
Virtual Screening and Quantum Chemistry Analysis for SARS-CoV-2 RNA-Dependent RNA Polymerase Using the ChEMBL Database: Reproduction of the Remdesivir-RTP and Favipiravir-RTP Binding Modes Obtained from Cryo-EM Experiments with High Binding Affinity


Abstract
:1. Introduction
2. Results and Discussion
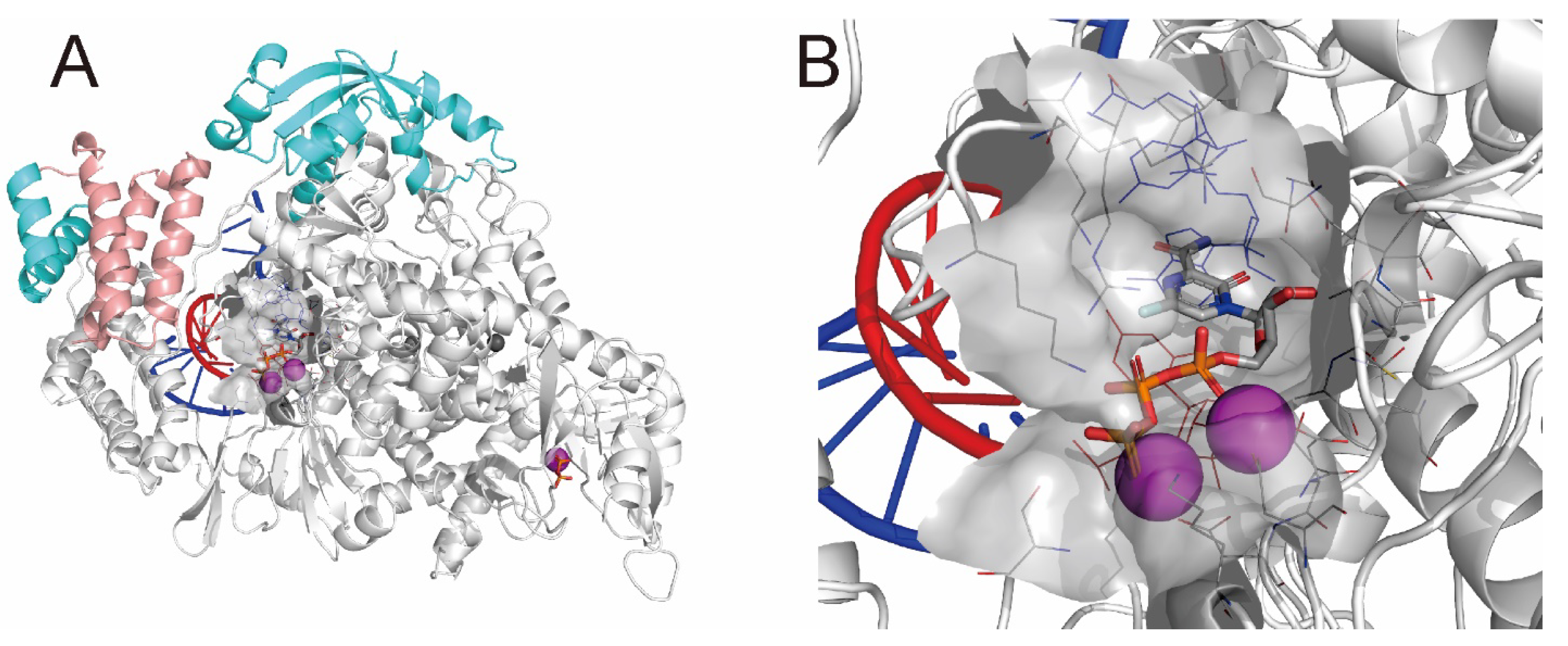
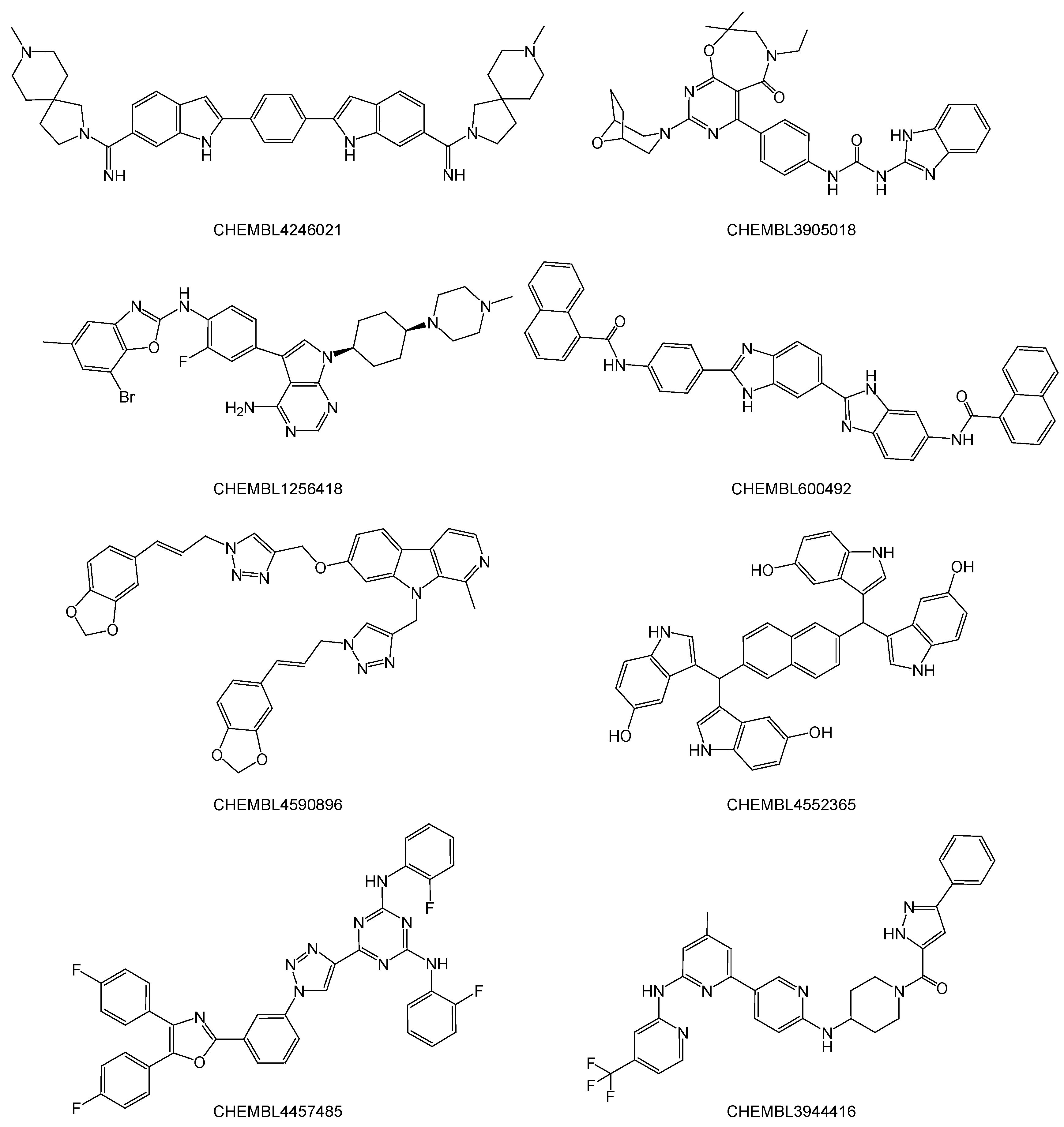
2.1. Structure-Based Virtual Screenings of the ChEMBL Database Predict Known Anti-SARS-CoV-2 Drugs
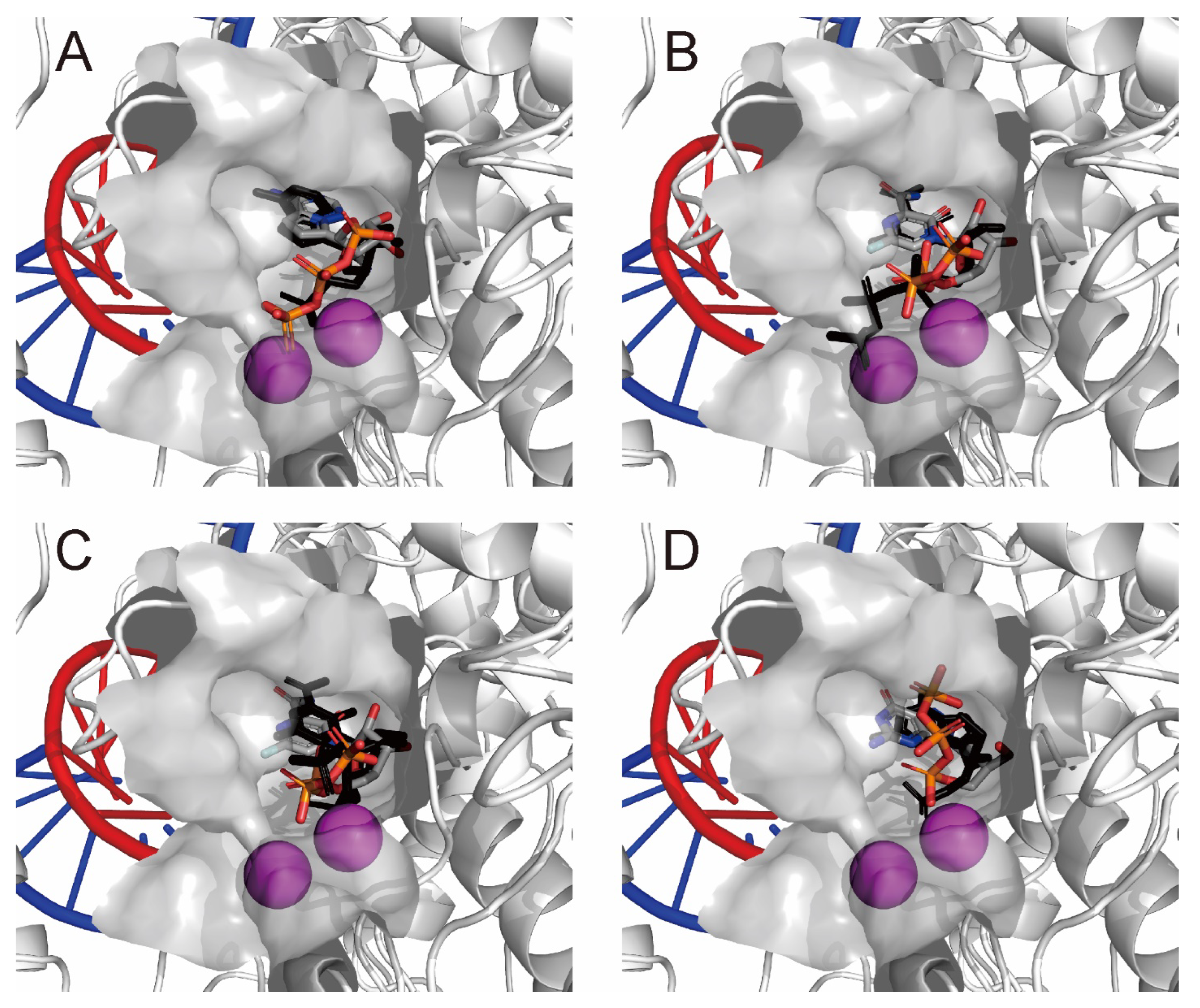
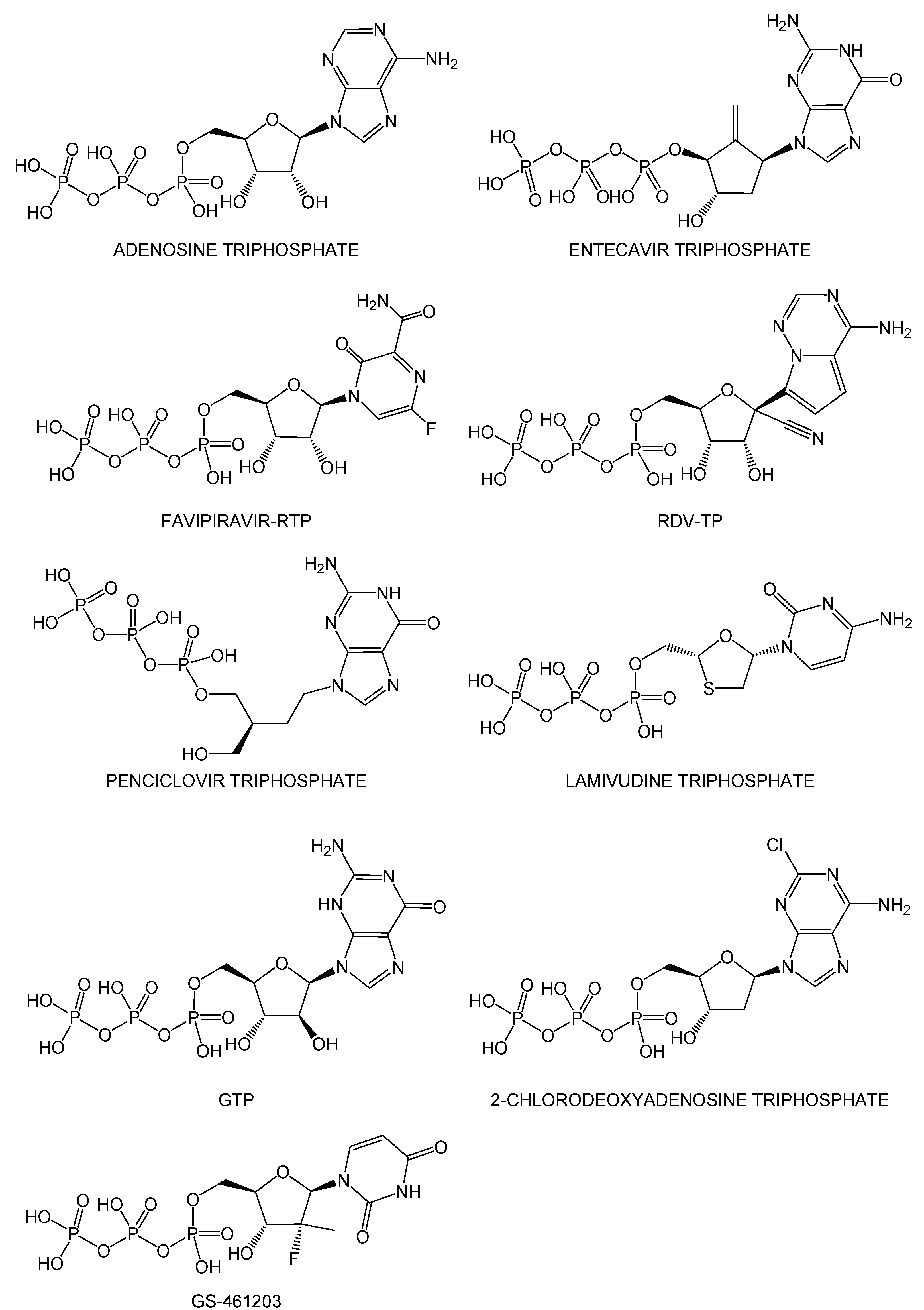
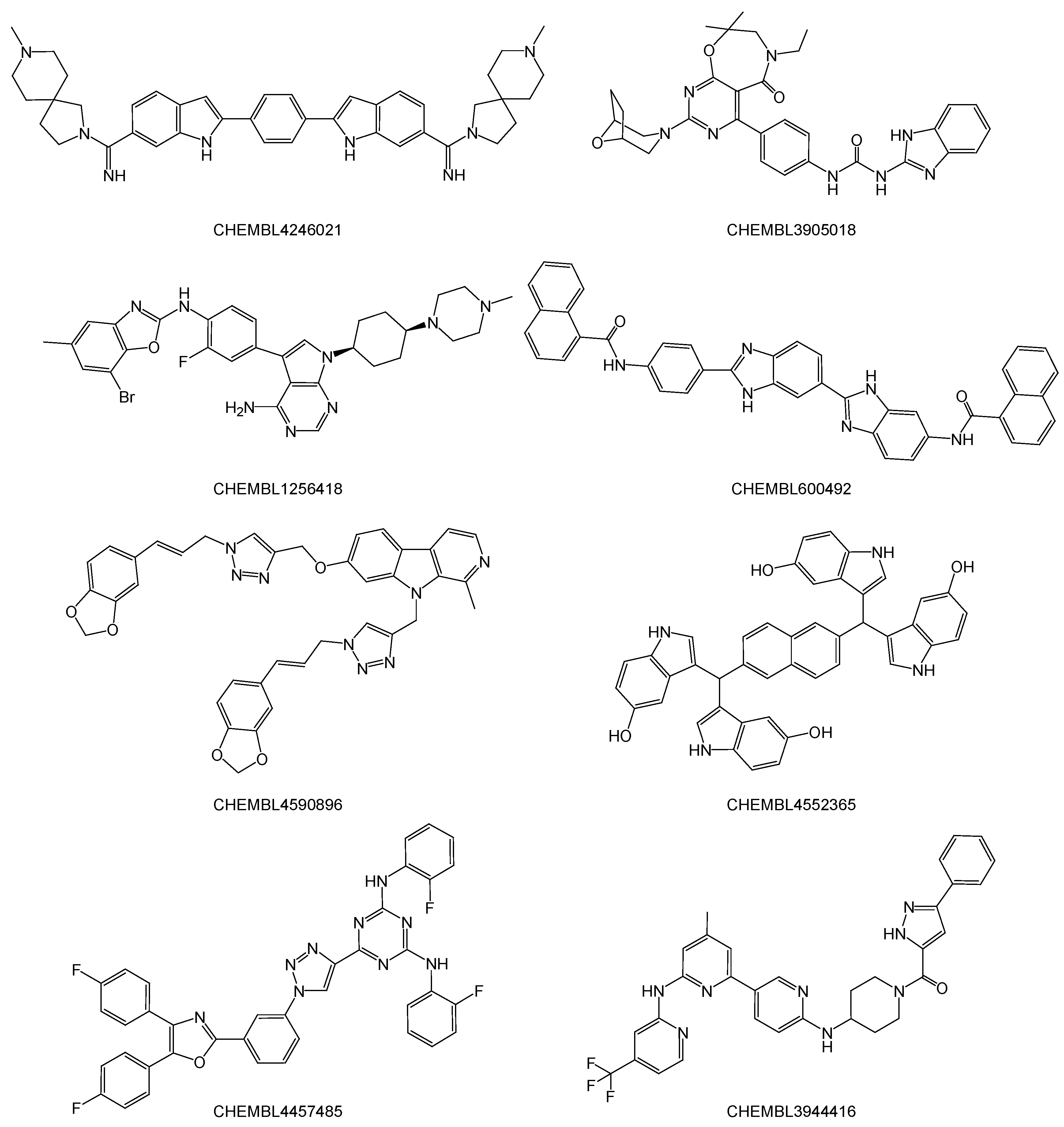
2.2. Noncovalent Bioactive Compounds Are Predicted as Potential Inhibitor Candidates for the SARS-CoV-2 RdRp Complex:dsRNA
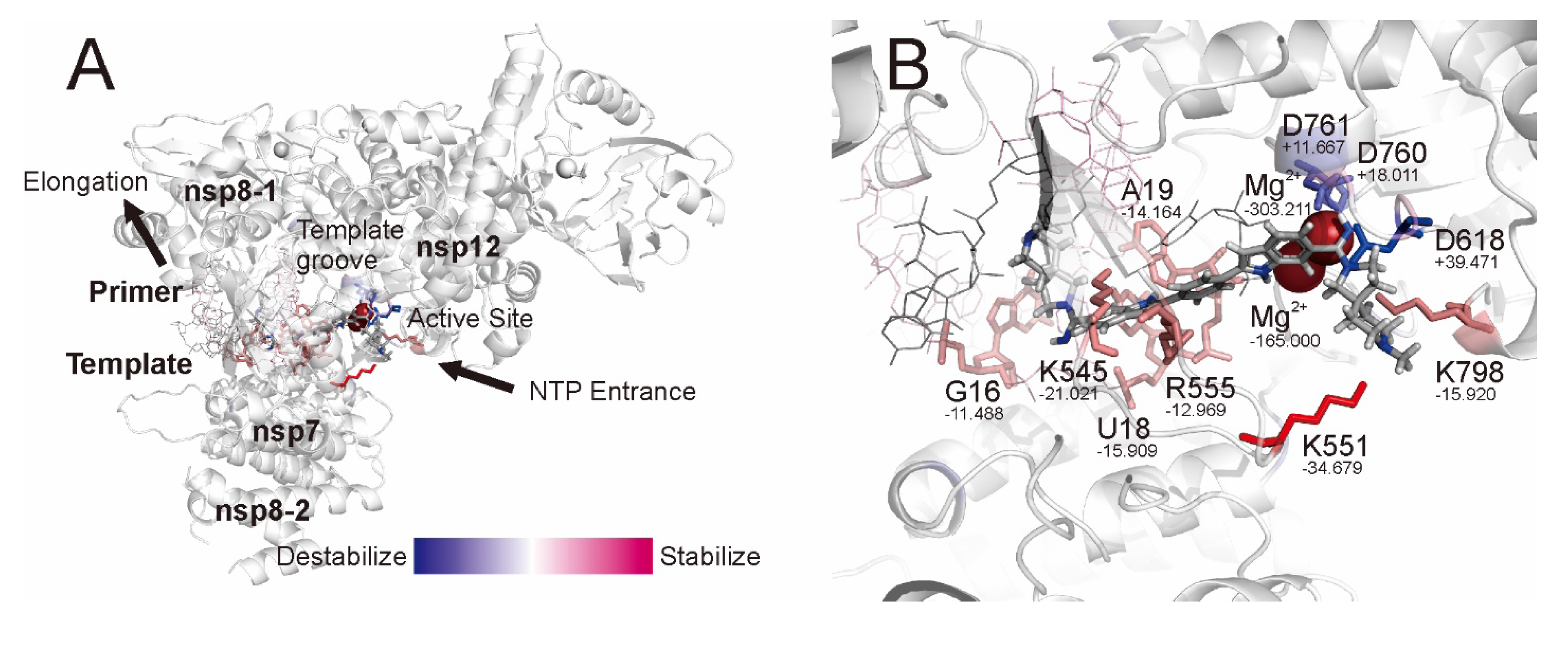
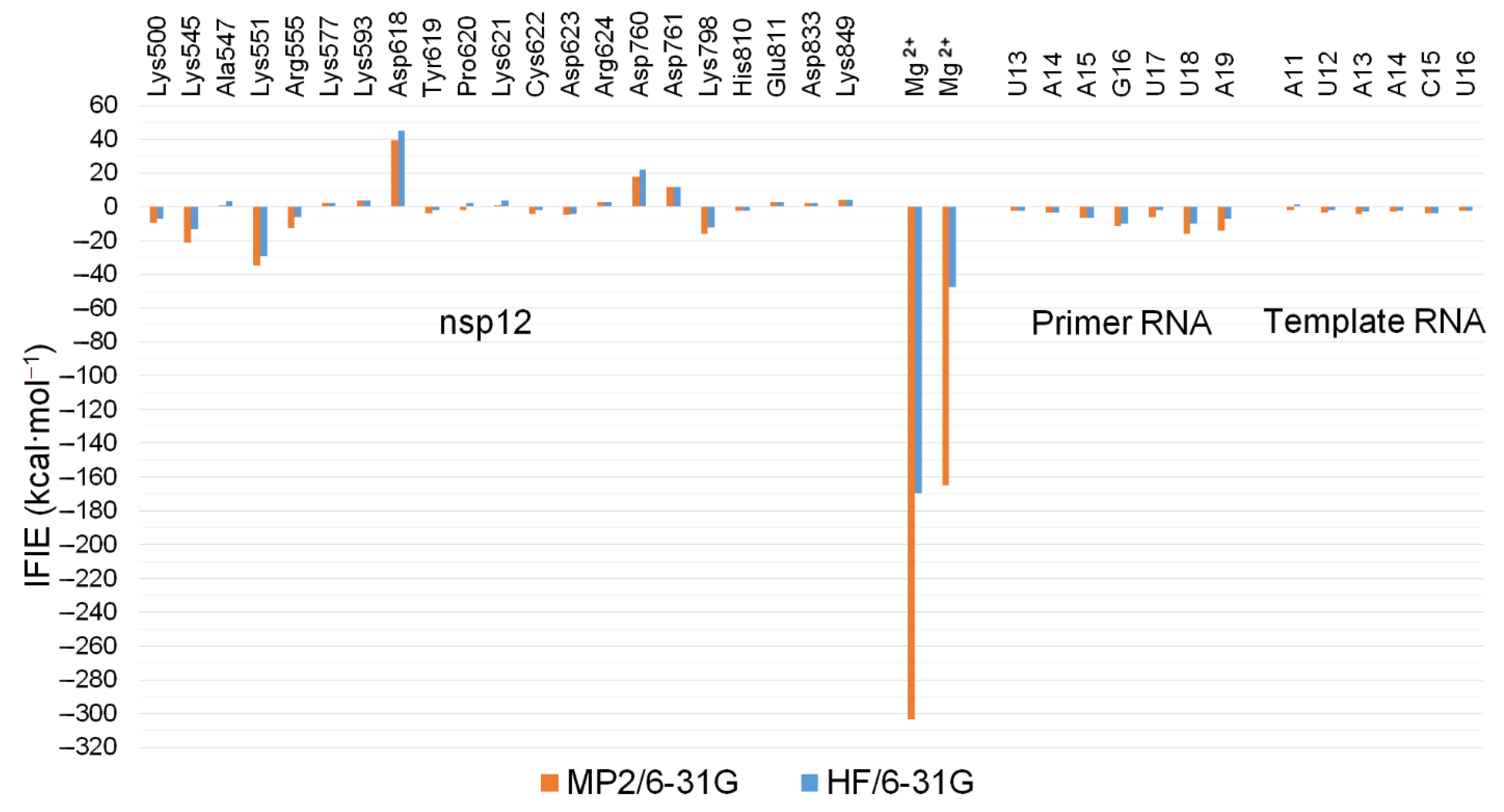
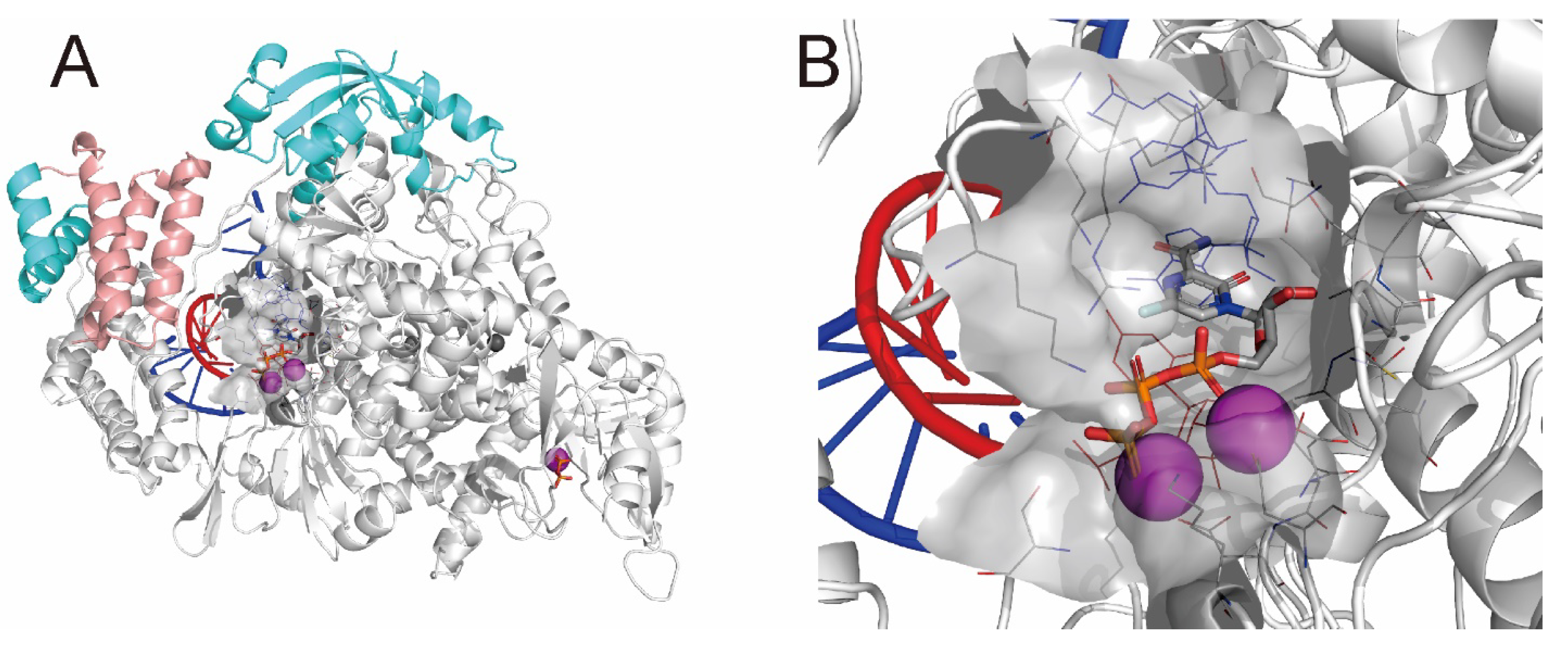
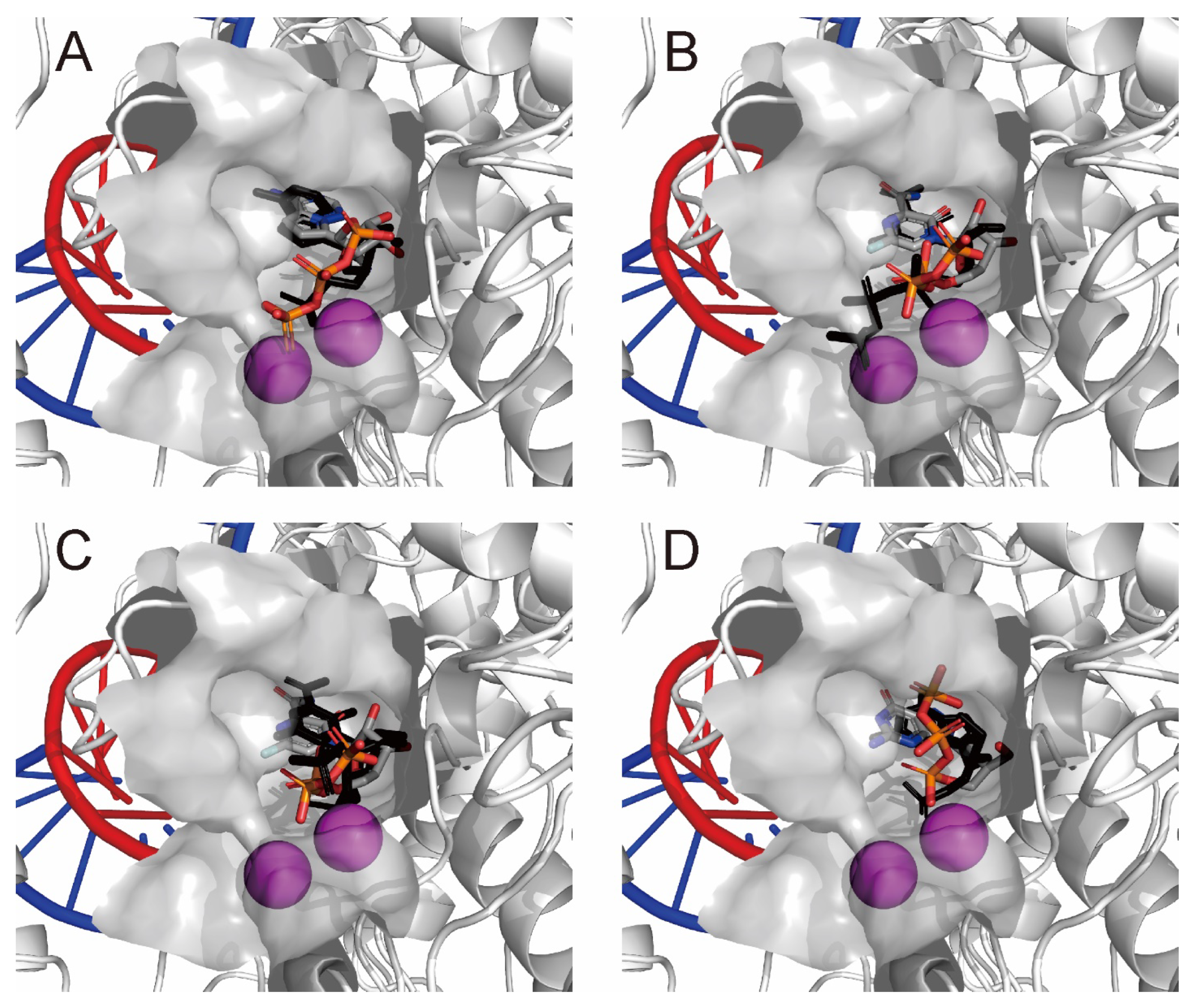
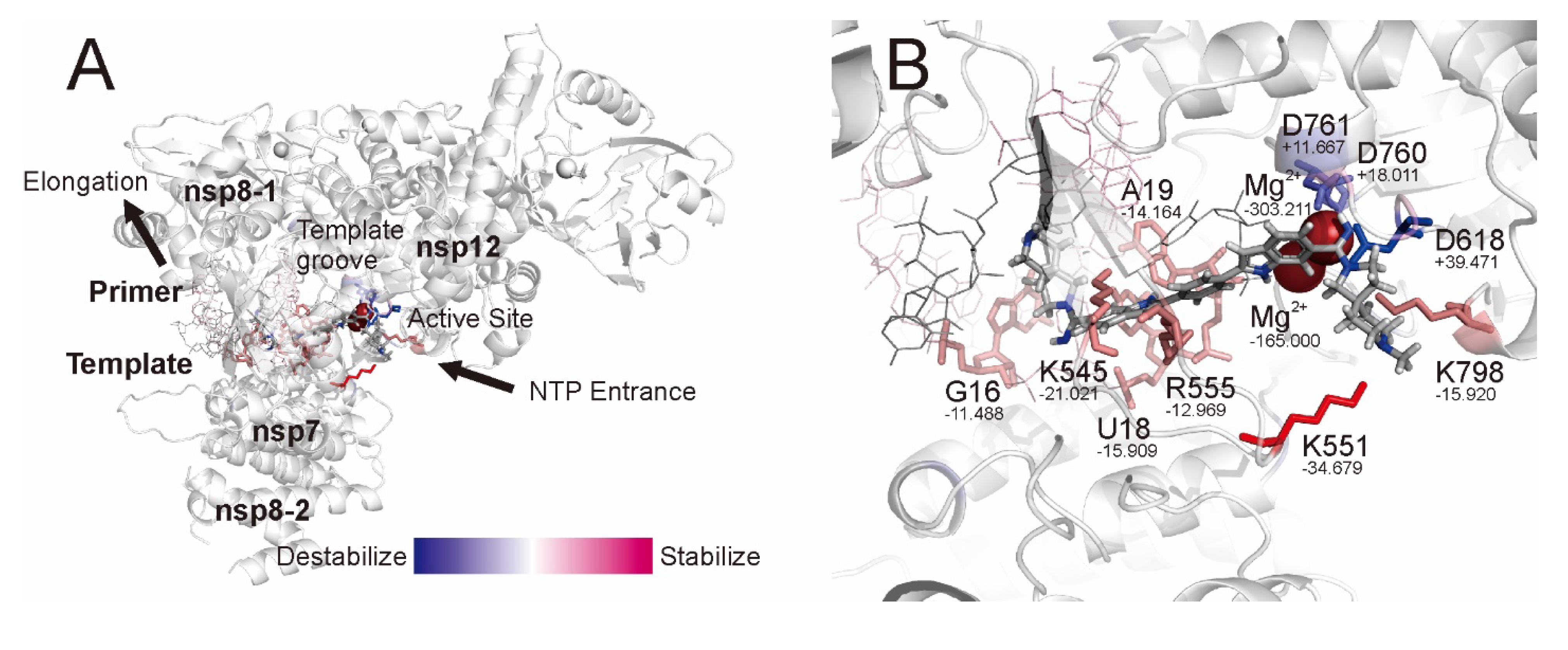
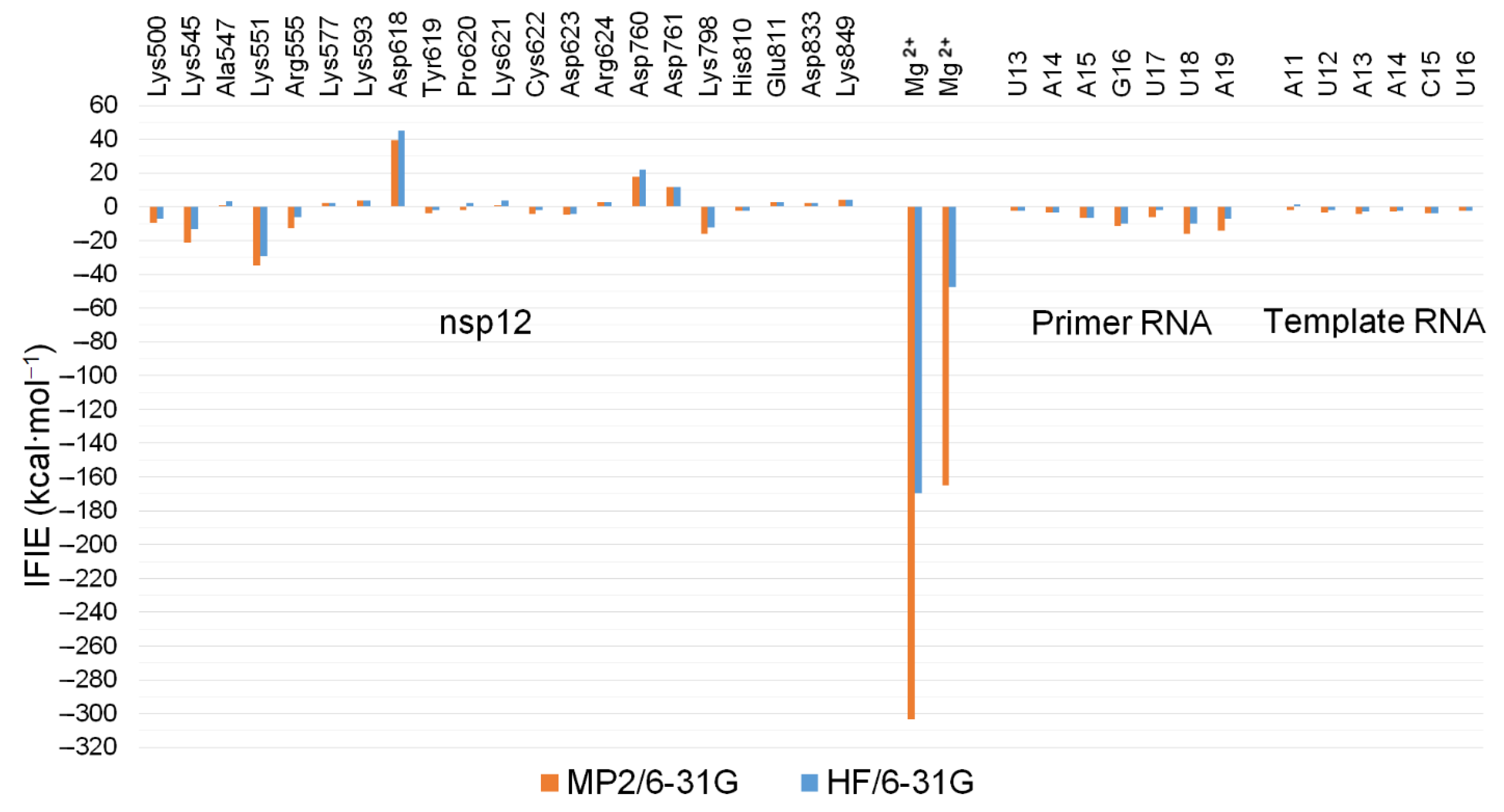
2.3. QM/MM ONIOM Geometry Optimization Calculations, Frequency Analyses, and FMO Calculations Are Useful for Improving Pharmacokinetic Properties and Developing Specific Anti-SARS-CoV-2 Drugs
3. Materials and Methods
3.1. Structural Refinement of the RdRp Complex:dsRNA
3.2. Preparation of the 3D Structure Database from the ChEMBL Database
3.3. Stepwise Structure-Based Virtual Screenings
3.4. ONIOM Geometry Optimization and Frequency Analysis Calculations
3.5. FMO Calculations
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavrius protease. FEBS Open Bio 2020, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 2020, 182, 417–428. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.P.K.; Dangerfield, T.L.; Taylor, D.W.; Johnson, K.A. Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol. Cell 2021, 81, 1548–1552. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural basis of SARS-CoV-2 polymerase inhibition by favipiravir. Innovation 2020, 2, 100080. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612-20. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 39, 3204–3212. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Janežič, D.; Bren, U. Potential novel thioether-amide or guanidine-linker class of SARS-CoV-2 virus RNA-dependent RNA polymerase inhibitors identified by high-throughput virtual screening coupled to free-energy calculations. Int. J. Mol. Sci. 2021, 22, 11143. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2021, 93, 389–400. [Google Scholar] [CrossRef]
- Singh, S.; Sk, M.F.; Sonawane, A.; Kar, P.; Sadhukhan, S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: An in-silico analysis. J. Biomol. Struct. Dyn. 2021, 39, 6249–6264. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Upadhyay, A.K.; Reddy, M.S. Screening of potent drug inhibitors against SARS-CoV-2 RNA polymerase: An in silico approach. 3 Biotech. 2021, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARSCoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 258, 118350. [Google Scholar] [CrossRef]
- Baby, K.; Maity, S.; Mehta, C.H.; Suresh, A.; Nayak, U.Y.; Nayak, Y. Targeting SARS-CoV-2 RNA-dependent RNA polymerase: An in silico drug repurposing for COVID-19. F1000Research 2020, 9, 1166. [Google Scholar] [CrossRef]
- Tsuji, M.; Shudo, K.; Kagechika, H. Identifying the receptor subtype selectivity of retinoid X and retinoic acid receptors via quantum mechanics. FEBS Open Bio 2017, 7, 391–396. [Google Scholar] [CrossRef]
- Tsuji, M. Antagonist-perturbation mechanism for activation function-2 fixed motifs: Active conformation and docking mode of retinoid X receptor antagonists. J. Comput. Aided Mol. Des. 2017, 31, 577–585. [Google Scholar] [CrossRef]
- Tsuji, M. Development of the structure-based drug design systems, HMHC and DSHC. Mol. Sci. 2007, 1, NP004. [Google Scholar]
- Tsuji, M. Homology Modeling Professional for HyperChem, Revision H1; Institute of Molecular Function: Saitama, Japan, 2018. [Google Scholar]
- HyperChem Professional, Version 8.0.10; Hypercube, Inc.: Gainesville, FL, USA, 2011.
- Tsuji, M.; Shudo, K.; Kagechika, H. Docking simulations suggest that all-trans retinoic acid could bind to retinoid X receptors. J. Comput. Aided Mol. Des. 2015, 29, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Sud, M. MayaChemTools: An open source package for computational drug discovery. J. Chem. Inf. Model 2016, 56, 2292–2297. [Google Scholar] [CrossRef] [PubMed]
- Vainio, M.J.; Johnson, M.S. Generating conformer ensembles using a multiobjective genetic algorithm. J. Chem. Inf. Model 2007, 47, 2462–2474. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Tsuji, M. Docking Study with HyperChem, Revision H1; Institute of Molecular Function: Saitama, Japan, 2018. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newslett. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein. Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian 98. Part 1. The calculation of energies, gradients and vibrational frequencies and electric field derivatives. J. Mol. Struct. 1999, 462, 1–21. [Google Scholar] [CrossRef]
- ABINIT-MP, Version 7; The University of Tokyo: Tokyo, Japan, 2013.
- Tanaka, S.; Mochizuki, Y.; Komeiji, Y.; Okiyama, Y.; Fukuzawa, K. Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems. Phys. Chem. Chem. Phys. 2014, 16, 10310–10344. [Google Scholar] [CrossRef]
- BioStation Viewer, Version 16.0; The University of Tokyo: Tokyo, Japan, 2014.
- Tateyama-Makino, R.; Abe-Yutori, M.; Iwamoto, T.; Tsutsumi, K.; Tsuji, M.; Morishita, S.; Kurita, K.; Yamamoto, Y.; Nishinaga, E.; Tsukinoki, K. The inhibitory effects of toothpaste and mouthwash ingredients on the interaction between the SARS-CoV-2 spike protein and ACE2, and the protease activity of TMPRSS2 in vitro. PLoS ONE 2021, 16, e0257705. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Method | G (Hartree) at 298.15 K and 1 atm | ΔGbind (kcal·mol−1) at 298.15 K and 1 atm |
|---|---|---|---|
| RdRp Complex:dsRNA−CHEMBL4246021 | MP2/6-31G:AMBER | −2017.589 | −157.799 |
| RdRp Complex:dsRNA | AMBER | 40.782 | |
| CHEMBL4246021 | MP2/6-31G | −2058.119 |
| Method | nsp12 | nsp8-1 (79–190) | nsp8-2 (85–110) | nsp7 | Primer RNA | Template RNA | Metal Ions | Pyrophosphate | Total |
|---|---|---|---|---|---|---|---|---|---|
| MP2/6-31G | −20.859 | 2.294 | 0.267 | 0.223 | −60.033 | −21.161 | −468.308 | 0.811 | −566.766 |
| HF/6-31G | 29.672 | 2.294 | 0.267 | 0.223 | −40.582 | −12.484 | −217.402 | 0.811 | −237.201 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji, M. Virtual Screening and Quantum Chemistry Analysis for SARS-CoV-2 RNA-Dependent RNA Polymerase Using the ChEMBL Database: Reproduction of the Remdesivir-RTP and Favipiravir-RTP Binding Modes Obtained from Cryo-EM Experiments with High Binding Affinity. Int. J. Mol. Sci. 2022, 23, 11009. https://doi.org/10.3390/ijms231911009
Tsuji M. Virtual Screening and Quantum Chemistry Analysis for SARS-CoV-2 RNA-Dependent RNA Polymerase Using the ChEMBL Database: Reproduction of the Remdesivir-RTP and Favipiravir-RTP Binding Modes Obtained from Cryo-EM Experiments with High Binding Affinity. International Journal of Molecular Sciences. 2022; 23(19):11009. https://doi.org/10.3390/ijms231911009
Chicago/Turabian StyleTsuji, Motonori. 2022. "Virtual Screening and Quantum Chemistry Analysis for SARS-CoV-2 RNA-Dependent RNA Polymerase Using the ChEMBL Database: Reproduction of the Remdesivir-RTP and Favipiravir-RTP Binding Modes Obtained from Cryo-EM Experiments with High Binding Affinity" International Journal of Molecular Sciences 23, no. 19: 11009. https://doi.org/10.3390/ijms231911009
APA StyleTsuji, M. (2022). Virtual Screening and Quantum Chemistry Analysis for SARS-CoV-2 RNA-Dependent RNA Polymerase Using the ChEMBL Database: Reproduction of the Remdesivir-RTP and Favipiravir-RTP Binding Modes Obtained from Cryo-EM Experiments with High Binding Affinity. International Journal of Molecular Sciences, 23(19), 11009. https://doi.org/10.3390/ijms231911009