. Effect of LPS-Neutralizing Peptide LL-37 on Senescent Cells
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Fazal, F. Hug tightly and say goodbye: Role of endothelial ICAM-1 in leukocyte transmigration. Antioxid. Redox Signal. 2009, 11, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef]
- Carnevale, R.; Nocella, C.; Petrozza, V.; Cammisotto, V.; Pacini, L.; Sorrentino, V.; Martinelli, O.; Irace, L.; Sciarretta, S.; Frati, G.; et al. Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci. Rep. 2018, 8, 3598. [Google Scholar] [CrossRef]
- Chiu, B.; Viira, E.; Tucker, W.; Fong, I.W. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation 1997, 96, 2144–2148. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (β)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Takahashi, M.; Oba, S.; Nishimatsu, H. Oncogene- and oxidative stress-induced cellular senescence shows distinct expression patterns of proinflammatory cytokines in vascular endothelial cells. Sci. World J. 2013, 2013, 754735. [Google Scholar] [CrossRef] [PubMed]
- Beyne-Rauzy, O.; Recher, C.; Dastugue, N.; Demur, C.; Pottier, G.; Laurent, G.; Sabatier, L.; Mansat-De Mas, V. Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene 2004, 23, 7507–7516. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Xu, Y.X.; Li, N.; Xiang, R.; Sun, P.Q. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-κB in Aging and Disease. Aging Dis. 2011, 2, 449–465. [Google Scholar]
- Vasile, E.; Tomita, Y.; Brown, L.F.; Kocher, O.; Dvorak, H.F. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: Evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001, 15, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B. Cellular senescence and atherosclerosis. Med. Hypotheses 1984, 14, 115–124. [Google Scholar] [CrossRef]
- Hansson, G.K.; Bondjers, G. Endothelial proliferation and atherogenesis in rabbits with moderate hypercholesterolemia. Artery 1980, 7, 316–329. [Google Scholar]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef]
- Honda, S.; Ikeda, K.; Urata, R.; Yamazaki, E.; Emoto, N.; Matoba, S. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 2021, 11, 14608. [Google Scholar] [CrossRef]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Hsiao, C.L.; Yoshida, Y.; Matsumoto, N.; Yoshida, Y.; Katayama, A.; Wada, J.; Seki, M.; et al. Glycoprotein nonmetastatic melanoma protein B regulates lysosomal integrity and lifespan of senescent cells. Sci. Rep. 2022, 12, 6522. [Google Scholar] [CrossRef]
- Lv, L.; Ye, M.; Duan, R.; Yuan, K.; Chen, J.; Liang, W.; Zhou, Z.; Zhang, L. Downregulation of Pin1 in human atherosclerosis and its association with vascular smooth muscle cell senescence. J. Vasc. Surg. 2018, 68, 873–883.875. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Newby, A.C.; George, S.J.; Ismail, Y.; Johnson, J.L.; Sala-Newby, G.B.; Thomas, A.C. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb. Haemost. 2009, 101, 1006–1011. [Google Scholar] [PubMed]
- Campbell, L.A.; Rosenfeld, M.E. Infection and Atherosclerosis Development. Arch. Med. Res. 2015, 46, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Stassen, F.R.; Vainas, T.; Bruggeman, C.A. Infection and atherosclerosis. An alternative view on an outdated hypothesis. Pharmacol. Rep. 2008, 60, 85–92. [Google Scholar] [PubMed]
- Epstein, S.E.; Zhou, Y.F.; Zhu, J. Infection and atherosclerosis: Emerging mechanistic paradigms. Circulation 1999, 100, e20–e28. [Google Scholar] [CrossRef]
- Haraszthy, V.I.; Zambon, J.J.; Trevisan, M.; Zeid, M.; Genco, R.J. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 2000, 71, 1554–1560. [Google Scholar] [CrossRef]
- Aimetti, M.; Romano, F.; Nessi, F. Microbiologic analysis of periodontal pockets and carotid atheromatous plaques in advanced chronic periodontitis patients. J. Periodontol. 2007, 78, 1718–1723. [Google Scholar] [CrossRef]
- Honarmand, H. Atherosclerosis Induced by Chlamydophila pneumoniae: A Controversial Theory. Interdiscip. Perspect. Infect. Dis. 2013, 2013, 941392. [Google Scholar] [CrossRef]
- Grayston, J.T. Background and current knowledge of Chlamydia pneumoniae and atherosclerosis. J. Infect. Dis. 2000, 181 (Suppl. S3), S402–S410. [Google Scholar] [CrossRef]
- He, C.; Yang, Z.; Lu, N.H. Helicobacter pylori-an infectious risk factor for atherosclerosis? J. Atheroscler. Thromb. 2014, 21, 1229–1242. [Google Scholar] [CrossRef][Green Version]
- Blessing, E.; Campbell, L.A.; Rosenfeld, M.E.; Chough, N.; Kuo, C.C. Chlamydia pneumoniae infection accelerates hyperlipidemia induced atherosclerotic lesion development in C57BL/6J mice. Atherosclerosis 2001, 158, 13–17. [Google Scholar] [CrossRef]
- Hayashi, C.; Viereck, J.; Hua, N.; Phinikaridou, A.; Madrigal, A.G.; Gibson, F.C., 3rd; Hamilton, J.A.; Genco, C.A. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 2011, 215, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Stoll, L.L.; Denning, G.M.; Weintraub, N.L. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.D.; Surani, S.; Horseman, M.A. Endotoxin, Toll-like Receptor-4, and Atherosclerotic Heart Disease. Curr. Cardiol. Rev. 2017, 13, 86–93. [Google Scholar] [CrossRef]
- Violi, F.; Cammisotto, V.; Bartimoccia, S.; Pignatelli, P.; Carnevale, R.; Nocella, C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat. Rev. Cardiol. 2022, 1–14. [Google Scholar] [CrossRef]
- Borel, N.; Pospischil, A.; Dowling, R.D.; Dumrese, C.; Gaydos, C.A.; Bunk, S.; Hermann, C.; Ramirez, J.A.; Summersgill, J.T. Antigens of persistent Chlamydia pneumoniae within coronary atheroma from patients undergoing heart transplantation. J. Clin. Pathol. 2012, 65, 171–177. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Tuomisto, K.; Jousilahti, P.; Havulinna, A.S.; Sundvall, J.; Salomaa, V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1433–1439. [Google Scholar] [CrossRef]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Sweet, M.J.; Hume, D.A. Endotoxin signal transduction in macrophages. J. Leukoc. Biol. 1996, 60, 8–26. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Mallat, Z.; Taleb, S.; Ait-Oufella, H.; Tedgui, A. The role of adaptive T cell immunity in atherosclerosis. J. Lipid Res. 2009, 50, S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Crockett-Torabi, E.; Sulenbarger, B.; Smith, C.W.; Fantone, J.C. Activation of human neutrophils through L-selectin and Mac-1 molecules. J. Immunol. 1995, 154, 2291–2302. [Google Scholar] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 2015, 349, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef]
- Koide, N.; Mu, M.M.; Hassan, F.; Islam, S.; Tumurkhuu, G.; Dagvadorj, J.; Naiki, Y.; Mori, I.; Yoshida, T.; Yokochi, T. Lipopolysaccharide enhances interferon-γ-induced nitric oxide (NO) production in murine vascular endothelial cells via augmentation of interferon regulatory factor-1 activation. J. Endotoxin Res. 2007, 13, 167–175. [Google Scholar] [CrossRef]
- Chakravortty, D.; Kato, Y.; Koide, N.; Sugiyama, T.; Kawai, M.; Fukada, M.; Yoshida, T.; Yokochi, T. Production of tissue factor in CD14-expressing human umbilical vein endothelial cells by lipopolysaccharide. FEMS Microbiol. Lett. 1999, 178, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Jiang, D.; Li, L.; Yang, Y.; Wu, P.; Luo, Y.; Yang, R.; Li, D. LPS Promotes Vascular Smooth Muscle Cells Proliferation Through the TLR4/Rac1/Akt Signalling Pathway. Cell. Physiol. Biochem. 2017, 44, 2189–2200. [Google Scholar] [CrossRef]
- Yang, X.; Coriolan, D.; Murthy, V.; Schultz, K.; Golenbock, D.T.; Beasley, D. Proinflammatory phenotype of vascular smooth muscle cells: Role of efficient Toll-like receptor 4 signaling. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1069–H1076. [Google Scholar] [CrossRef]
- Detmer, K.; Wang, Z.; Warejcka, D.; Leeper-Woodford, S.K.; Newman, W.H. Endotoxin stimulated cytokine production in rat vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H661–H668. [Google Scholar] [CrossRef]
- Yin, K.; Tang, S.L.; Yu, X.H.; Tu, G.H.; He, R.F.; Li, J.F.; Xie, D.; Gui, Q.J.; Fu, Y.C.; Jiang, Z.S.; et al. Apolipoprotein A-I inhibits LPS-induced atherosclerosis in ApoE(−/−) mice possibly via activated STAT3-mediated upregulation of tristetraprolin. Acta Pharmacol. Sin. 2013, 34, 837–846. [Google Scholar] [CrossRef]
- Ni, J.Q.; Ouyang, Q.; Lin, L.; Huang, Z.; Lu, H.; Chen, X.; Lin, H.; Wang, Z.; Xu, D.; Zhang, Y. Role of toll-like receptor 4 on lupus lung injury and atherosclerosis in LPS-challenge ApoE−/− mice. Clin. Dev. Immunol. 2013, 2013, 476856. [Google Scholar] [CrossRef]
- Suh, J.S.; Kim, S.; Boström, K.I.; Wang, C.Y.; Kim, R.H.; Park, N.H. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial-mesenchymal transition in mice. Int. J. Oral Sci. 2019, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Björkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 2004, 10, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Tobias, P.S.; Curtiss, L.K. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Investig. 2005, 115, 3149–3156. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Deng, T.; Zhang, Y.; Su, S.; Wei, C.; Han, D. Lipopolysaccharide inhibits macrophage phagocytosis of apoptotic neutrophils by regulating the production of tumour necrosis factor α and growth arrest-specific gene 6. Immunology 2011, 132, 287–295. [Google Scholar] [CrossRef]
- Feng, X.; Yuan, Y.; Wang, C.; Feng, J.; Yuan, Z.; Zhang, X.; Sui, W.; Hu, P.; Zheng, P.; Ye, J. Autophagy involved in lipopolysaccharide-induced foam cell formation is mediated by adipose differentiation-related protein. Lipids Health Dis. 2014, 13, 10. [Google Scholar] [CrossRef]
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci. Rep. 2016, 6, 20931. [Google Scholar] [CrossRef]
- Gong, H.; Liu, J.; Xue, Z.; Wang, W.; Li, C.; Xu, F.; Du, Y.; Lyu, X. SIRT3 attenuates coronary atherosclerosis in diabetic patients by regulating endothelial cell function. J. Clin. Lab. Anal. 2022, 36, e24586. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, X.; Pang, M.; Sun, Z.; Qian, Y.; Xue, W.; Wang, Z.; Li, L. Role of histone deacetylase Sirt3 in the development and regression of atherosclerosis. Life Sci. 2021, 272, 119178. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sukhorukov, V.N.; Zhuravlev, A.; Orekhov, N.A.; Kalmykov, V.; Orekhov, A.N. Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis. Int J. Mol. Sci. 2022, 23, 1153. [Google Scholar] [CrossRef] [PubMed]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.C.; Peruchetti, D.B.; Silva, L.S.; Silva-Filho, J.L.; Souza, M.C.; Henriques, M.D.G.; Caruso-Neves, C.; Pinheiro, A.A.S. LPS Induces mTORC1 and mTORC2 Activation During Monocyte Adhesion. Front. Mol. Biosci. 2018, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- Xu, X.H.; Shah, P.K.; Faure, E.; Equils, O.; Thomas, L.; Fishbein, M.C.; Luthringer, D.; Xu, X.P.; Rajavashisth, T.B.; Yano, J.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. [Google Scholar] [CrossRef]
- Malgor, R.; Bhatt, P.M.; Connolly, B.A.; Jacoby, D.L.; Feldmann, K.J.; Silver, M.J.; Nakazawa, M.; McCall, K.D.; Goetz, D.J. Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflamm. Res. 2014, 63, 277–285. [Google Scholar] [CrossRef][Green Version]
- Westerterp, M.; Berbée, J.F.; Pires, N.M.; van Mierlo, G.J.; Kleemann, R.; Romijn, J.A.; Havekes, L.M.; Rensen, P.C. Apolipoprotein C-I is crucially involved in lipopolysaccharide-induced atherosclerosis development in apolipoprotein E-knockout mice. Circulation 2007, 116, 2173–2181. [Google Scholar] [CrossRef]
- Kosma, P. Chlamydial lipopolysaccharide. Biochim. Biophys. Acta 1999, 1455, 387–402. [Google Scholar] [CrossRef]
- Muotiala, A.; Helander, I.M.; Pyhälä, L.; Kosunen, T.U.; Moran, A.P. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect. Immun. 1992, 60, 1714–1716. [Google Scholar] [CrossRef]
- Ogawa, T.; Uchida, H.; Amino, K. Immunobiological activities of chemically defined lipid A from lipopolysaccharides of Porphyromonas gingivalis. Microbiology 1994, 140, 1209–1216. [Google Scholar] [CrossRef]
- Triantafilou, M.; Gamper, F.G.; Lepper, P.M.; Mouratis, M.A.; Schumann, C.; Harokopakis, E.; Schifferle, R.E.; Hajishengallis, G.; Triantafilou, K. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell. Microbiol. 2007, 9, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi-Ishii, Y.; Shimada, K.; Daida, H.; Toman, R.; Nagaoka, I. Low potency of Chlamydophila LPS to activate human mononuclear cells due to its reduced affinities for CD14 and LPS-binding protein. Int. Immunol. 2008, 20, 199–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, C.O.; Huh, A.J.; Han, S.H.; Kim, J.M. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch. Gerontol. Geriatr. 2012, 54, e35–e41. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef]
- Yu, H.M.; Zhao, Y.M.; Luo, X.G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.Y.; Luo, X.M.; Chen, S.D.; Wang, X.Y. Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef]
- Feng, G.; Zheng, K.; Cao, T.; Zhang, J.; Lian, M.; Huang, D.; Wei, C.; Gu, Z.; Feng, X. Repeated stimulation by LPS promotes the senescence of DPSCs via TLR4/MyD88-NF-κB-p53/p21 signaling. Cytotechnology 2018, 70, 1023–1035. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, X. Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E334–E344. [Google Scholar] [CrossRef]
- Wang, H.; Fu, H.; Zhu, R.; Wu, X.; Ji, X.; Li, X.; Jiang, H.; Lin, Z.; Tang, X.; Sun, S.; et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 2020, 12, 9240–9259. [Google Scholar] [CrossRef]
- Xuan, H.; Yuan, W.; Chang, H.; Liu, M.; Hu, F. Anti-inflammatory effects of Chinese propolis in lipopolysaccharide-stimulated human umbilical vein endothelial cells by suppressing autophagy and MAPK/NF-κB signaling pathway. Inflammopharmacology 2019, 27, 561–571. [Google Scholar] [CrossRef]
- Sagiv, A.; Bar-Shai, A.; Levi, N.; Hatzav, M.; Zada, L.; Ovadya, Y.; Roitman, L.; Manella, G.; Regev, O.; Majewska, J.; et al. p53 in Bronchial Club Cells Facilitates Chronic Lung Inflammation by Promoting Senescence. Cell Rep. 2018, 22, 3468–3479. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Shi, Y.; Liu, Y.; Pan, X.H.; Zhang, K.X. Telomere shortening activates TGF-β/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol. Sin. 2018, 39, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Pratsinis, H.; Zacharatos, P.; Demoliou, C.; Sigala, F.; Asimacopoulos, P.J.; Papavassiliou, A.G.; Kletsas, D. p53-dependent ICAM-1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab. Investig. 2005, 85, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohkuma, M.; Nagaoka, I. Bacterial lipopolysaccharide and antimicrobial LL-37 enhance ICAM-1 expression and NF-κB p65 phosphorylation in senescent endothelial cells. Int. J. Mol. Med. 2019, 44, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, M.; Honma, T.; Sato, K.; Shinohara, N.; Ito, J.; Tanaka, Y.; Tsuduki, T.; Ikeda, I. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci. Biotechnol. Biochem. 2011, 75, 1098–1103. [Google Scholar] [CrossRef]
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12. [Google Scholar] [CrossRef]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. Regulation of NF-κB signaling by the A20 deubiquitinase. Cell. Mol. Immunol. 2012, 9, 123–130. [Google Scholar] [CrossRef]
- Budamagunta, V.; Manohar-Sindhu, S.; Yang, Y.; He, Y.; Traktuev, D.O.; Foster, T.C.; Zhou, D. Senescence-associated hyper-activation to inflammatory stimuli in vitro. Aging 2021, 13, 19088–19107. [Google Scholar] [CrossRef]
- Ogura, N.; Matsuda, U.; Tanaka, F.; Shibata, Y.; Takiguchi, H.; Abiko, Y. In vitro senescence enhances IL-6 production in human gingival fibroblasts induced by lipopolysaccharide from Campylobacter rectus. Mech. Ageing Dev. 1996, 87, 47–59. [Google Scholar] [CrossRef]
- Mochizuki, K.; Yamaguchi, M.; Abiko, Y. Enhancement of LPS-stimulated plasminogen activator production in aged gingival fibroblasts. J. Periodontal Res. 1999, 34, 251–260. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, 295. [Google Scholar] [CrossRef]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging 2019, 11, 3574–3584. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, I.; Hirota, S.; Niyonsaba, F.; Hirata, M.; Adachi, Y.; Tamura, H.; Tanaka, S.; Heumann, D. Augmentation of the lipopolysaccharide-neutralizing activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by replacement with hydrophobic and cationic amino acid residues. Clin. Diagn. Lab. Immunol. 2002, 9, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Murakami, T.; Suzuki, K.; Tamura, H.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I. Antimicrobial Cathelicidin Peptide LL-37 Inhibits the LPS/ATP-Induced Pyroptosis of Macrophages by Dual Mechanism. PLoS ONE 2014, 9, e85765. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Murakami, T.; Kuwahara-Arai, K.; Tamura, H.; Hiramatsu, K.; Nagaoka, I. Human anti-microbial cathelicidin peptide LL-37 suppresses the LPS-induced apoptosis of endothelial cells. Int. Immunol. 2011, 23, 185–193. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species of LPS | Condition | Target Cells | Responses | Ref. | |||
|---|---|---|---|---|---|---|---|
| SA-β-Gal Staining | Induction of SASP Factors | Induction of Molecules for Cell Cycle Arrest | Other Responses | ||||
| P. gingivalis | 10 ng/mL, 6 days | Mouse periodontal alveolar osteocytes | + | ICAM-1, IL-1β, IL-6, IL-8, MCP1, MMP12, MMP13 | p16, p21, p53 | Disordered distribution of F-actin | [84] |
| E. coli | 10 ng/mL, 6 days (3 or 6 times) | Human dental pulp stem cells | + | NA | p21, p53 | Disordered distribution of F-actin Increased nuclear localization of NF-κB p65 | [86] |
| Species not described | 10 ng/mL, 6 days (3 or 6 times) | BV2 mouse microglial cells | + | NA | p53 | Increased SAHF formation | [85] |
| Species not described | 15 µg/mL, 7 days | A549 human pulmonary alveolar epithelial cells | + | NA | NA | Increased lysosomal content Telomere shortening | [83] |
| Species not described | 0.2 µg/mL, 24 h | Mouse adipocyte progenitor cells | + | TNF-α, IL-6, MCP1, VEGF-A, HIF-1α | NA | Increased expression of C/EBPβ, p38 MAPK and NF-κB p65 | [87] |
| Species not described | 1 µg/mL, 24 h | THP-1 human macrophage-like cells | + | IL-6, TNF-α, CXCL1 | p16, p21, p53 | Increased expression of NF-κB | [88] |
| Species not described | 1 µg/mL, 24 h | HUVECs | NA | NA | p21, p53 | Increased expression of NF-κB p65 | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, K.; Susaki, E.A.; Nagaoka, I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 11148. https://doi.org/10.3390/ijms231911148
Suzuki K, Susaki EA, Nagaoka I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(19):11148. https://doi.org/10.3390/ijms231911148
Chicago/Turabian StyleSuzuki, Kaori, Etsuo A. Susaki, and Isao Nagaoka. 2022. "Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis" International Journal of Molecular Sciences 23, no. 19: 11148. https://doi.org/10.3390/ijms231911148
APA StyleSuzuki, K., Susaki, E. A., & Nagaoka, I. (2022). Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. International Journal of Molecular Sciences, 23(19), 11148. https://doi.org/10.3390/ijms231911148