
Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Patients’ Characteristics
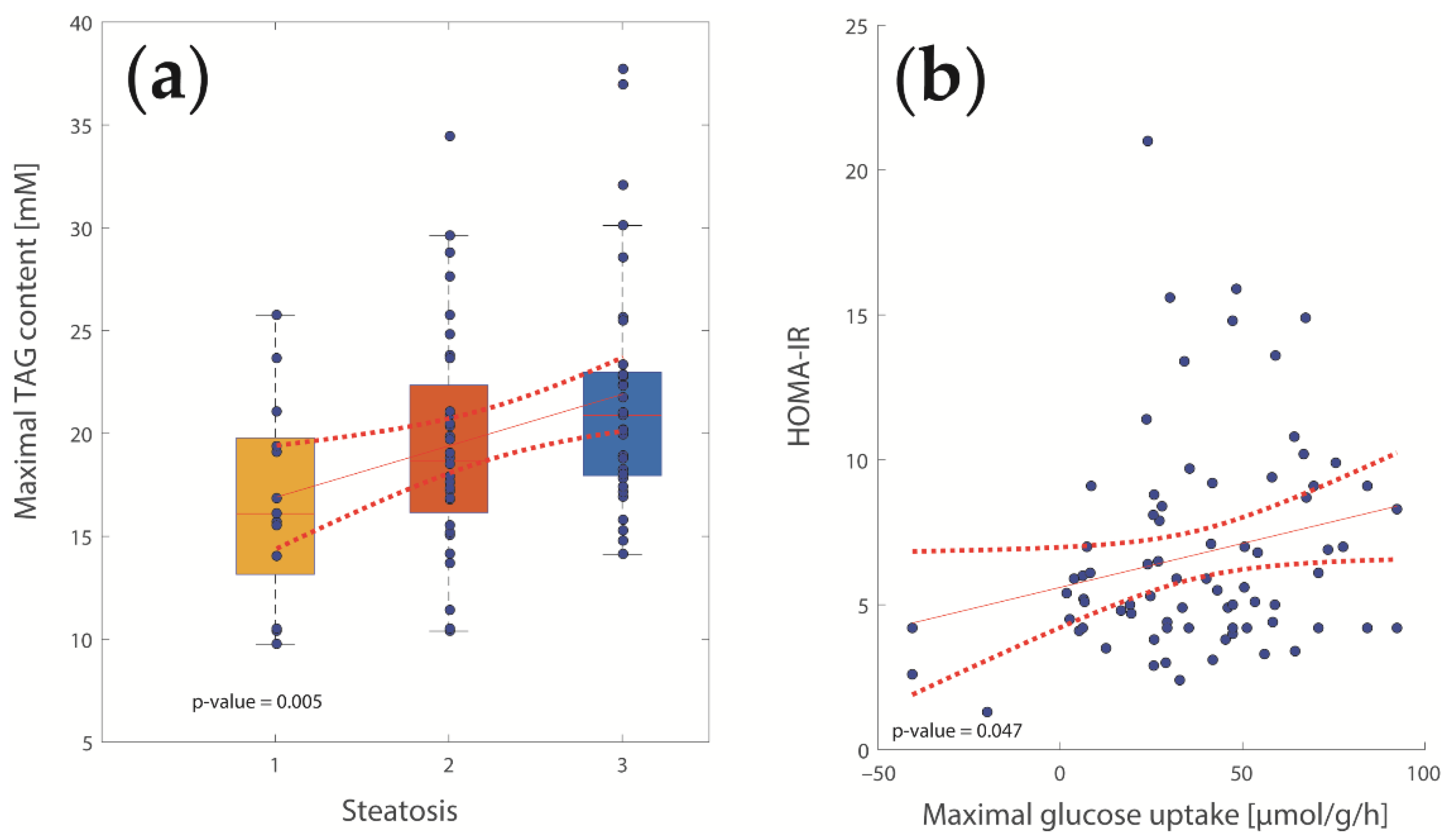
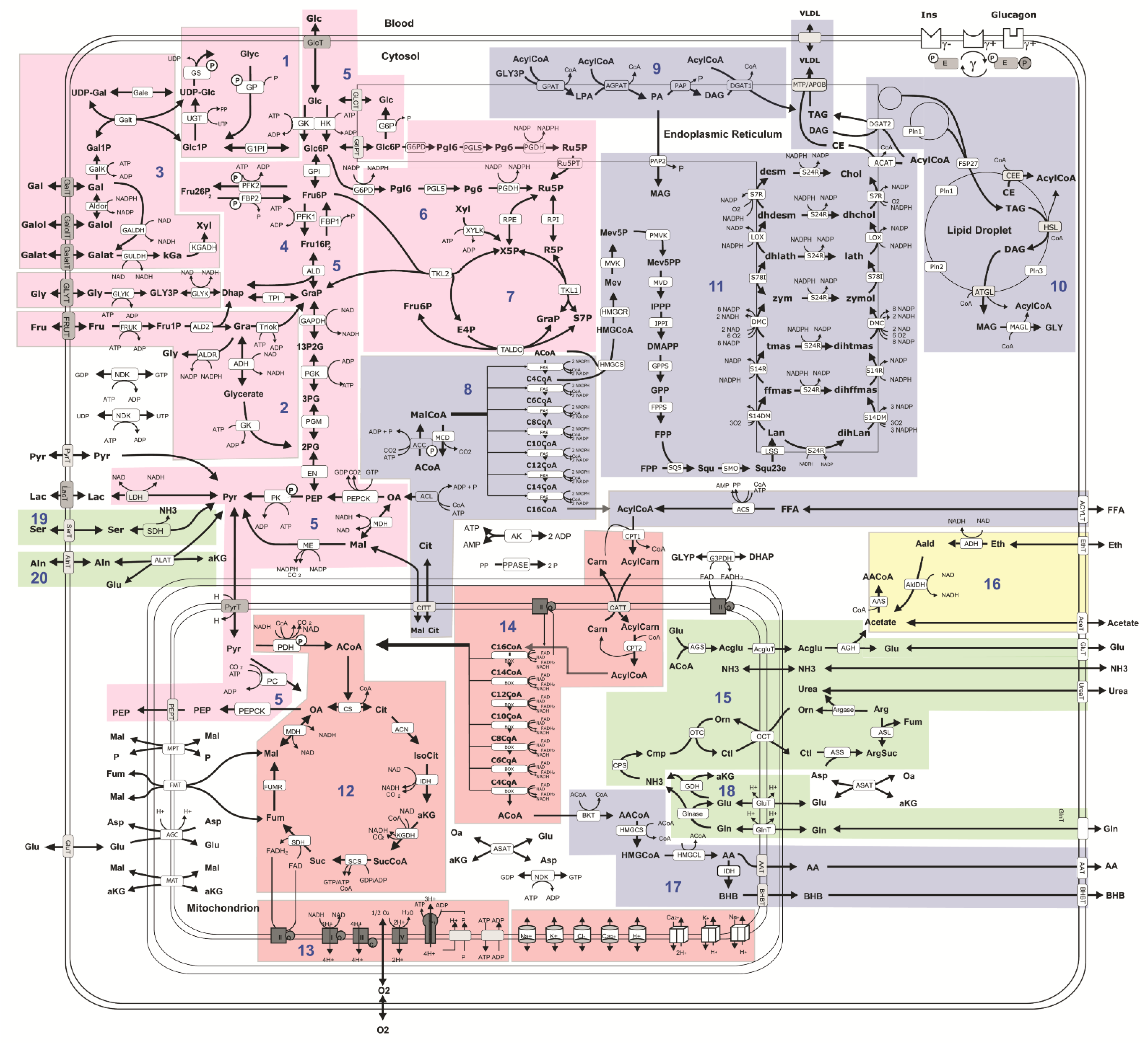
2.2. Computational Assessment of Metabolic Functions and Maximal Metabolic Capacities
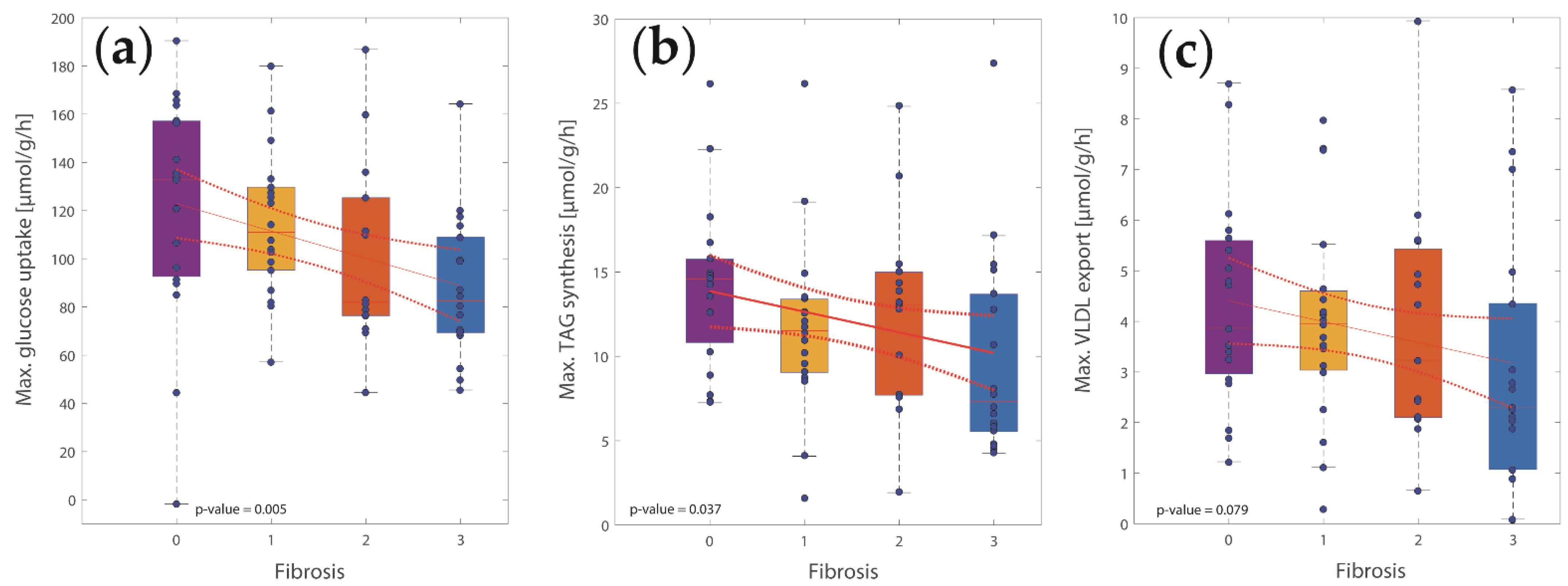
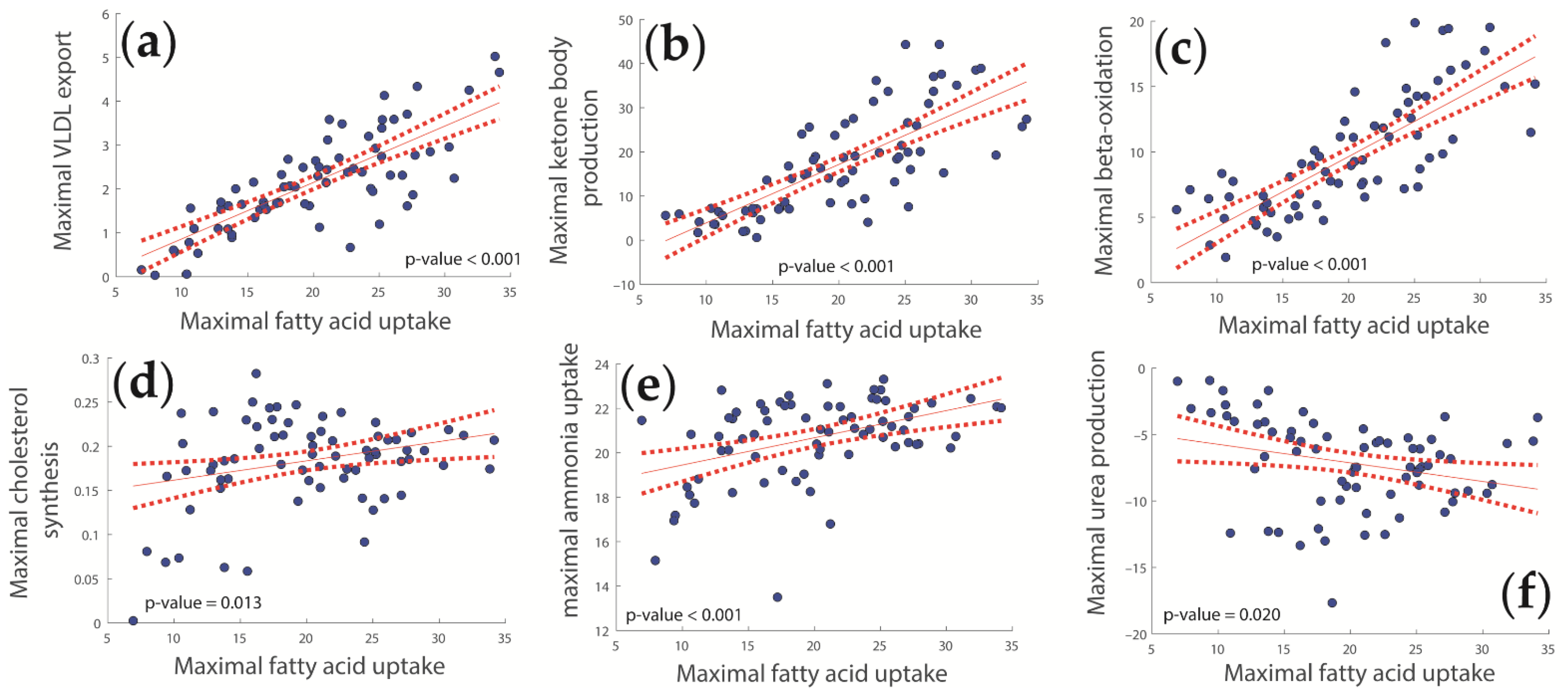
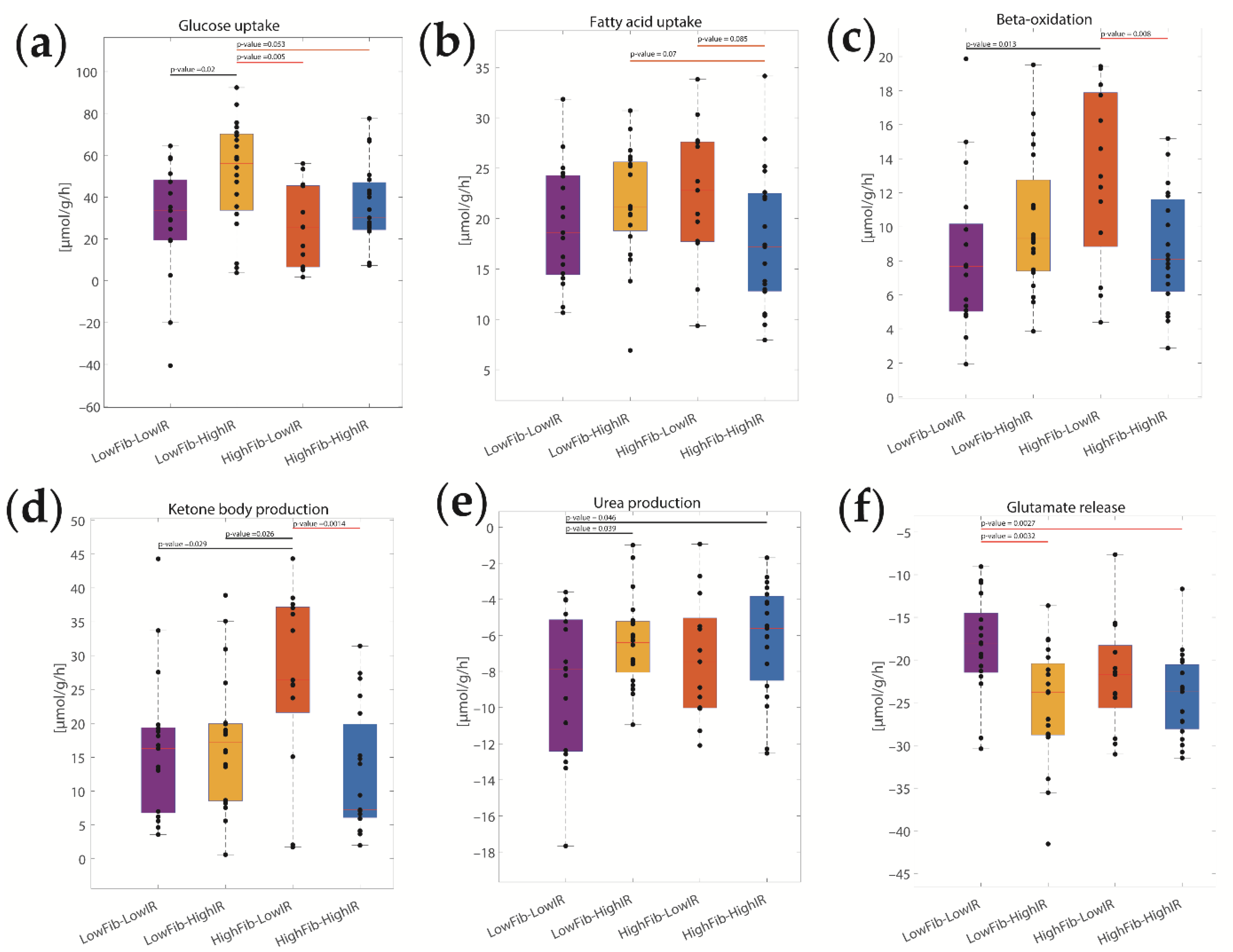
2.3. Concerted Metabolic Alterations in NASH Livers
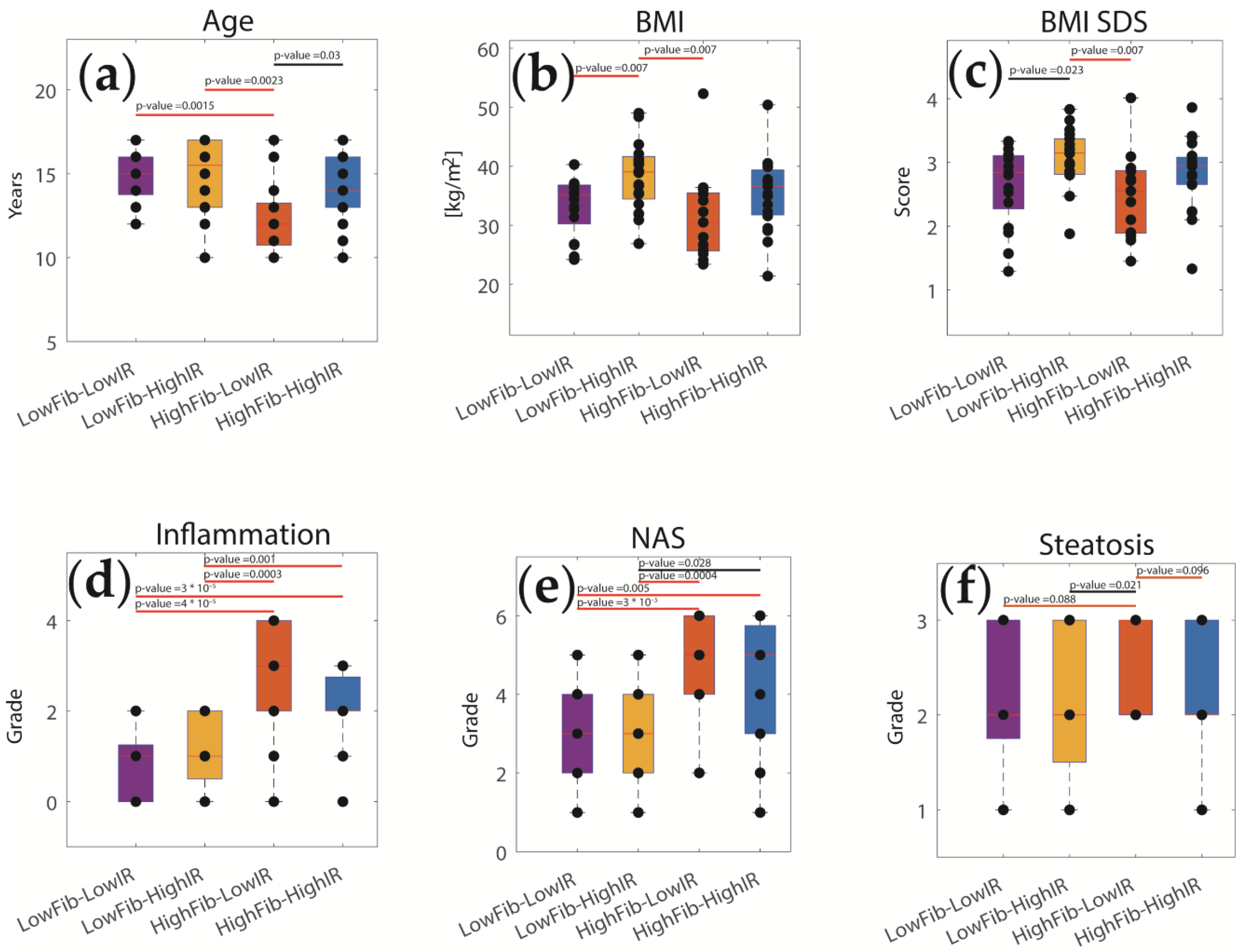
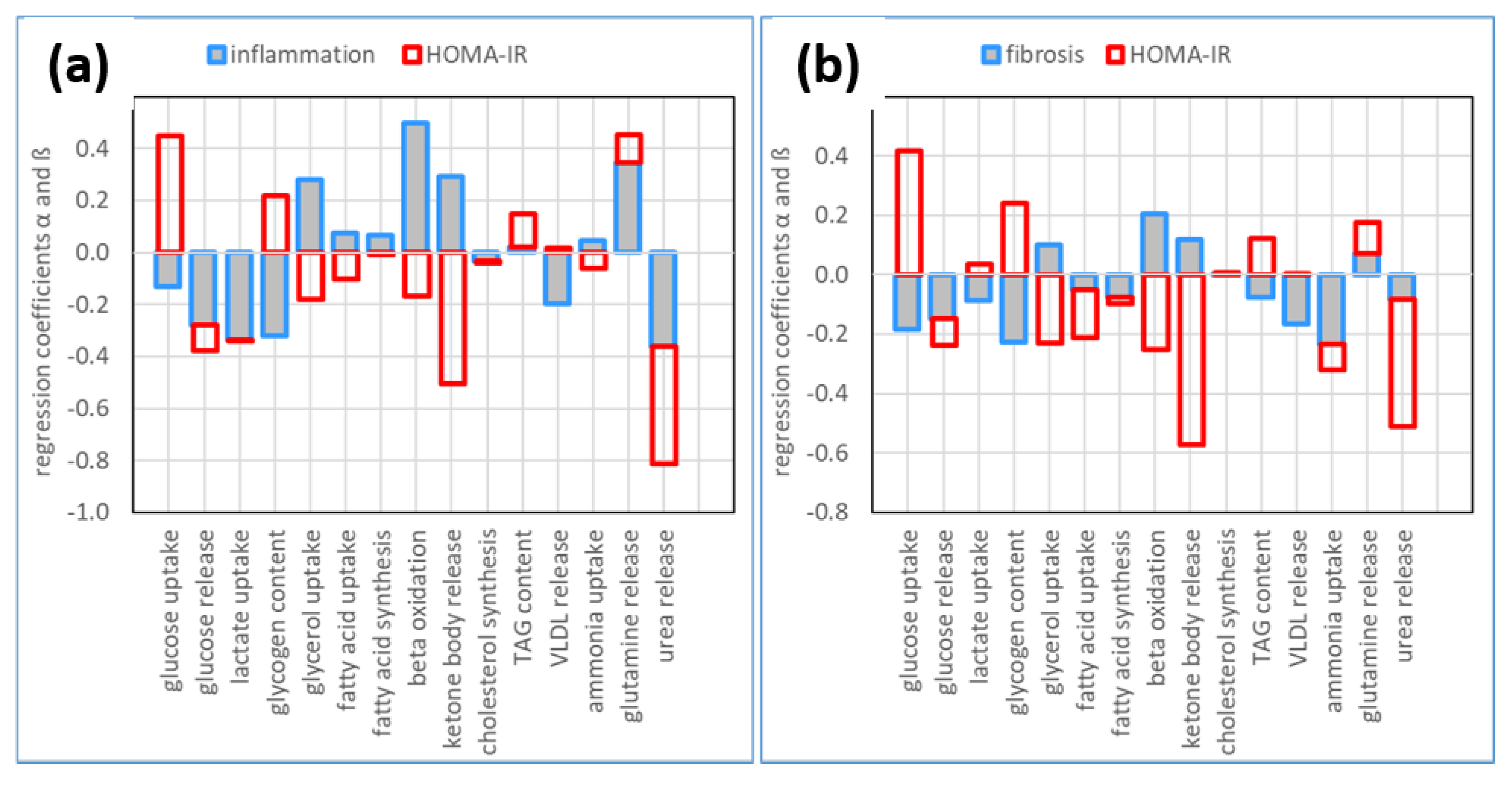
2.4. The Relative Impact of NASH and IR on Alterations of Hepatic Metabolism
3. Discussion
3.1. Association of Metabolic Capacities and Clinical Markers of NASH and IR
3.2. Hepatic Metabolic Alterations in NASH and IR
3.3. Lipid Metabolism
3.4. Glucose Metabolism
3.5. Ammonia Metabolism
3.6. Limitations of Our Approach
4. Materials and Methods
4.1. Study Design and Patients
4.2. Histological Assessment
4.3. Liquid Chromatography-Mass Spectrometry (MS) Instrument Settings for Shotgun Proteome Profiling and Data Analysis
4.4. Computational Assessment of Metabolic Capacities
4.5. Disease Classes Based on the Severity of NNASH and IR
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiegand, S.; Keller, K.M.; Röbl, M.; Reinehr, T.; Widhalm, K.; Holl, R.W. Obese boys at increased risk for nonalcoholic liver disease: Evaluation of 16,390 overweight or obese children and adolescents. Int. J. Obes. 2010, 34, 1468–1474. [Google Scholar] [CrossRef]
- Kohli, R.; Sunduram, S.; Mouzaki, M.; Ali, S.; Sathya, P.; Abrams, S.; Xanthakos, S.A.; Vos, M.; Schwimmer, J.B. Pediatric Nonalcoholic Fatty Liver Disease: A Report from the Expert Committee on Nonalcoholic Fatty Liver Disease (ECON). J. Pediatr. 2016, 172, 9–13. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef]
- Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. Int. J. Mol. Sci. 2016, 17, 947. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D. Nonalcoholic fatty liver disease: A novel cardiometabolic risk factor for type 2 diabetes and its complications. J. Clin. Endocrinol. Metab. 2013, 98, 483–495. [Google Scholar] [CrossRef]
- Nobili, V.; Mantovani, A.; Cianfarani, S.; Alisi, A.; Mosca, A.; Sartorelli, M.R.; Maffeis, C.; Loomba, R.; Byrne, C.D.; Targher, G. Prevalence of prediabetes and diabetes in children and adolescents with biopsy-proven non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 802–810. [Google Scholar] [CrossRef]
- Dharmalingam, M.; Yamasandhi, P. Nonalcoholic fatty liver disease and type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Gastaldelli, A. Nonalcoholic fatty liver disease and type 2 diabetes: Common pathophysiologic mechanisms. Curr. Diabetes Rep. 2015, 15, 1–13. [Google Scholar] [CrossRef]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and diabetes: Two sides of the same coin? Rationale for gene-based personalized NAFLD treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Hansmannel, F.; Mordier, S.; Iynedjian, P.B. Insulin induction of glucokinase and fatty acid synthase in hepatocytes: Analysis of the roles of sterol-regulatory-element-binding protein-1c and liver X receptor. Biochem. J. 2006, 399, 275–283. [Google Scholar] [CrossRef]
- Klover, P.J.; Zimmers, T.A.; Koniaris, L.G.; Mooney, R.A. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 2003, 52, 2784–2789. [Google Scholar] [CrossRef]
- Berger, D.; Desai, V.; Janardhan, S. Con: Liver biopsy remains the gold standard to evaluate fibrosis in patients with nonalcoholic fatty liver disease. Clin. Liver Dis. 2019, 13, 114. [Google Scholar] [CrossRef]
- Kaikkonen, J.E.; Würtz, P.; Suomela, E.; Lehtovirta, M.; Kangas, A.J.; Jula, A.; Mikkilä, V.; Viikari, J.S.; Juonala, M.; Rönnemaa, T.; et al. Metabolic profiling of fatty liver in young and middle-aged adults: Cross-sectional and prospective analyses of the Young Finns Study. Hepatology 2017, 65, 491–500. [Google Scholar] [CrossRef]
- Berndt, N.; Bulik, S.; Wallach, I.; Wünsch, T.; König, M.; Stockmann, M.; Meierhofer, D.; Holzhütter, H.-G. HEPATOKIN1 is a biochemistry-based model of liver metabolism for applications in medicine and pharmacology. Nat. Commun. 2018, 9, 2386. [Google Scholar] [CrossRef]
- Wallstab, C.; Eleftheriadou, D.; Schulz, T.; Damm, G.; Seehofer, D.; Borlak, J.; Holzhütter, H.-G.; Berndt, N. A unifying mathematical model of lipid droplet metabolism reveals key molecular players in the development of hepatic steatosis. FEBS J. 2017, 284, 3245–3261. [Google Scholar] [CrossRef]
- Chandar, V.; Gidvani, C.; Gupta, A.; Wilson, C.; Sharma, Y. Lipid Profile in Normal Healthy Children. Med. J. Armed. Forces. India 1994, 50, 101–104. [Google Scholar] [CrossRef]
- Peplies, J.; Jiménez-Pavón, D.; Savva, S.C.; Buck, C.; Günther, K.; Fraterman, A.; Russo, P.; Iacoviello, L.; Veidebaum, T.; Tornaritis, M.; et al. Percentiles of fasting serum insulin, glucose, HbA1c and HOMA-IR in pre-pubertal normal weight European children from the IDEFICS cohort. Int. J. Obes. 2014, 38 (Suppl. S2), S39–S47. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Argo, C.K.; Ikura, Y.; Lackner, C.; Caldwell, S.H. The fat droplet in hepatocellular ballooning and implications for scoring nonalcoholic steatohepatitis therapeutic response. Hepatology 2016, 63, 1056–1057. [Google Scholar] [CrossRef]
- Berndt, N.; Holzhütter, H.-G. Dynamic Metabolic Zonation of the Hepatic Glucose Metabolism Is Accomplished by Sinusoidal Plasma Gradients of Nutrients and Hormones. Front. Physiol. 2018, 9, 1786. [Google Scholar] [CrossRef]
- Loomis, A.K.; Kabadi, S.; Preiss, D.; Hyde, C.; Bonato, V.; Louis, M.S.; Desai, J.; Gill, J.M.R.; Welsh, P.; Waterworth, D.; et al. Body Mass Index and Risk of Nonalcoholic Fatty Liver Disease: Two Electronic Health Record Prospective Studies. J. Clin. Endocrinol. Metab. 2016, 101, 945–952. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, M.; Hu, Z.; Shrestha, U.K. Prevalence of nonalcoholic fatty liver disease and its metabolic risk factors in women of different ages and body mass index. Menopause 2015, 22, 667–673. [Google Scholar] [CrossRef]
- Parks, E.J.; Hellerstein, M.K. Thematic review series: Patient-oriented research. Recent advances in liver triacylglycerol and fatty acid metabolism using stable isotope labeling techniques. J. Lipid Res. 2006, 47, 1651–1660. [Google Scholar] [CrossRef]
- Reisz, J.A.; D’Alessandro, A. Measurement of metabolic fluxes using stable isotope tracers in whole animals and human patients. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 366–374. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Bawden, S.; Aithal, G. Metabolic Imaging in Non-Alcoholic Fatty Liver Disease: Applications of Magnetic Resonance Spectroscopy. J. Clin. Med. 2021, 10, 632. [Google Scholar] [CrossRef]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2009, 16, 893–901. [Google Scholar] [CrossRef]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investiig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Randle, P.; Garland, P.; Hales, C.; Newsholme, E. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef]
- Lopes, R.; Santana, M.S.; Da Cruz, C.R.; Fulindi, R.B.; Gaspar, A.M.M.; Da Costa, P.I. Central cellular signaling pathways involved with the regulation of lipid metabolism in the liver: A review. Acta Scientiarum. Biol. Sci. 2020, 42, e51151. [Google Scholar] [CrossRef]
- Hijona, E.; Hijona, L.; Arenas, J.I.; Bujanda, L. Inflammatory mediators of hepatic steatosis. Mediat. Inflamm. 2010, 2010, 837419. [Google Scholar] [CrossRef]
- Memon, R.A.; Grunfeld, C.; Moser, A.H.; Feingold, K.R. Tumor necrosis factor mediates the effects of endotoxin on cholesterol and triglyceride metabolism in mice. Endocrinology 1993, 132, 2246–2253. [Google Scholar] [CrossRef]
- Anderson, S.W.; Soto, J.A.; Milch, H.N.; Ozonoff, A.; O'Brien, M.; Hamilton, J.A.; Jara, H.J. Effect of disease progression on liver apparent diffusion coefficient values in a murine model of NASH at 11.7 Tesla MRI. J. Magn. Reson. Imaging 2011, 33, 882–888. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of ketone body metabolism and the role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4, e127737. [Google Scholar] [CrossRef]
- Cali, A.M.; De Oliveira, A.M.; Kim, H.; Chen, S.; Reyes-Mugica, M.; Escalera, S.; Dziura, J.; Taksali, S.E.; Kursawe, R.; Shaw, M.; et al. Glucose dysregulation and hepatic steatosis in obese adolescents: Is there a link? Hepatology 2009, 49, 1896–1903. [Google Scholar] [CrossRef]
- Bulik, S.; Holzhütter, H.-G.; Berndt, N. The relative importance of kinetic mechanisms and variable enzyme abundances for the regulation of hepatic glucose metabolism--insights from mathematical modeling. BMC Biol. 2016, 14, 15. [Google Scholar] [CrossRef]
- König, M.; Bulik, S.; Holzhütter, H.-G. Quantifying the contribution of the liver to glucose homeostasis: A detailed kinetic model of human hepatic glucose metabolism. PLoS Comput. Biol. 2012, 8, e1002577. [Google Scholar] [CrossRef]
- De Chiara, F.; Heebøll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Grønbæk, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef]
- Du, K.; Chitneni, S.K.; Suzuki, A.; Wang, Y.; Henao, R.; Hyun, J.; Premont, R.T.; Naggie, S.; Moylan, C.A.; Bashir, M.R.; et al. Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef]
- Rosario, A.S.; Kurth, B.-M.; Stolzenberg, H.; Ellert, U.; Neuhauser, H. Body mass index percentiles for children and adolescents in Germany based on a nationally representative sample (KiGGS 2003–2006). Eur. J. Clin. Nutr. 2010, 64, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Gielisch, I.; Meierhofer, D. Metabolome and proteome profiling of complex I deficiency induced by rotenone. J. Proteome Res. 2015, 14, 224–235. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhütter, H.-G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Mean ± SD |
|---|---|
| Age (years) | 14.2 (11–17) |
| Male sex, n (%) | 55 (77.5) |
| BMI | 34.9 ± 6.4 |
| BMI z-score | 2.77 ± 0.59 |
| CRP (mg/L) | 4.7 ± 6.6 |
| ALT (U/L) | 108 ± 68 |
| AST (U/L) | 63 ± 41 |
| Triacylglycerol (mg/dL) | 136 ± 72 |
| Total cholesterol (mg/dL) | 165 ± 29 |
| HOMA-IR | 7.0 ± 3.7 |
| HbA1c (%) | 5.4 ± 1.0 |
| Grade/Stage | Steatosis | Portal Inflammation | Lobular Inflammation | Ballooning | Fibrosis |
|---|---|---|---|---|---|
| 0 | - | 26 (36.6) | 22 (31.0) | 37 (52.1) | 19 (26.8) |
| 1 | 13 (18.3) | 37 (52.1) | 39 (54.9) | 27 (38.0) | 19 (26.8) |
| 2 | 25 (35.2) | 8 (11.3) | 10 (14.1) | 7 (9.9) | 15 (21.1) |
| 3 | 33 (46.5) | - | - | - | 18 (25.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berndt, N.; Hudert, C.A.; Eckstein, J.; Loddenkemper, C.; Henning, S.; Bufler, P.; Meierhofer, D.; Sack, I.; Wiegand, S.; Wallach, I.; et al. Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 11072. https://doi.org/10.3390/ijms231911072
Berndt N, Hudert CA, Eckstein J, Loddenkemper C, Henning S, Bufler P, Meierhofer D, Sack I, Wiegand S, Wallach I, et al. Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(19):11072. https://doi.org/10.3390/ijms231911072
Chicago/Turabian StyleBerndt, Nikolaus, Christian A. Hudert, Johannes Eckstein, Christoph Loddenkemper, Stephan Henning, Philip Bufler, David Meierhofer, Ingolf Sack, Susanna Wiegand, Iwona Wallach, and et al. 2022. "Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 19: 11072. https://doi.org/10.3390/ijms231911072
APA StyleBerndt, N., Hudert, C. A., Eckstein, J., Loddenkemper, C., Henning, S., Bufler, P., Meierhofer, D., Sack, I., Wiegand, S., Wallach, I., & Holzhütter, H.-G. (2022). Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 23(19), 11072. https://doi.org/10.3390/ijms231911072