
Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene



Abstract
1. Introduction
1.1. PSEN1 Structure and Functions
1.2. PSEN1 Mutations and Their Classification
1.3. PSEN1 Mutations and Significant Residues at Each Domain
2. Cell Models and Possible Biomarkers of PSEN1 Mutation Pathogenicity
3. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; An, S.S.A.; Van Giau, V.; Shim, K.; Youn, Y.C.; Bagyinszky, E.; Kim, S. Identification of a novel PSEN1 mutation (Leu232Pro) in a Korean patient with early-onset Alzheimer’s disease and a family history of dementia. Neurobiol. Aging 2017, 56, 212.e11–212.e17. [Google Scholar] [CrossRef]
- Laudon, H.; Hansson, E.M.; Melén, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; von Heijne, G.; Näslund, J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Zhang, Y.W.; Xu, H.; Thinakaran, G. Pathological and physiological fun.nctions of presenilins. Mol. Neurodegener. 2006, 1, 4. [Google Scholar] [CrossRef]
- Brunkan, A.L.; Martinez, M.; Wang, J.; Walker, E.S.; Beher, D.; Shearman, M.S.; Goate, A.M. Two domains within the first putative transmembrane domain of presenilin 1 differentially influence presenilinase and gamma-secretase activity. J. Neurochem. 2005, 94, 1315–1328. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Craessaerts, K.; Bammens, L.; Bentahir, M.; Borgions, F.; Herdewijn, P.; Staes, A.; Timmerman, E.; Vandekerckhove, J.; Rubinstein, E.; et al. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 2009, 11, 1340–1346. [Google Scholar]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Tolia, A.; De Strooper, B. Structure and function of gamma-secretase. Semin. Cell Dev. Biol. 2009, 20, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Vetrivel, K.S.; Nguyen, P.D.; Meckler, X.; Cheng, H.; Kounnas, M.Z.; Wagner, S.L.; Parent, A.T.; Thinakaran, G. Mutation analysis of the presenilin 1 N-terminal domain reveals a broad spectrum of gamma-secretase activity toward amyloid precursor protein and other substrates. J. Biol. Chem. 2010, 285, 38042–38052. [Google Scholar] [CrossRef]
- Somavarapu, A.K.; Kepp, K.P. Membrane Dynamics of gamma-Secretase Provides a Molecular Basis for beta-Amyloid Binding and Processing. ACS Chem. Neurosci. 2017, 8, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.S.; Song, B.; Koulen, P. Presenilins as Drug Targets for Alzheimer’s Disease-Recent Insights from Cell Biology and Electrophysiology as Novel Opportunities in Drug Development. Int. J. Mol. Sci. 2018, 19, 1621. [Google Scholar]
- Watanabe, N.; Tomita, T.; Sato, C.; Kitamura, T.; Morohashi, Y.; Iwatsubo, T. Pen-2 is incorporated into the gamma-secretase complex through binding to transmembrane domain 4 of presenilin 1. J. Biol. Chem. 2005, 280, 41967–41975. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Wolfe, M.S.; De Los Angeles, J.; Miller, D.D.; Xia, W.; Selkoe, D.J. Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry 1999, 38, 11223–11230. [Google Scholar]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and gamma-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar]
- Zhang, Z.; Hartmann, H.; Do, V.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; van de Wetering, M.; Clevers, H.; Saftig, P.; et al. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395, 698–702. [Google Scholar]
- Boonen, R.A.; van Tijn, P.; Zivkovic, D. Wnt signaling in Alzheimer’s disease: Up or down, that is the question. Ageing Res. Rev. 2009, 8, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Thakar, A.; Santoro, S.W.; Pratt, K.G. Presenilin Regulates Retinotectal Synapse Formation through EphB2 Receptor Processing. Dev. Neurobiol. 2018, 78, 1171–1190. [Google Scholar] [CrossRef]
- Repetto, E.; Yoon, I.S.; Zheng, H.; Kang, D.E. Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal-lysosomal pathway. J. Biol. Chem. 2007, 282, 31504–31516. [Google Scholar] [CrossRef] [PubMed]
- Dehvari, N.; Cedazo-Minguez, A.; Isacsson, O.; Nilsson, T.; Winblad, B.; Karlström, H.; Benedikz, E.; Cowburn, R.F. Presenilin dependence of phospholipase C and protein kinase C signaling. J. Neurochem. 2007, 102, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yamaguchi, H.; Lai, F.A.; Shen, J. Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Wan, K.; Ma, Z.J.; Zhou, X.; Zhang, Y.M.; Yu, X.F.; You, M.Z.; Huang, C.J.; Zhang, W.; Sun, Z.W. A Novel Probable Pathogenic PSEN2 Mutation p.Phe369Ser Associated With Early-Onset Alzheimer’s Disease in a Chinese Han Family: A Case Report. Front. Aging Neurosci. 2021, 13, 710075. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef] [PubMed]
- Acx, H.; Chávez-Gutiérrez, L.; Serneels, L.; Lismont, S.; Benurwar, M.; Elad, N.; De Strooper, B. Signature amyloid beta profiles are produced by different gamma-secretase complexes. J. Biol. Chem. 2014, 289, 4346–4355. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Steiner, H.; Duff, K.; Capell, A.; Romig, H.; Grim, M.G.; Lincoln, S.; Hardy, J.; Yu, X.; Picciano, M.; Fechteler, K.; et al. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J. Biol. Chem. 1999, 274, 28669–28673. [Google Scholar] [CrossRef]
- Watanabe, H.; Imaizumi, K.; Cai, T.; Zhou, Z.; Tomita, T.; Okano, H. Flexible and Accurate Substrate Processing with Distinct Presenilin/gamma-Secretases in Human Cortical Neurons. eNeuro 2021, 8, ENEURO.0500-20.2021. [Google Scholar] [PubMed]
- Yonemura, Y.; Futai, E.; Yagishita, S.; Suo, S.; Tomita, T.; Iwatsubo, T.; Ishiura, S. Comparison of presenilin 1 and presenilin 2 gamma-secretase activities using a yeast reconstitution system. J. Biol. Chem. 2011, 286, 44569–44575. [Google Scholar] [PubMed]
- Pintchovski, S.A.; Schenk, D.B.; Basi, G.S. Evidence that enzyme processivity mediates differential Abeta production by PS1 and PS2. Curr. Alzheimer Res. 2013, 10, 4–10. [Google Scholar] [PubMed]
- Newman, M.; Wilson, L.; Verdile, G.; Lim, A.; Khan, I.; Moussavi Nik, S.H.; Pursglove, S.; Chapman, G.; Martins, R.N.; Lardelli, M. Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum. Mol. Genet. 2014, 23, 602–617. [Google Scholar]
- Giau, V.V.; Bagyinszky, E.; Youn, Y.C.; An, S.; Kim, S. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar]
- Guerreiro, R.J.; Baquero, M.; Blesa, R.; Boada, M.; Brás, J.M.; Bullido, M.J.; Calado, A.; Crook, R.; Ferreira, C.; Frank, A.; et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 2010, 31, 725–731. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar]
- Hsu, S.; Pimenova, A.A.; Hayes, K.; Villa, J.A.; Rosene, M.J.; Jere, M.; Goate, A.M.; Karch, C.M. Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol. Dis. 2020, 139, 104817. [Google Scholar]
- Nygaard, H.B.; Lippa, C.F.; Mehdi, D.; Baehring, J.M. A Novel Presenilin 1 Mutation in Early-Onset Alzheimer’s Disease With Prominent Frontal Features. Am. J. Alzheimers Dis. Other Dement. 2014, 29, 433–435. [Google Scholar] [CrossRef]
- Perrone, F.; Bjerke, M.; Hens, E.; Sieben, A.; Timmers, M.; De Roeck, A.; Vandenberghe, R.; Sleegers, K.; Martin, J.J.; De Deyn, P.P.; et al. Amyloid-beta1-43 cerebrospinal fluid levels and the interpretation of APP, PSEN1 and PSEN2 mutations. Alzheimers Res. Ther. 2020, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; van Duijn, C.M.; Backhovens, H.; Van den Broeck, M.; Wehnert, A.; Serneels, S.; Sherrington, R.; Hutton, M.; Hardy, J.; St George-Hyslop, P.H.; et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 1998, 7, 43–51. [Google Scholar] [PubMed]
- Finckh, U.; Müller-Thomsen, T.; Mann, U.; Eggers, C.; Marksteiner, J.; Meins, W.; Binetti, G.; Alberici, A.; Hock, C.; Nitsch, R.M.; et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am. J. Hum. Genet. 2000, 66, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kauwe, J.S.; Jacquart, S.; Chakraverty, S.; Wang, J.; Mayo, K.; Fagan, A.M.; Holtzman, D.M.; Morris, J.C.; Goate, A.M. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann. Neurol. 2007, 61, 446–453. [Google Scholar] [PubMed]
- Cruchaga, C.; Haller, G.; Chakraverty, S.; Mayo, K.; Vallania, F.L.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar]
- Ibanez, L.; Dube, U.; Davis, A.A.; Fernandez, M.V.; Budde, J.; Cooper, B.; Diez-Fairen, M.; Ortega-Cubero, S.; Pastor, P.; Perlmutter, J.S.; et al. Pleiotropic Effects of Variants in Dementia Genes in Parkinson Disease. Front. Neurosci. 2018, 12, 230. [Google Scholar] [CrossRef]
- Meeus, B.; Verstraeten, A.; Crosiers, D.; Engelborghs, S.; Van den Broeck, M.; Mattheijssens, M.; Peeters, K.; Corsmit, E.; Elinck, E.; Pickut, B.; et al. DLB and PDD: A role for mutations in dementia and Parkinson disease genes? Neurobiol. Aging 2012, 33, 629.e5–629.e18. [Google Scholar]
- Kumar-Singh, S.; Theuns, J.; Van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; Van Broeckhoven, C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat. 2006, 27, 686–695. [Google Scholar] [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimers Dement. 2020, 16, 178–191. [Google Scholar]
- Nicolas, G.; Wallon, D.; Charbonnier, C.; Quenez, O.; Rousseau, S.; Richard, A.C.; Rovelet-Lecrux, A.; Coutant, S.; Le Guennec, K.; Bacq, D.; et al. Screening of dementia genes by whole-exome sequencing in early-onset Alzheimer disease: Input and lessons. Eur. J. Hum. Genet. 2016, 24, 710–716. [Google Scholar]
- Mao, C.; Li, J.; Dong, L.; Huang, X.; Lei, D.; Wang, J.; Chu, S.; Liu, C.; Peng, B.; Román, G.C.; et al. Clinical Phenotype and Mutation Spectrum of Alzheimer’s Disease with Causative Genetic Mutation in a Chinese Cohort. Curr. Alzheimer Res. 2021, 18, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Koriath, C.; Kenny, J.; Adamson, G.; Druyeh, R.; Taylor, W.; Beck, J.; Quinn, L.; Mok, T.H.; Dimitriadis, A.; Norsworthy, P.; et al. Predictors for a dementia gene mutation based on gene-panel next-generation sequencing of a large dementia referral series. Mol. Psychiatry 2020, 25, 3399–3412. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Rojas, G.J.; Nemirovsky, S.I.; Da Prat, G.; Persi, G.; Cesarini, M.; Etcheverry, J.L.; Rojas, N.G.; Parisi, V.; Cordoba, M.; et al. A novel mutation in PSEN1 (p.Arg41Ser) in an Argentinian woman with early onset Parkinsonism. Parkinsonism Relat. Disord. 2020, 77, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Ch’ng, G.S.; Chan, M.Y.; An, S.; Kim, S. A Pathogenic Presenilin-1 Val96Phe Mutation from a Malaysian Family. Brain Sci. 2021, 11, 1328. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Tominaga, A.; Sato, C.; Tomita, T.; Iwatsubo, T. Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the gamma-secretase. J. Neurosci. 2010, 30, 15943–15950. [Google Scholar] [CrossRef]
- Queralt, R.; Ezquerra, M.; Lleó, A.; Castellví, M.; Gelpí, J.; Ferrer, I.; Acarín, N.; Pasarín, L.; Blesa, R.; Oliva, R. A novel mutation (V89L) in the presenilin 1 gene in a family with early onset Alzheimer’s disease and marked behavioural disturbances. J. Neurol. Neurosurg. Psychiatry 2002, 72, 266–269. [Google Scholar] [CrossRef]
- Liu, L.; Lauro, B.M.; Wolfe, M.S.; Selkoe, D.J. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate gamma-secretase function in generating Alzheimer-causing Abeta peptides. J. Biol. Chem. 2021, 296, 100393. [Google Scholar] [CrossRef]
- Sato, C.; Takagi, S.; Tomita, T.; Iwatsubo, T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J. Neurosci. 2008, 28, 6264–6271. [Google Scholar] [CrossRef]
- Sato, C.; Morohashi, Y.; Tomita, T.; Iwatsubo, T. Structure of the catalytic pore of gamma-secretase probed by the accessibility of substituted cysteines. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 12081–12088. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Islam, S.; Sun, Y.; Gao, Y.; Nakamura, T.; Noorani, A.A.; Li, T.; Wong, P.C.; Kimura, N.; Matsubara, E.; Kasuga, K.; et al. Presenilin Is Essential for ApoE Secretion, a Novel Role of Presenilin Involved in Alzheimer’s Disease Pathogenesis. J. Neurosci. 2022, 42, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Ataka, S.; Tomiyama, T.; Takuma, H.; Yamashita, T.; Shimada, H.; Tsutada, T.; Kawabata, K.; Mori, H.; Miki, T. A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch. Neurol. 2004, 61, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- López-García, S.; Jiménez-Bonilla, J.; López Delgado, A.; Orizaola Balaguer, P.; Infante Ceberio, J.; Banzo Marraco, I.; Rodríguez Rodríguez, E.; Sánchez-Juan, P. A Rare PSEN1 (Leu85Pro) Mutation Causing Alzheimer’s Disease in a 29-Year-Old Woman Presenting as Corticobasal Syndrome. J. Alzheimers Dis. 2019, 70, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Ohki, Y.; Tomita, T.; Osawa, S.; Reed, B.R.; Jagust, W.; Van Berlo, V.; Jin, L.W.; Chui, H.C.; Coppola, G.; et al. Two Novel Mutations in the First Transmembrane Domain of Presenilin1 Cause Young-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1035–1041. [Google Scholar] [CrossRef]
- Houlden, H.; Baker, M.; McGowan, E.; Lewis, P.; Hutton, M.; Crook, R.; Wood, N.W.; Kumar-Singh, S.; Geddes, J.; Swash, M.; et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann. Neurol. 2000, 48, 806–808. [Google Scholar] [CrossRef]
- Jia, J.; Xu, E.; Shao, Y.; Jia, J.; Sun, Y.; Li, D. One novel presenilin-1 gene mutation in a Chinese pedigree of familial Alzheimer’s disease. J. Alzheimers Dis. 2005, 7, 119–124; discussion 173–180. [Google Scholar] [CrossRef]
- Gallo, M.; Frangipane, F.; Cupidi, C.; De Bartolo, M.; Turone, S.; Ferrari, C.; Nacmias, B.; Grimaldi, G.; Laganà, V.; Colao, R.; et al. The novel PSEN1 M84V mutation associated to frontal dysexecutive syndrome, spastic paraparesis, and cerebellar atrophy in a dominant Alzheimer’s disease family. Neurobiol. Aging 2017, 56, 213.e7–213.e12. [Google Scholar] [CrossRef]
- Fray, S.; Ali, N.B.; Rassas, A.A.; Kechaou, M.; Oudiaa, N.; Cherif, A.; Echebbi, S.; Messaoud, T.; Belal, S. Early psychiatrics symptoms in familial Alzheimer’s disease with presenilin 1 mutation (I83T). J. Neural Trans. 2016, 123, 451–453. [Google Scholar] [CrossRef]
- Hsu, S.; Gordon, B.A.; Hornbeck, R.; Norton, J.B.; Levitch, D.; Louden, A.; Ziegemeier, E.; Laforce, R., Jr.; Chhatwal, J.; Day, G.S.; et al. Discovery and validation of autosomal dominant Alzheimer’s disease mutations. Alzheimers Res. Ther. 2018, 10, 67. [Google Scholar] [CrossRef]
- Tedde, A.; Nacmias, B.; Ciantelli, M.; Forleo, P.; Cellini, E.; Bagnoli, S.; Piccini, C.; Caffarra, P.; Ghidoni, E.; Paganini, M.; et al. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch. Neurol. 2003, 60, 1541–1544. [Google Scholar] [CrossRef]
- Fray, S.; Rassas, A.; Messaoud, T.; Belal, S. Refractory epilepsy in PSEN 1 mutation (I83T). Neurocase 2020, 26, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Lee, H.M.; Van Giau, V.; Koh, S.B.; Jeong, J.H.; An, S.; Kim, S. PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 2604. [Google Scholar]
- Lindquist, S.G.; Schwartz, M.; Batbayli, M.; Waldemar, G.; Nielsen, J.E. Genetic testing in familial AD and FTD: Mutation and phenotype spectrum in a Danish cohort. Clin. Genet. 2009, 76, 205–209. [Google Scholar] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Abeta Length by Destabilizing gamma-Secretase-Abetan Interactions. Cell 2017, 170, 443–456.e14. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Veugelen, S.; Benurwar, M.; Lismont, S.; Sepulveda-Falla, D.; Lleo, A.; Ryan, N.S.; Lashley, T.; Fox, N.C.; Murayama, S.; et al. Qualitative changes in human gamma-secretase underlie familial Alzheimer’s disease. J. Exp. Med. 2015, 212, 2003–2013. [Google Scholar] [CrossRef]
- Li, L.; Roh, J.H.; Chang, E.H.; Lee, Y.; Lee, S.; Kim, M.; Koh, W.; Chang, J.W.; Kim, H.J.; Nakanishi, M.; et al. iPSC Modeling of Presenilin1 Mutation in Alzheimer’s Disease with Cerebellar Ataxia. Exp. Neurobiol. 2018, 27, 350–364. [Google Scholar]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: A case series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar]
- Dobricic, V.; Stefanova, E.; Jankovic, M.; Gurunlian, N.; Novakovic, I.; Hardy, J.; Kostic, V.; Guerreiro, R. Genetic testing in familial and young-onset Alzheimer’s disease: Mutation spectrum in a Serbian cohort. Neurobiol. Aging 2012, 33, 1481.e7–1481.e12. [Google Scholar]
- Wisniewski, T.; Dowjat, W.K.; Buxbaum, J.D.; Khorkova, O.; Efthimiopoulos, S.; Kulczycki, J.; Lojkowska, W.; Wegiel, J.; Wisniewski, H.M.; Frangione, B. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport 1998, 9, 217–221. [Google Scholar] [CrossRef]
- Alberici, A.; Bonato, C.; Borroni, B.; Cotelli, M.; Mattioli, F.; Binetti, G.; Gennarelli, M.; Luca, M.D.; Simonati, A.; Perani, D.; et al. Dementia, delusions and seizures: Storage disease or genetic AD? Eur. J. Neurol. 2007, 14, 1057–1059. [Google Scholar]
- Dowjat, W.K.; Kuchna, I.; Wisniewski, T.; Wegiel, J. A novel highly pathogenic Alzheimer presenilin-1 mutation in codon 117 (Pro117Ser): Comparison of clinical, neuropathological and cell culture phenotypes of Pro117Leu and Pro117Ser mutations. J. Alzheimers Dis. 2004, 6, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Lee, H.; Pyun, J.M.; Suh, J.; Kang, M.J.; Vo, V.G.; An, S.; Park, K.H.; Kim, S. Pathogenic PSEN1 Thr119Ile Mutation in Two Korean Patients with Early-Onset Alzheimer’s Disease. Diagnostics 2020, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, X.; Zhang, L.; Meng, X.; Ma, L.; Zhang, G.; Wu, H.; Liang, L.; Cao, M.; Mei, F. Identification of a Rare PSEN1 Mutation (Thr119Ile) in Late-Onset Alzheimer’s Disease With Early Presentation of Behavioral Disturbance. Front. Psychiatry 2020, 11, 347. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Cho, H.; Kim, H.J.; Na, D.L.; Seo, S.W.; Ki, C.S. PSEN1 variants in Korean patients with clinically suspicious early-onset familial Alzheimer’s disease. Sci. Rep. 2020, 10, 3480. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Maeda, K.; Hashimoto, M.; Yamashita, H.; Ikejiri, Y.; Bird, T.D.; Tanaka, C.; Schellenberg, G.D. A pedigree with a novel presenilin 1 mutation at a residue that is not conserved in presenilin 2. Arch. Neurol. 1999, 56, 65–69. [Google Scholar] [CrossRef]
- Blanco, J.A.; Alonso, A.; Blanco, J.; Rojo, E.; Tellería, J.J.; Torres, M.A.; Uribe, F. Novel presenilin 1 mutation (p.Thr-Pro116-117Ser-Thr) in a Spanish family with early-onset Alzheimer’s disease. Neurobiol. Aging 2019, 84, 238.e19–238.e24. [Google Scholar]
- Tysoe, C.; Whittaker, J.; Xuereb, J.; Cairns, N.J.; Cruts, M.; Van Broeckhoven, C.; Wilcock, G.; Rubinsztein, D.C. A presenilin-1 truncating mutation is present in two cases with autopsy-confirmed early-onset Alzheimer disease. Am. J. Hum. Genet. 1998, 62, 70–76. [Google Scholar]
- Anheim, M.; Hannequin, D.; Boulay, C.; Martin, C.; Campion, D.; Tranchant, C. Ataxic variant of Alzheimer’s disease caused by Pro117Ala PSEN1 mutation. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1414–1415. [Google Scholar] [PubMed]
- Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G.V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics 2005, 6, 85–89. [Google Scholar] [CrossRef]
- Reznik-Wolf, H.; Treves, T.A.; Davidson, M.; Aharon-Peretz, J.; St George Hyslop, P.H.; Chapman, J.; Korczyn, A.D.; Goldman, B.; Friedman, E. A novel mutation of presenilin 1 in familial Alzheimer’s disease in Israel detected by denaturing gradient gel electrophoresis. Hum. Genet. 1996, 98, 700–702. [Google Scholar] [CrossRef]
- Gómez-Tortosa, E.; Barquero, S.; Barón, M.; Gil-Neciga, E.; Castellanos, F.; Zurdo, M.; Manzano, S.; Muñoz, D.G.; Jiménez-Huete, A.; Rábano, A.; et al. Clinical-genetic correlations in familial Alzheimer’s disease caused by presenilin 1 mutations. J. Alzheimers Dis. 2010, 19, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Cheng, C.Y.; Liao, Y.C.; Hong, C.J.; Fuh, J.L. Mutational analysis in familial Alzheimer’s disease of Han Chinese in Taiwan with a predominant mutation PSEN1 p.Met146Ile. Sci. Rep. 2020, 10, 19769. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, S.; Smolek, T.; Turcani, P.; Petrovic, R.; Brandoburova, P.; Jadhav, S.; Novak, P.; Attems, J.; Zilka, N. Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by presenilin-1 missense mutation Thr116Asn. J. Neural Transm. 2018, 125, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef]
- Arber, C.; Lovejoy, C.; Harris, L.; Willumsen, N.; Alatza, A.; Casey, J.M.; Lines, G.; Kerins, C.; Mueller, A.K.; Zetterberg, H.; et al. Familial Alzheimer’s Disease Mutations in PSEN1 Lead to Premature Human Stem Cell Neurogenesis. Cell Rep. 2021, 34, 108615. [Google Scholar] [CrossRef]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote gamma-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef]
- Hung, C.O.Y.; Livesey, F.J. Altered gamma-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018, 25, 3647–3660.e2. [Google Scholar] [CrossRef]
- Al Rahim, M.; Yoon, Y.; Dimovasili, C.; Shao, Z.; Huang, Q.; Zhang, E.; Kezunovic, N.; Chen, L.; Schaffner, A.; Huntley, G.W.; et al. Presenilin1 familial Alzheimer disease mutants inactivate EFNB1- and BDNF-dependent neuroprotection against excitotoxicity by affecting neuroprotective complexes of N-methyl-d-aspartate receptor. Brain Commun. 2020, 2, fcaa100. [Google Scholar] [CrossRef]
- Yoon, Y.; Voloudakis, G.; Doran, N.; Zhang, E.; Dimovasili, C.; Chen, L.; Shao, Z.; Darmanis, S.; Tang, C.; Tang, J.; et al. PS1 FAD mutants decrease ephrinB2-regulated angiogenic functions, ischemia-induced brain neovascularization and neuronal survival. Mol. Psychiatry 2021, 26, 1996–2012. [Google Scholar] [CrossRef]
- Larner, A.J.; Plessis, D.G.d. Early-onset Alzheimer’s disease with presenilin-1 M139V mutation: Clinical, neuropsychological and neuropathological study. Eur. J. Neurol. 2003, 10, 319–323. [Google Scholar] [CrossRef]
- Riudavets, M.A.; Bartoloni, L.; Troncoso, J.C.; Pletnikova, O.; St George-Hyslop, P.; Schultz, M.; Sevlever, G.; Allegri, R.F. Familial dementia with frontotemporal features associated with M146V presenilin-1 mutation. Brain Pathol. 2013, 23, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Song, Y.J.; Lepar, G.; Brooks, W.S.; Kwok, J.B.; Kersaitis, C.; Gregory, G.; Shepherd, C.E.; Rahimi, F.; Schofield, P.R.; et al. Pick bodies in a family with presenilin-1 Alzheimer’s disease. Ann. Neurol. 2005, 57, 139–143. [Google Scholar] [CrossRef]
- Rudzinski, L.A.; Fletcher, R.M.; Dickson, D.W.; Crook, R.; Hutton, M.L.; Adamson, J.; Graff-Radford, N.R. Early onset familial Alzheimer Disease with spastic paraparesis, dysarthria, and seizures and N135S mutation in PSEN1. Alzheimer Dis. Assoc. Disord. 2008, 22, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Zekanowski, C.; Styczyńska, M.; Pepłońska, B.; Gabryelewicz, T.; Religa, D.; Ilkowski, J.; Kijanowska-Haładyna, B.; Kotapka-Minc, S.; Mikkelsen, S.; Pfeffer, A.; et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp. Neurol. 2003, 184, 991–996. [Google Scholar] [CrossRef]
- Jiao, B.; Liu, H.; Guo, L.; Xiao, X.; Liao, X.; Zhou, Y.; Weng, L.; Zhou, L.; Wang, X.; Jiang, Y.; et al. The role of genetics in neurodegenerative dementia: A large cohort study in South China. NPJ Genom. Med. 2021, 6, 69. [Google Scholar] [CrossRef]
- Wang, J.C.; Alinaghi, S.; Tafakhori, A.; Sikora, E.; Azcona, L.J.; Karkheiran, S.; Goate, A.; Paisán-Ruiz, C.; Darvish, H. Genetic screening in two Iranian families with early-onset Alzheimer’s disease identified a novel PSEN1 mutation. Neurobiol. Aging 2018, 62, 244.e15–244.e17. [Google Scholar] [CrossRef]
- Crook, R.; Ellis, R.; Shanks, M.; Thal, L.J.; Perez-Tur, J.; Baker, M.; Hutton, M.; Haltia, T.; Hardy, J.; Galasko, D. Early-onset Alzheimer’s disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann. Neurol. 1997, 42, 124–128. [Google Scholar] [CrossRef]
- Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Gen. 1995, 11, 219–222. [Google Scholar] [CrossRef]
- Janssen, J.C.; Lantos, P.L.; Fox, N.C.; Harvey, R.J.; Beck, J.; Dickinson, A.; Campbell, T.A.; Collinge, J.; Hanger, D.P.; Cipolotti, L.; et al. Autopsy-confirmed familial early-onset Alzheimer disease caused by the l153V presenilin 1 mutation. Arch. Neurol. 2001, 58, 953–958. [Google Scholar] [CrossRef][Green Version]
- Sandbrink, R.; Zhang, D.; Schaeffer, S.; Masters, C.L.; Bauer, J.; Förstl, H.; Beyreuther, K. Missense mutations of the PS-1/S182 gene in German early-onset Alzheimer’s disease patients. Ann. Neurol. 1996, 40, 265–266. [Google Scholar] [CrossRef]
- Keller, L.; Welander, H.; Chiang, H.H.; Tjernberg, L.O.; Nennesmo, I.; Wallin, A.K.; Graff, C. The PSEN1 I143T mutation in a Swedish family with Alzheimer’s disease: Clinical report and quantification of Aβ in different brain regions. Eur. J. Hum. Genet. 2010, 18, 1202–1208. [Google Scholar] [CrossRef]
- Arai, N.; Kishino, A.; Takahashi, Y.; Morita, D.; Nakamura, K.; Yokoyama, T.; Watanabe, T.; Ida, M.; Goto, J.; Tsuji, S. Familial cases presenting very early onset autosomal dominant Alzheimer’s disease with I143T in presenilin-1 gene: Implication for genotype-phenotype correlation. Neurogenetics 2008, 9, 65–67. [Google Scholar] [CrossRef]
- Morelli, L.; Prat, M.I.; Levy, E.; Mangone, C.A.; Castaño, E.M. Presenilin 1 Met146Leu variant due to an A --> T transversion in an early-onset familial Alzheimer’s disease pedigree from Argentina. Clin. Genet. 1998, 53, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.M.; Newman, S.K.; Frackowiak, R.S.; Cunningham, V.J.; Roques, P.; Stevens, J.; Neary, D.; Bruton, C.J.; Warrington, E.K.; Rossor, M.N. Chromosome 14 linked familial Alzheimer’s disease. A clinico-pathological study of a single pedigree. Brain 1995, 118 Pt 1, 185–205. [Google Scholar] [CrossRef]
- Denvir, J.; Neitch, S.; Fan, J.; Niles, R.M.; Boskovic, G.; Schreurs, B.G.; Primerano, D.A.; Alkon, D.L. Identification of the PS1 Thr147Ile Variant in a Family with Very Early Onset Dementia and Expressive Aphasia. J. Alzheimers Dis. 2015, 46, 483–490. [Google Scholar] [CrossRef]
- Hattori, S.; Sakuma, K.; Wakutani, Y.; Wada, K.; Shimoda, M.; Urakami, K.; Kowa, H.; Nakashima, K. A novel presenilin 1 mutation (Y154N) in a patient with early onset Alzheimer’s disease with spastic paraparesis. Neurosci. Lett. 2004, 368, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Zarea, A.; Charbonnier, C.; Rovelet-Lecrux, A.; Nicolas, G.; Rousseau, S.; Borden, A.; Pariente, J.; Le Ber, I.; Pasquier, F.; Formaglio, M.; et al. Seizures in dominantly inherited Alzheimer disease. Neurology 2016, 87, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Raux, G.; Gantier, R.; Martin, C.; Pothin, Y.; Brice, A.; Frebourg, T.; Campion, D. A novel presenilin 1 missense mutation (L153V) segregating with early-onset autosomal dominant Alzheimer’s disease. Hum. Mutat. 2000, 16, 95. [Google Scholar] [CrossRef]
- Qiu, Q.; Shen, L.; Jia, L.; Wang, Q.; Li, F.; Li, Y.; Jia, J. A Novel PSEN1 M139L Mutation Found in a Chinese Pedigree with Early-Onset Alzheimer’s Disease Increases Abeta42/Abeta40 ratio. J. Alzheimers Dis. 2019, 69, 199–212. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Holbrook, K. Novel presenilin-1 Y159F sequence variant associated with early-onset Alzheimer’s disease. Neurosci. Lett. 2012, 531, 142–144. [Google Scholar] [CrossRef]
- Cornejo-Olivas, M.R.; Yu, C.E.; Mazzetti, P.; Mata, I.F.; Meza, M.; Lindo-Samanamud, S.; Leverenz, J.B.; Bird, T.D. Clinical and molecular studies reveal a PSEN1 mutation (L153V) in a Peruvian family with early-onset Alzheimer’s disease. Neurosci. Lett. 2014, 563, 140–143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gallo, M.; Marcello, N.; Curcio, S.A.; Colao, R.; Geracitano, S.; Bernardi, L.; Anfossi, M.; Puccio, G.; Frangipane, F.; Clodomiro, A.; et al. A novel pathogenic PSEN1 mutation in a family with Alzheimer’s disease: Phenotypical and neuropathological features. J. Alzheimers Dis. 2011, 25, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Senanarong, V.; An, S.; Vo Van, G.; Limwongse, C.; Bagyinszky, E.; Kim, S. Pathogenic PSEN1 Glu184Gly Mutation in a Family from Thailand with Probable Autosomal Dominant Early Onset Alzheimer’s Disease. Diagnostics 2020, 10, 135. [Google Scholar] [CrossRef]
- Cai, T.; Morishima, K.; Takagi-Niidome, S.; Tominaga, A.; Tomita, T. Conformational Dynamics of Transmembrane Domain 3 of Presenilin 1 Is Associated with the Trimming Activity of gamma-Secretase. J. Neurosci. 2019, 39, 8600–8610. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.A.; Servian-Morilla, E.; Scholl, F.G. Presenilin/gamma-secretase regulates neurexin processing at synapses. PLoS ONE 2011, 6, e19430. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Park, H.; Maharana, C.; Gwon, A.R.; Park, J.; Baek, S.H.; Bae, H.G.; Cho, Y.; Kim, H.K.; Sul, J.H.; et al. Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 2021, 11, 8855–8873. [Google Scholar] [CrossRef]
- Moehlmann, T.; Winkler, E.; Xia, X.; Edbauer, D.; Murrell, J.; Capell, A.; Kaether, C.; Zheng, H.; Ghetti, B.; Haass, C.; et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc. Natl. Acad. Sci. USA 2002, 99, 8025–8030. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 2019, 104, 256–270.e5. [Google Scholar] [CrossRef]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef]
- Li, L.; Kim, H.J.; Roh, J.H.; Kim, M.; Koh, W.; Kim, Y.; Heo, H.; Chung, J.; Nakanishi, M.; Yoon, T.; et al. Pathological manifestation of the induced pluripotent stem cell-derived cortical neurons from an early-onset Alzheimer’s disease patient carrying a presenilin-1 mutation (S170F). Cell Prolif. 2020, 53, e12798. [Google Scholar] [CrossRef]
- Takao, M.; Ghetti, B.; Murrell, J.R.; Unverzagt, F.W.; Giaccone, G.; Tagliavini, F.; Bugiani, O.; Piccardo, P.; Hulette, C.M.; Crain, B.J.; et al. Ectopic white matter neurons, a developmental abnormality that may be caused by the PSEN1 S169L mutation in a case of familial AD with myoclonus and seizures. J. Neuropathol. Exp. Neurol. 2001, 60, 1137–1152. [Google Scholar] [CrossRef][Green Version]
- Dermaut, B.; Kumar-Singh, S.; Engelborghs, S.; Theuns, J.; Rademakers, R.; Saerens, J.; Pickut, B.A.; Peeters, K.; van den Broeck, M.; Vennekens, K.; et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann. Neurol. 2004, 55, 617–626. [Google Scholar] [CrossRef]
- Lyoo, C.H.; Cho, H.; Choi, J.Y.; Hwang, M.S.; Hong, S.K.; Kim, Y.J.; Ryu, Y.H.; Lee, M.S. Tau Accumulation in Primary Motor Cortex of Variant Alzheimer’s Disease with Spastic Paraparesis. J. Alzheimers Dis. 2016, 51, 671–675. [Google Scholar] [CrossRef]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; McCarty-Wood, E.; Van Deerlin, V.M.; Lee, V.M.; Trojanowski, J.Q. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am. J. Pathol. 2013, 183, 344–351. [Google Scholar] [CrossRef]
- Ehling, R.; Nosková, L.; Stránecký, V.; Hartmannová, H.; Přistoupilová, A.; Hodaňová, K.; Benke, T.; Kovacs, G.G.; Ströbel, T.; Niedermüller, U.; et al. Cerebellar dysfunction in a family harboring the PSEN1 mutation co-segregating with a cathepsin D variant p.A58V. J. Neurol. Sci. 2013, 326, 75–82. [Google Scholar] [CrossRef]
- Roeber, S.; Müller-Sarnowski, F.; Kress, J.; Edbauer, D.; Kuhlmann, T.; Tüttelmann, F.; Schindler, C.; Winter, P.; Arzberger, T.; Müller, U.; et al. Three novel presenilin 1 mutations marking the wide spectrum of age at onset and clinical patterns in familial Alzheimer’s disease. J. Neural Transm. 2015, 122, 1715–1719. [Google Scholar] [CrossRef]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Al-Sarraj, S.; Niblock, M.; Gallo, J.M.; Adnan, J.; et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2881.e1–2881.e6. [Google Scholar] [CrossRef]
- Colacicco, A.M.; Panza, F.; Basile, A.M.; Solfrizzi, V.; Capurso, C.; D’Introno, A.; Torres, F.; Capurso, S.; Cozza, S.; Flora, R.; et al. F175S change and a novel polymorphism in presenilin-1 gene in late-onset familial Alzheimer’s disease. Eur. Neurol. 2002, 47, 209–213. [Google Scholar] [CrossRef]
- Kasuga, K.; Ohno, T.; Ishihara, T.; Miyashita, A.; Kuwano, R.; Onodera, O.; Nishizawa, M.; Ikeuchi, T. Depression and psychiatric symptoms preceding onset of dementia in a family with early-onset Alzheimer disease with a novel PSEN1 mutation. J. Neurol. 2009, 256, 1351–1353. [Google Scholar] [CrossRef]
- Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Quillard-Muraine, M.; Guyant-Maréchal, L.; Martinaud, O.; Pariente, J.; Puel, M.; Rollin-Sillaire, A.; Pasquier, F.; et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: Mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimers Dis. 2012, 30, 847–856. [Google Scholar] [CrossRef]
- Klünemann, H.H.; Rogaeva, E.; Neumann, M.; Kretzschmar, H.A.; Kandel, M.; Toulina, A.; Sato, C.; Salehi-Rad, S.; Pfister, K.; Klein, H.E.; et al. Novel PS1 mutation in a Bavarian kindred with familial Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2004, 18, 256–258. [Google Scholar]
- Yasuda, M.; Maeda, K.; Ikejiri, Y.; Kawamata, T.; Kuroda, S.; Tanaka, C. A novel missense mutation in the presenilin-1 gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci. Lett. 1997, 232, 29–32. [Google Scholar] [CrossRef]
- Park, J.E.; Kim, H.J.; Kim, Y.E.; Jang, H.; Cho, S.H.; Kim, S.J.; Na, D.L.; Won, H.H.; Ki, C.S.; Seo, S.W. Analysis of dementia-related gene variants in APOE epsilon4 noncarrying Korean patients with early-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155.e5–155.e8. [Google Scholar] [CrossRef]
- Kim, J.; Bagyinszky, E.; Chang, Y.H.; Choe, G.; Choi, B.O.; An, S.S.; Kim, S. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: Clinical, neuroimaging, and neuropathological findings. Neurosci. Lett. 2012, 530, 109–114. [Google Scholar] [CrossRef]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; Medway, C.; et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2422.e13–2422.e16. [Google Scholar] [CrossRef]
- Muller, U.; Winter, P.; Graeber, M.B. A presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet. Neurol. 2013, 12, 129–130. [Google Scholar] [CrossRef]
- Jiang, B.; Bi, M.; Li, J.; Liu, Q.; Xiao, N.A.; Fang, J.; Shi, M.Y.; Yu, Z.W.; Ma, Q.L.; Tong, S.J.; et al. A Pathogenic Variant p.Phe177Val in PSEN1 Causes Early-Onset Alzheimer’s Disease in a Chinese Family. Front. Genet. 2020, 11, 713. [Google Scholar]
- Tominaga, A.; Cai, T.; Takagi-Niidome, S.; Iwatsubo, T.; Tomita, T. Conformational Changes in Transmembrane Domain 4 of Presenilin 1 Are Associated with Altered Amyloid-beta 42 Production. J. Neurosci. 2016, 36, 1362–1372. [Google Scholar] [PubMed]
- Dong, H.; Sharma, M.; Zhou, H.X.; Cross, T.A. Glycines: Role in alpha-helical membrane protein structures and a potential indicator of native conformation. Biochemistry 2012, 51, 4779–4789. [Google Scholar]
- Chen, W.T.; Hsieh, Y.F.; Huang, Y.J.; Lin, C.C.; Lin, Y.T.; Liu, Y.C.; Lien, C.C.; Cheng, I.H. G206D Mutation of Presenilin-1 Reduces Pen2 Interaction, Increases Abeta42/Abeta40 Ratio and Elevates ER Ca(2+) Accumulation. Mol. Neurobiol. 2015, 52, 1835–1849. [Google Scholar] [CrossRef]
- Kamino, K.; Sato, S.; Sakaki, Y.; Yoshiiwa, A.; Nishiwaki, Y.; Takeda, M.; Tanabe, H.; Nishimura, T.; Ii, K.; St George-Hyslop, P.H.; et al. Three different mutations of presenilin 1 gene in early-onset Alzheimer’s disease families. Neurosci. Lett. 1996, 208, 195–198. [Google Scholar] [CrossRef]
- Miranda, A.M.; Herman, M.; Cheng, R.; Nahmani, E.; Barrett, G.; Micevska, E.; Fontaine, G.; Potier, M.C.; Head, E.; Schmitt, F.A.; et al. Excess Synaptojanin 1 Contributes to Place Cell Dysfunction and Memory Deficits in the Aging Hippocampus in Three Types of Alzheimer’s Disease. Cell. Rep. 2018, 23, 2967–2975. [Google Scholar] [CrossRef]
- Couthouis, J.; Raphael, A.R.; Daneshjou, R.; Gitler, A.D. Targeted exon capture and sequencing in sporadic amyotrophic lateral sclerosis. PLoS Genet. 2014, 10, e1004704. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Cheng, I.H.; Lee, C.C.; Chiu, M.J.; Lee, M.J.; Chen, T.F.; Hsu, J.L. Clinical phenotype of G206D mutation in the presenilin 1 gene in pathologically confirmed familial Alzheimer’s disease. J. Alzheimers Dis. 2011, 25, 145–150. [Google Scholar] [CrossRef]
- Lohmann, E.; Guerreiro, R.J.; Erginel-Unaltuna, N.; Gurunlian, N.; Bilgic, B.; Gurvit, H.; Hanagasi, H.A.; Luu, N.; Emre, M.; Singleton, A. Identification of PSEN1 and PSEN2 gene mutations and variants in Turkish dementia patients. Neurobiol. Aging 2012, 33, 1850.e17–1850.e27. [Google Scholar] [CrossRef] [PubMed]
- Marui, W.; Iseki, E.; Sugiyama, N.; Matsumura, T.; Suzuki, K.; Odawara, T.; Hino, H.; Kosaka, K. A Japanese family with familial Alzheimer’s disease associated with presenilin 1 mutation: Relationship between younger age of onset and ApoE gene polymorphism. No To Shinkei 2003, 55, 349–353. [Google Scholar]
- Li, Y.S.; Yang, Z.H.; Zhang, Y.; Yang, J.; Shang, D.D.; Zhang, S.Y.; Wu, J.; Ji, Y.; Zhao, L.; Shi, C.H.; et al. Two Novel Mutations and a de novo Mutation in PSEN1 in Early-onset Alzheimer’s Disease. Aging Dis. 2019, 10, 908–914. [Google Scholar] [CrossRef]
- Norton, J.B.; Cairns, N.J.; Chakraverty, S.; Wang, J.; Levitch, D.; Galvin, J.E.; Goate, A. Presenilin1 G217R mutation linked to Alzheimer disease with cotton wool plaques. Neurology 2009, 73, 480–482. [Google Scholar] [CrossRef]
- Smith, M.J.; Gardner, R.J.; Knight, M.A.; Forrest, S.M.; Beyreuther, K.; Storey, E.; McLean, C.A.; Cotton, R.G.; Cappal, R.; Masters, C.L. Early-onset Alzheimer’s disease caused by a novel mutation at codon 219 of the presenilin-1 gene. Neuroreport 1999, 10, 503–507. [Google Scholar] [CrossRef]
- Goldman, J.S.; Reed, B.; Gearhart, R.; Kramer, J.H.; Miller, B.L. Very early-onset familial Alzheimer’s disease: A novel presenilin 1 mutation. Int. J. Geriatr. Psychiatry 2002, 17, 649–651. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.; Shen, J.; Tian, W.; Fang, R.; Li, B.; Ma, J.; Cao, L.; Chen, S.; Li, G.; et al. The Whole Exome Sequencing Clarifies the Genotype- Phenotype Correlations in Patients with Early-Onset Dementia. Aging Dis. 2018, 9, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Yonemura, K.; Kakuda, S.; Tashiro, Y.; Fujita, Y.; Takai, E.; Hashimoto, Y.; Makioka, K.; Furuta, N.; Ishiguro, K.; et al. Cerebrospinal fluid levels of phosphorylated tau and Abeta1-38/Abeta1-40/Abeta1-42 in Alzheimer’s disease with PS1 mutations. Amyloid 2013, 20, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Church, A.; Prescott, J.; Lillis, S.; Rees, J.; Chance, P.; Williamson, K.; Morris, H.R. A novel presenilin 1 mutation, I202F occurring at a previously predicted pathogenic site causing autosomal dominant Alzheimer’s disease. Neurobiol. Aging 2011, 32, 556.e1–556.e2. [Google Scholar] [CrossRef]
- Takao, M.; Ghetti, B.; Hayakawa, I.; Ikeda, E.; Fukuuchi, Y.; Miravalle, L.; Piccardo, P.; Murrell, J.R.; Glazier, B.S.; Koto, A. A novel mutation (G217D) in the Presenilin 1 gene ( PSEN1) in a Japanese family: Presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol. 2002, 104, 155–170. [Google Scholar] [CrossRef]
- Ringman, J.M.; Gylys, K.H.; Medina, L.D.; Fox, M.; Kepe, V.; Flores, D.L.; Apostolova, L.G.; Barrio, J.R.; Small, G.; Silverman, D.H.; et al. Biochemical, neuropathological, and neuroimaging characteristics of early-onset Alzheimer’s disease due to a novel PSEN1 mutation. Neurosci. Lett. 2011, 487, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, K.; Qiu, Y.; Ren, Z.; Dai, R.; Deng, Y.; Qing, H. Effect of Presenilin Mutations on APP Cleavage; Insights into the Pathogenesis of FAD. Front. Aging Neurosci. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Pennington, P.R.; Wei, Z.; Rui, L.; Doig, J.A.; Graham, B.; Kuski, K.; Gabriel, G.G.; Mousseau, D.D. Alzheimer disease-related presenilin-1 variants exert distinct effects on monoamine oxidase-A activity in vitro. J. Neural Trans. 2011, 118, 987–995. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Liu, J.; Li, G.; Yang, E. Increased phosphorylation of tau and synaptic protein loss in the aged transgenic mice expressing familiar Alzheimer’s disease-linked presenilin 1 mutation. Neurochem. Res. 2012, 37, 15–22. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Gamma-secretase gene mutations in familial acne inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef]
- Li, A.; Peng, Y.; Taiclet, L.M.; Tanzi, R.E. Analysis of hidradenitis suppurativa-linked mutations in four genes and the effects of PSEN1-P242LfsX11 on cytokine and chemokine expression in macrophages. Hum. Mol. Genet. 2019, 28, 1173–1182. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Guella, I.; Lehman, A.; Foti, D.; Farrer, M.J. PSEN1 p.Met233Val in a Complex Neurodegenerative Movement and Neuropsychiatric Disorder. J. Mov. Disord. 2018, 11, 45–48. [Google Scholar] [CrossRef]
- Seliverstov, Y.; Kanivets, I.; Illarioshkin, S. Spinocerebellar Ataxia-Like Presentation of the M233V PSEN1 Mutation. Cerebellum 2020, 19, 744–747. [Google Scholar] [CrossRef]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Zekanowski, C.; Golan, M.P.; Krzyśko, K.A.; Lipczyńska-Łojkowska, W.; Filipek, S.; Kowalska, A.; Rossa, G.; Pepłońska, B.; Styczyńska, M.; Maruszak, A.; et al. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: Genetic and bioinformatic assessment. Exp. Neurol. 2006, 200, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F.; McMurtray, A. Frontotemporal dementia-like phenotypes associated with presenilin-1 mutations. Am. J. Alzheimers Dis. Other Dement. 2006, 21, 281–286. [Google Scholar] [CrossRef]
- Uttner, I.; Kirchheiner, J.; Tumani, H.; Mottaghy, F.M.; Lebedeva, E.; Ozer, E.; Ludolph, A.C.; Huber, R.; von Arnim, C.A. A novel presenilin1 mutation (Q223R) associated with early onset Alzheimer’s disease, dysarthria and spastic paraparesis and decreased Abeta levels in CSF. Eur. J. Neurol. 2010, 17, 631–633. [Google Scholar] [CrossRef]
- Ringman, J.M.; Casado, M.; Van Berlo, V.; Pa, J.; Joseph-Mathurin, N.; Fagan, A.M.; Benzinger, T.; Bateman, R.J.; Morris, J.C. A novel PSEN1 (S230N) mutation causing early-onset Alzheimer’s Disease associated with prosopagnosia, hoarding, and Parkinsonism. Neurosci. Lett. 2017, 657, 11–15. [Google Scholar] [CrossRef]
- Antonell, A.; Balasa, M.; Oliva, R.; Lladó, A.; Bosch, B.; Fabregat, N.; Fortea, J.; Molinuevo, J.L.; Sánchez-Valle, R. A novel PSEN1 gene mutation (L235R) associated with familial early-onset Alzheimer’s disease. Neurosci. Lett. 2011, 496, 40–42. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Q.; Jing, D.; Gao, R.; Zhang, J.; Cui, C.; Qiao, H.; Liang, Z.; Wang, C.; Rosa-Neto, P.; et al. Diagnostic Approach of Early-Onset Dementia with Negative Family History: Implications from Two Cases of Early-Onset Alzheimer’s Disease with De Novo PSEN1 Mutation. J. Alzheimers Dis. 2019, 68, 551–558. [Google Scholar] [CrossRef]
- Navarro, E.; De Andrés, C.; Guerrero, C.; Giménez-Roldán, S. Corticobasal Syndrome in a Family with Early-Onset Alzheimer’s Disease Linked to a Presenilin-1 Gene Mutation. Mov. Disord. Clin. Pract. 2015, 2, 388–394. [Google Scholar] [CrossRef][Green Version]
- Revesz, T.; McLaughlin, J.L.; Rossor, M.N.; Lantos, P.L. Pathology of familial Alzheimer’s disease with Lewy bodies. J. Neural Trans. Suppl. 1997, 51, 121–135. [Google Scholar]
- Saura, C.A.; Tomita, T.; Soriano, S.; Takahashi, M.; Leem, J.Y.; Honda, T.; Koo, E.H.; Iwatsubo, T.; Thinakaran, G. The nonconserved hydrophilic loop domain of presenilin (PS) is not required for PS endoproteolysis or enhanced abeta 42 production mediated by familial early onset Alzheimer’s disease-linked PS variants. J. Biol. Chem. 2000, 275, 17136–17142. [Google Scholar] [CrossRef] [PubMed]
- Fukumori, A.; Steiner, H. Substrate recruitment of gamma-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 2016, 35, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- Trambauer, J.; Rodríguez Sarmiento, R.M.; Fukumori, A.; Feederle, R.; Baumann, K.; Steiner, H. Abeta43-producing PS1 FAD mutants cause altered substrate interactions and respond to gamma-secretase modulation. EMBO Rep. 2020, 21, e47996. [Google Scholar] [CrossRef]
- Armijo, E.; Gonzalez, C.; Shahnawaz, M.; Flores, A.; Davis, B.; Soto, C. Increased susceptibility to Abeta toxicity in neuronal cultures derived from familial Alzheimer’s disease (PSEN1-A246E) induced pluripotent stem cells. Neurosci. Lett. 2017, 639, 74–81. [Google Scholar] [CrossRef]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; ASproul, A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef]
- Ben-Gedalya, T.; Moll, L.; Bejerano-Sagie, M.; Frere, S.; Cabral, W.A.; Friedmann-Morvinski, D.; Slutsky, I.; Burstyn-Cohen, T.; Marini, J.C.; Cohen, E. Alzheimer’s disease-causing proline substitutions lead to presenilin 1 aggregation and malfunction. EMBO J. 2015, 34, 2820–2839. [Google Scholar] [CrossRef]
- Kwok, J.B.; Halliday, G.M.; Brooks, W.S.; Dolios, G.; Laudon, H.; Murayama, O.; Hallupp, M.; Badenhop, R.F.; Vickers, J.; Wang, R.; et al. Presenilin-1 mutation L271V results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. J. Biol. Chem. 2003, 278, 6748–6754. [Google Scholar] [CrossRef]
- Wang, W.; Moerman-Herzog, A.M.; Slaton, A.; Barger, S.W. Presenilin 1 mutations influence processing and trafficking of the ApoE receptor apoER2. Neurobiol. Aging 2017, 49, 145–153. [Google Scholar] [CrossRef]
- Elsworthy, R.J.; King, M.C.; Grainger, A.; Fisher, E.; Crowe, J.A.; Alqattan, S.; Ludlam, A.; Hill, D.E.J.; Aldred, S. Amyloid-beta precursor protein processing and oxidative stress are altered in human iPSC-derived neuron and astrocyte co-cultures carrying presenillin-1 gene mutations following spontaneous differentiation. Mol. Cell Neurosci. 2021, 114, 103631. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Gardner, M.; Lintott, C.; Anderson, T. Novel Presenilin-1 Mutation (Ala275Ser) Associated With Clinical Features of Dementia With Lewy Bodies. Alzheimer Dis. Assoc. Disord. 2021, 35, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Mengel, D.; Liu, L.; Yamamoto, R.; Zülow, S.; Deuschl, C.; Hermann, D.M.; Zerr, I.; Selkoe, D.J.; Dodel, R. A novel V272D presenilin mutation associated with logopenia, disorientation, and apraxia in an autosomal-dominant Alzheimer’s disease family. Neurobiol. Aging 2020, 85, 154.e5–154.e7. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Nakamura, M.; Ichiba, M.; Fujita, S.; Takeuchi, K.; Fujimoto, T.; Sano, A. Different clinical phenotypes in siblings with a presenilin-1 P264L mutation. Different clinical phenotypes in siblings with a presenilin-1 P264L mutation. Dement. Geriatr. Cogn. Disord. 2012, 33, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, C.J.; Downey, L.E.; Beck, J.; Liang, Y.; Mead, S.; Perry, R.J.; Warren, J.D. The presenilin 1 P264L mutation presenting as non-fluent/agrammatic primary progressive aphasia. J. Alzheimers Dis. 2013, 36, 239–243. [Google Scholar] [CrossRef]
- Perez-Tur, J.; Croxton, R.; Wright, K.; Phillips, H.; Zehr, C.; Crook, R.; Hutton, M.; Hardy, J.; Karran, E.; Roberts, G.W.; et al. A further presenilin 1 mutation in the exon 8 cluster in familial Alzheimer’s disease. Neurodegeneration 1996, 5, 207–212. [Google Scholar] [CrossRef]
- Tortelli, R.; Seripa, D.; Zecca, C.; Dell’Abate, M.T.; Bisceglia, P.; Barulli, M.R.; De Blasi, R.; Logroscino, G. A New Presenilin 1 (Psen1) Mutation (p.Cys263Trp) as a Cause of Both Early and Late-Onset Alzheimer’s Disease in a Large Italian Family. Int. J. Mol. Sci. 2021, 22, 6215. [Google Scholar] [CrossRef]
- Jimenez-Escrig, A.; Rabano, A.; Guerrero, C.; Simon, J.; Barquero, M.S.; Güell, I.; Ginestal, R.C.; Montero, T.; Orensanz, L. New V272A presenilin 1 mutation with very early onset subcortical dementia and parkinsonism. Eur. J. Neurol. 2004, 11, 663–669. [Google Scholar] [CrossRef]
- Raman, A.; Lin, X.; Suri, M.; Hewitt, M.; Constantinescu, C.S.; Phillips, M.F. A presenilin 1 mutation (Arg278Ser) associated with early onset Alzheimer’s disease and spastic paraparesis. J. Neurol. Sci. 2007, 260, 78–82. [Google Scholar] [CrossRef]
- Kwok, J.B.; Taddei, K.; Hallupp, M.; Fisher, C.; Brooks, W.S.; Broe, G.A.; Hardy, J.; Fulham, M.J.; Nicholson, G.A.; Stell, R.; et al. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer’s disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport 1997, 8, 1537–1542. [Google Scholar] [CrossRef]
- Assini, A.; Terreni, L.; Borghi, R.; Giliberto, L.; Piccini, A.; Loqui, D.; Fogliarino, S.; Forloni, G.; Tabaton, M. Pure spastic paraparesis associated with a novel presenilin 1 R278K mutation. Neurology 2003, 60, 150. [Google Scholar] [CrossRef] [PubMed]
- Matsubara-Tsutsui, M.; Yasuda, M.; Yamagata, H.; Nomura, T.; Taguchi, K.; Kohara, K.; Miyoshi, K.; Miki, T. Molecular evidence of presenilin 1 mutation in familial early onset dementia. Am. J. Med. Genet. 2002, 114, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Yasuda, M.; Terasawa, K.J.; Tanaka, K.; Murai, H.; Kira, J.; Ohyagi, Y. A novel mutation (L250V) in the presenilin 1 gene in a Japanese familial Alzheimer’s disease with myoclonus and generalized convulsion. J. Neurol. Sci. 2003, 209, 75–77. [Google Scholar] [CrossRef]
- Hutton, M.; Busfield, F.; Wragg, M.; Crook, R.; Perez-Tur, J.; Clark, R.F.; Prihar, G.; Talbot, C.; Phillips, H.; Wright, K.; et al. Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. Neuroreport 1996, 7, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef]
- Steiner, H.; Romig, H.; Pesold, B.; Philipp, U.; Baader, M.; Citron, M.; Loetscher, H.; Jacobsen, H.; Haass, C. Amyloidogenic function of the Alzheimer’s disease-associated presenilin 1 in the absence of endoproteolysis. Biochemistry 1999, 38, 14600–14605. [Google Scholar] [CrossRef]
- Dehury, B.; Somavarapu, A.K.; Kepp, K.P. A computer-simulated mechanism of familial Alzheimer’s disease: Mutations enhance thermal dynamics and favor looser substrate-binding to gamma-secretase. J. Struct. Biol. 2020, 212, 107648. [Google Scholar] [CrossRef]
- Rojas-Charry, L.; Calero-Martinez, S.; Morganti, C.; Morciano, G.; Park, K.; Hagel, C.; Marciniak, S.J.; Glatzel, M.; Pinton, P.; Sepulveda-Falla, D. Susceptibility to cellular stress in PS1 mutant N2a cells is associated with mitochondrial defects and altered calcium homeostasis. Sci. Rep. 2020, 10, 6455. [Google Scholar] [CrossRef]
- Soto-Mercado, V.; Mendivil-Perez, M.; Velez-Pardo, C.; Lopera, F.; Jimenez-Del-Rio, M. Cholinergic-like neurons carrying PSEN1 E280A mutation from familial Alzheimer’s disease reveal intraneuronal sAPPbeta fragments accumulation, hyperphosphorylation of TAU, oxidative stress, apoptosis and Ca2+ dysregulation: Therapeutic implications. PLoS ONE 2020, 15, e0221669. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Liang, G.; Xu, Z.; Chu, C.T.; Wei, H. Alzheimer’s Disease Presenilin-1 Mutation Sensitizes Neurons to Impaired Autophagy Flux and Propofol Neurotoxicity: Role of Calcium Dysregulation. J. Alzheimers Dis. 2019, 67, 137–147. [Google Scholar] [CrossRef]
- Lee, M.K.; Borchelt, D.R.; Kim, G.; Thinakaran, G.; Slunt, H.H.; Ratovitski, T.; Martin, L.J.; Kittur, A.; Gandy, S.; Levey, A.I.; et al. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat. Med. 1997, 3, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef]
- Knappenberger, K.S.; Tian, G.; Ye, X.; Sobotka-Briner, C.; Ghanekar, S.V.; Greenberg, B.D.; Scott, C.W. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar]
- Landman, N.; Jeong, S.Y.; Shin, S.Y.; Voronov, S.V.; Serban, G.; Kang, M.S.; Park, M.K.; Di Paolo, G.; Chung, S.; Kim, T.W. Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 19524–19529. [Google Scholar] [CrossRef]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef]
- Oh, H.G.; Chung, S. Activation of transient receptor potential melastatin 7 (TRPM7) channel increases basal autophagy and reduces amyloid beta-peptide. Biochem. Biophys. Res. Commun. 2017, 493, 494–499. [Google Scholar] [CrossRef]
- O’Riordan, S.; McMonagle, P.; Janssen, J.C.; Fox, N.C.; Farrell, M.; Collinge, J.; Rossor, M.N.; Hutchinson, M. Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities. Neurology 2002, 59, 1108–1110. [Google Scholar] [CrossRef]
- Tabira, T.; Chui, D.H.; Nakayama, H.; Kuroda, S.; Shibuya, M. Alzheimer’s disease with spastic paresis and cotton wool type plaques. J. Neurosci. Res. 2002, 70, 367–372. [Google Scholar] [CrossRef]
- Sánchez-Valle, R.; Lladó, A.; Ezquerra, M.; Rey, M.J.; Rami, L.; Molinuevo, J.L. A novel mutation in the PSEN1 gene (L286P) associated with familial early-onset dementia of Alzheimer type and lobar haematomas. Eur. J. Neurol. 2007, 14, 1409–1412. [Google Scholar] [CrossRef]
- Dumanchin, C.; Tournier, I.; Martin, C.; Didic, M.; Belliard, S.; Carlander, B.; Rouhart, F.; Duyckaerts, C.; Pellissier, J.F.; Latouche, J.B.; et al. Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum. Mutat. 2006, 27, 1063. [Google Scholar] [CrossRef]
- Hiltunen, M.; Helisalmi, S.; Mannermaa, A.; Alafuzoff, I.; Koivisto, A.M.; Lehtovirta, M.; Pirskanen, M.; Sulkava, R.; Verkkoniemi, A.; Soininen, H. Identification of a novel 4.6-kb genomic deletion in presenilin-1 gene which results in exclusion of exon 9 in a Finnish early onset Alzheimer’s disease family: An Alu core sequence-stimulated recombination? Eur. J. Hum. Genet. 2000, 8, 259–266. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perez-Tur, J.; Froelich, S.; Prihar, G.; Crook, R.; Baker, M.; Duff, K.; Wragg, M.; Busfield, F.; Lendon, C.; Clark, R.F.; et al. A mutation in Alzheimer’s disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport 1995, 7, 297–301. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Zeng, W.; Deng, L.; Lei, Z.; Gao, X.; Zhang, V.W.; Wang, Y. Identification and Clinical Analysis of the First Nonsense Mutation in the PSEN1 Gene in a Family With Acute Encephalopathy and Retinitis Pigmentosa. Front. Neurol 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Palmieri, I.; Valente, M.; Farina, L.M.; Gana, S.; Minafra, B.; Zangaglia, R.; Pansarasa, O.; Sproviero, D.; Costa, A.; Pacchetti, C.; et al. PSEN1 Compound Heterozygous Mutations Associated with Cerebral Amyloid Angiopathy and Cognitive Decline Phenotype. Int. J. Mol. Sci. 2021, 22, 3870. [Google Scholar] [CrossRef]
- Boeve, B.F.; Baker, M.; Dickson, D.W.; Parisi, J.E.; Giannini, C.; Josephs, K.A.; Hutton, M.; Pickering-Brown, S.M.; Rademakers, R.; Tang-Wai, D.; et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G->A mutation in progranulin: A clinicopathologic study. Brain 2006, 129 Pt 11, 3103–3114. [Google Scholar] [CrossRef]
- Monacelli, F.; Martella, L.; Parodi, M.N.; Odetti, P.; Fanelli, F.; Tabaton, M. Frontal Variant of Alzheimer’s Disease: A Report of a Novel PSEN1 Mutation. J. Alzheimers Dis. 2019, 70, 11–15. [Google Scholar] [CrossRef]
- Abdala, B.B.; Dos Santos, J.M.; Gonçalves, A.P.; da Motta, L.B.; Laks, J.; de Borges, M.B.; Gonçalves Pimentel, M.M.; Santos-Rebouças, C.B. Influence of low frequency PSEN1 variants on familial Alzheimer’s disease risk in Brazil. Neurosci. Lett. 2017, 653, 341–345. [Google Scholar] [CrossRef]
- Rogaeva, E.A.; Fafel, K.C.; Song, Y.Q.; Medeiros, H.; Sato, C.; Liang, Y.; Richard, E.; Rogaev, E.I.; Frommelt, P.; Sadovnick, A.D.; et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology 2001, 57, 621–625. [Google Scholar] [CrossRef]
- Ikeuchi, T.; Kaneko, H.; Miyashita, A.; Nozaki, H.; Kasuga, K.; Tsukie, T.; Tsuchiya, M.; Imamura, T.; Ishizu, H.; Aoki, K.; et al. Mutational analysis in early-onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement. Geriatr. Cogn. Disord. 2008, 26, 43–49. [Google Scholar] [CrossRef]
- Shea, Y.F.; Chan, A.O.; Chu, L.W.; Lee, S.C.; Law, C.Y.; See, C.H.; Yiu, K.L.; Chiu, P.K. Novel presenilin 1 mutation (p.F386I) in a Chinese family with early-onset Alzheimer’s disease. Neurobiol. Aging 2017, 50, 168.e9–168.e11. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Revuelta, B.I.; Fukumori, A.; Lammich, S.; Yamasaki, A.; Haass, C.; Steiner, H. Requirement for small side chain residues within the GxGD-motif of presenilin for gamma-secretase substrate cleavage. J. Neurochem. 2010, 112, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Kostka, M.; Romig, H.; Basset, G.; Pesold, B.; Hardy, J.; Capell, A.; Meyn, L.; Grim, M.L.; Baumeister, R.; et al. Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. Cell Biol. 2000, 2, 848–851. [Google Scholar] [CrossRef]
- Lou, F.; Luo, X.; Li, M.; Ren, Y.; He, Z. Very early-onset sporadic Alzheimer’s disease with a de novo mutation in the PSEN1 gene. Neurobiol. Aging 2017, 53, 193.e1–193.e5. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Pilotto, A.; Bonvicini, C.; Archetti, S.; Alberici, A.; Lupi, A.; Gennarelli, M.; Padovani, A. Atypical presentation of a novel Presenilin 1 R377W mutation: Sporadic, late-onset Alzheimer disease with epilepsy and frontotemporal atrophy. Neurol. Sci. 2012, 33, 375–378. [Google Scholar] [CrossRef]
- Scarioni, M.; Arighi, A.; Fenoglio, C.; Sorrentino, F.; Serpente, M.; Rotondo, E.; Mercurio, M.; Marotta, G.; Dijkstra, A.A.; Pijnenburg, Y.A.L.; et al. Late-onset presentation and phenotypic heterogeneity of the rare R377W PSEN1 mutation. Eur. J. Neurol. 2020, 27, 2630–2634. [Google Scholar] [CrossRef]
- Llibre-Guerra, J.J.; Li, Y.; Allegri, R.F.; Mendez, P.C.; Surace, E.I.; Llibre-Rodriguez, J.J.; Sosa, A.L.; Aláez-Verson, C.; Longoria, E.M.; Tellez, A.; et al. Dominantly inherited Alzheimer’s disease in Latin America: Genetic heterogeneity and clinical phenotypes. Alzheimers Dement. 2021, 17, 653–664. [Google Scholar] [CrossRef]
- Jin, S.C.; Pastor, P.; Cooper, B.; Cervantes, S.; Benitez, B.A.; Razquin, C.; Goate, A. Ibero-American Alzheimer Disease Genetics Group Researchers, Cruchaga C, Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther 2012, 4, 34. [Google Scholar] [CrossRef]
- Ramos-Campoy, O.; Antonell, A.; Falgàs, N.; Balasa, M.; Borrego-Écija, S.; Rodríguez-Santiago, B.; Datta, D.; Armengol, L.; Fernández-Villullas, G.; Bosch, B.; et al. Screening of dementia genes by whole-exome sequencing in Spanish patients with early-onset dementia: Likely pathogenic, uncertain significance and risk variants. Neurobiol. Aging 2020, 93, e1–e9. [Google Scholar] [CrossRef]
- Zhan, Y.; Zheng, H.; Wang, C.; Rong, Z.; Xiao, N.; Ma, Q.; Zhang, Y.W. A novel presenilin 1 mutation (F388L) identified in a Chinese family with early-onset Alzheimer’s disease. Neurobiol. Aging 2017, 50, 168.e1–168.e4. [Google Scholar] [CrossRef]
- Dintchov Traykov, L.; Mehrabian, S.; Van den Broeck, M.; Radoslavova Raycheva, M.; Cruts, M.; Kirilova Jordanova, A.; Van Broeckhoven, C. Novel PSEN1 mutation in a Bulgarian patient with very early-onset Alzheimer’s disease, spastic paraparesis, and extrapyramidal signs. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Maeda, S.; Kawamata, T.; Tamaoka, A.; Yamamoto, Y.; Kuroda, S.; Maeda, K.; Tanaka, C. Novel presenilin-1 mutation with widespread cortical amyloid deposition but limited cerebral amyloid angiopathy. J. Neurol. Neurosurg. Psychiatry 2000, 68, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.; Khan, A.; Keith, J.; Rogaeva, E.; Bilbao, J.; St George-Hyslop, P.; Ghani, M.; Freedman, M.; Stuss, D.T.; Chow, T.; et al. Characterizing familial corticobasal syndrome due to Alzheimer’s disease pathology and PSEN1 mutations. Alzheimers Dement. 2017, 13, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Dolzhanskaya, N.; Gonzalez, M.A.; Sperziani, F.; Stefl, S.; Messing, J.; Wen, G.Y.; Alexov, E.; Zuchner, S.; Velinov, M. A novel p.Leu(381)Phe mutation in presenilin 1 is associated with very early onset and unusually fast progressing dementia as well as lysosomal inclusions typically seen in Kufs disease. J. Alzheimers Dis. 2014, 39, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and Function of the gamma-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.; Tasiaux, B.; Opsomer, R.; Hage, S.; Sodero, A.O.; Dewachter, I.; Octave, J.N.; Smith, S.O.; Constantinescu, S.N.; Kienlen-Campard, P. Presenilin transmembrane domain 8 conserved AXXXAXXXG motifs are required for the activity of the gamma-secretase complex. J. Biol. Chem. 2015, 290, 7169–7184. [Google Scholar] [CrossRef]
- Guven, G.; Erginel-Unaltuna, N.; Samanci, B.; Gulec, C.; Hanagasi, H.; Bilgic, B. A patient with early-onset Alzheimer’s disease with a novel PSEN1 p.Leu424Pro mutation. Neurobiol. Aging 2019, 84, 238.e1–238.e4. [Google Scholar] [CrossRef]
- Xia, D.; Kelleher, R.J., 3rd; Shen, J. Loss of Abeta43 Production Caused by Presenilin-1 Mutations in the Knockin Mouse Brain. Neuron 2016, 90, 417–422. [Google Scholar] [CrossRef][Green Version]
- Song, W.; Nadeau, P.; Yuan, M.; Yang, X.; Shen, J.; Yankner, B.A. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 6959–6963. [Google Scholar] [CrossRef]
- Ramirez Aguilar, L.; Acosta-Uribe, J.; Giraldo, M.M.; Moreno, S.; Baena, A.; Alzate, D.; Cuastumal, R.; Aguillón, D.; Madrigal, L.; Saldarriaga, A.; et al. Genetic origin of a large family with a novel PSEN1 mutation (Ile416Thr). Alzheimers Dement. 2019, 15, 709–719. [Google Scholar] [CrossRef]
- Giau, V.V.; Wang, M.J.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. Novel PSEN1 p.Gly417Ala mutation in a Korean patient with early-onset Alzheimer’s disease with parkinsonism. Neurobiol. Aging 2018, 72, 188.e13–188.e17. [Google Scholar] [CrossRef] [PubMed]
- Leverenz, J.B.; Fishel, M.A.; Peskind, E.R.; Montine, T.J.; Nochlin, D.; Steinbart, E.; Raskind, M.A.; Schellenberg, G.D.; Bird, T.D.; Tsuang, D. Lewy body pathology in familial Alzheimer disease: Evidence for disease- and mutation-specific pathologic phenotype. Arch. Neurol. 2006, 63, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, A.; Pruchnik-Wolińska, D.; Florczak, J.; Modestowicz, R.; Szczech, J.; Kozubski, W.; Rossa, G.; Wender, M. Genetic study of familial cases of Alzheimer’s disease. Acta Biochim. Pol. 2004, 51, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Yokota, O.; Haraguchi, T.; Ikeuchi, T.; Zhu, B.; Takenoshita, S.; Terada, S.; Yamada, N. Young adult-onset, very slowly progressive cognitive decline with spastic paraparesis in Alzheimer’s disease with cotton wool plaques due to a novel presenilin1 G417S mutation. Acta Neuropathol. Commun. 2019, 7, 19. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ohta, Y.; Sasaki, R.; Tadokoro, K.; Sato, K.; Shang, J.; Takemoto, M.; Hishikawa, N.; Yamashita, T.; Haraguchi, T.; et al. A novel presenilin 1 mutation (Leu418Trp) associated with spasticity, parkinsonism, and white matter lesion in a dominant Alzheimer’s family. J. Neurol. Sci. 2018, 387, 166–169. [Google Scholar] [CrossRef]
- Tedde, A.; Bartoli, A.; Piaceri, I.; Ferrara, S.; Bagnoli, S.; Serio, A.; Sorbi, S.; Nacmias, B. Novel presenilin 1 mutation (Ile408Thr) in an Italian family with late-onset Alzheimer’s disease. Neurosci. Lett. 2016, 610, 150–153. [Google Scholar] [CrossRef]
- Niwa, A.; Matsuo, K.; Shindo, A.; Yata, K.; Shiraishi, T.; Tomimoto, H. Clinical and neuropathological findings in a patient with familial Alzheimer disease showing a mutation in the PSEN1 gene. Neuropathology 2013, 33, 199–203. [Google Scholar] [CrossRef]
- Yescas, P.; Huertas-Vazquez, A.; Villarreal-Molina, M.T.; Rasmussen, A.; Tusié-Luna, M.T.; López, M.; Canizales-Quinteros, S.; Alonso, M.E. Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early-onset Alzheimer’s disease in Mexican families. Neurogenetics 2006, 7, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Tomaino, C.; Anfossi, M.; Gallo, M.; Geracitano, S.; Costanzo, A.; Colao, R.; Puccio, G.; Frangipane, F.; Curcio, S.A.; et al. Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol. Aging 2009, 30, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.; Sobrido, M.J.; García-Murias, M.; Prieto, J.M.; Lema, M.; Santos, D.; Paramo, M. Clinical picture of a patient with a novel PSEN1 mutation (L424V). Am. J. Alzheimers Dis. Other Dement. 2009, 24, 40–45. [Google Scholar] [CrossRef]
- de Bot, S.T.; Kremer, H.P.; Dooijes, D.; Verbeek, M.M. CSF studies facilitate DNA diagnosis in familial Alzheimer’s disease due to a presenilin-1 mutation. J. Alzheimers Dis. 2009, 17, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Mozaffar, T.; Messmore, A.; Deignan, J.L.; Kimonis, V.E.; Ringman, J.M. Homozygosity for the A431E mutation in PSEN1 presenting with a relatively aggressive phenotype. Neurosci. Lett. 2019, 699, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Shrimpton, A.E.; Schelper, R.L.; Linke, R.P.; Hardy, J.; Crook, R.; Dickson, D.W.; Ishizawa, T.; Davis, R.L. A presenilin 1 mutation (L420R) in a family with early onset Alzheimer disease, seizures and cotton wool plaques, but not spastic paraparesis. Neuropathology 2007, 27, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Chang, S.; Xia, W.; Wong, S.T. Molecular dynamics simulation study reveals potential substrate entry path into gamma-secretase/presenilin-1. J. Struct. Biol. 2015, 191, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qin, W.; Wu, L.; Zhou, A.; Tang, Y.; Wang, Q.; Jia, L.; Jia, J. Two novel presenilin-1 mutations (I249L and P433S) in early onset Chinese Alzheimer’s pedigrees and their functional characterization. Biochem. Biophys. Res. Commun. 2019, 516, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human gamma-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.; Fotiou, A.; Jyrinji, D.; Tycko, B.; DeArmand, S.; Rogaeva, E.; Song, Y.Q.; Medieros, H.; Liang, Y.; Orlacchio, A.; et al. Novel presenilin 1 mutations associated with early onset of dementia in a family with both early-onset and late-onset Alzheimer disease. Arch. Neurol. 2000, 57, 1454–1457. [Google Scholar] [CrossRef]
- Taddei, K.; Kwok, J.B.; Kril, J.J.; Halliday, G.M.; Creasey, H.; Hallupp, M.; Fisher, C.; Brooks, W.S.; Chung, C.; Andrews, C.; et al. Two novel presenilin-1 mutations (Ser169Leu and Pro436Gln) associated with very early onset Alzheimer’s disease. Neuroreport 1998, 9, 3335–3339. [Google Scholar] [CrossRef]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Uphill, J.B.; Adamson, G.; Geddes, J.F.; Revesz, T.; Davis, M.B.; Wood, N.W.; Collinge, J.; et al. Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Kim, M.; Park, S.; Park, J.E.; Cho, S.H.; Kim, S.J.; Jang, H.; Jung, Y.H.; Kim, J.; Na, D.L.; et al. Dopa Responsive Parkinsonism in an Early Onset Alzheimer’s Disease Patient with a Presenilin 1 Mutation (A434T). J. Alzheimers Dis. 2019, 71, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Tang, B.; Liu, X.; Xu, J.; Wang, Y.; Zhou, L.; Zhang, F.; Yan, X.; Zhou, Y.; Shen, L. Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol. Aging 2014, 35, 1957.e1–1957.e6. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Piao, Y.S.; Miyashita, A.; Kuwano, R.; Onodera, O.; Ohtake, H.; Suzuki, M.; Nishizawa, M.; Takahashi, H. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann. Neurol. 2005, 57, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Lee, H.; Shim, K.H.; Bagyinszky, E.; An, S.S.A. Genome-editing applications of CRISPR-Cas9 to promote in vitro studies of Alzheimer’s disease. Clin. Interv. Aging 2018, 13, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol 2020, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Murayama, O.; Tomita, T.; Nihonmatsu, N.; Murayama, M.; Sun, X.; Honda, T.; Iwatsubo, T.; Takashima, A. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci. Lett. 1999, 265, 61–63. [Google Scholar] [CrossRef]
- Qi, Y.; Morishima-Kawashima, M.; Sato, T.; Mitsumori, R.; Ihara, Y. Distinct mechanisms by mutant presenilin 1 and 2 leading to increased intracellular levels of amyloid beta-protein 42 in Chinese hamster ovary cells. Biochemistry 2003, 42, 1042–1052. [Google Scholar] [CrossRef]
- Shioi, J.; Georgakopoulos, A.; Mehta, P.; Kouchi, Z.; Litterst, C.M.; Baki, L.; Robakis, N.K. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J. Neurochem. 2007, 101, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Qi-Takahara, Y.; Morishima-Kawashima, M.; Tanimura, Y.; Dolios, G.; Hirotani, N.; Horikoshi, Y.; Kametani, F.; Maeda, M.; Saido, T.C.; Wang, R.; et al. Longer forms of amyloid beta protein: Implications for the mechanism of intramembrane cleavage by gamma-secretase. J. Neurosci. 2005, 25, 436–445. [Google Scholar] [CrossRef]
- Petit, D.; Fernández, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Abeta profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Shen, J.; Kelleher, R.J. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Beglopoulos, V.; Sun, X.; Saura, C.A.; Lemere, C.A.; Kim, R.D.; Shen, J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J. Biol. Chem. 2004, 279, 46907–46914. [Google Scholar] [CrossRef]
- Saura, C.A.; Choi, S.Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chattarji, S.; Kelleher, R.J., 3rd; Kandel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef]
- Wines-Samuelson, M.; Schulte, E.C.; Smith, M.J.; Aoki, C.; Liu, X.; Kelleher, R.J., 3rd; Shen, J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS ONE 2010, 5, e10195. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Brayne, C. Understanding the roles of mutations in the amyloid precursor protein in Alzheimer disease. Mol. Psychiatry 2018, 23, 81–93. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Shea, Y.F.; Chu, L.W.; Chan, A.O.; Ha, J.; Li, Y.; Song, Y.Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Itzcovich, T.; Chrem-Méndez, P.; Vázquez, S.; Barbieri-Kennedy, M.; Niikado, M.; Martinetto, H.; Allegri, R.; Sevlever, G.; Surace, E.I. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early- and late-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155.e9–155.e12. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cheng, R.; Vardarajan, B.; Lantigua, R.; Reyes-Dumeyer, D.; Ortmann, W.; Graham, R.R.; Bhangale, T.; Behrens, T.W.; Medrano, M.; et al. Genetic Modifiers of Age at Onset in Carriers of the G206A Mutation in PSEN1 With Familial Alzheimer Disease Among Caribbean Hispanics. JAMA Neurol. 2015, 72, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- Vélez, J.I.; Lopera, F.; Silva, C.T.; Villegas, A.; Espinosa, L.G.; Vidal, O.M.; Mastronardi, C.A.; Arcos-Burgos, M. Familial Alzheimer’s Disease and Recessive Modifiers. Mol. Neurobiol. 2020, 57, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Stepanichev, M. Gene Editing and Alzheimer’s Disease: Is There Light at the End of the Tunnel? Front. Genome Ed. 2020, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Acuña-Hidalgo, R.; Keogh, M.J.; Quenez, O.; Steehouwer, M.; Lelieveld, S.; Rousseau, S.; Richard, A.C.; Oud, M.S.; Marguet, F.; et al. Somatic variants in autosomal dominant genes are a rare cause of sporadic Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Calero, M.; Gómez-Ramos, A.; Calero, O.; Soriano, E.; Avila, J.; Medina, M. Additional mechanisms conferring genetic susceptibility to Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 138. [Google Scholar] [CrossRef] [PubMed]














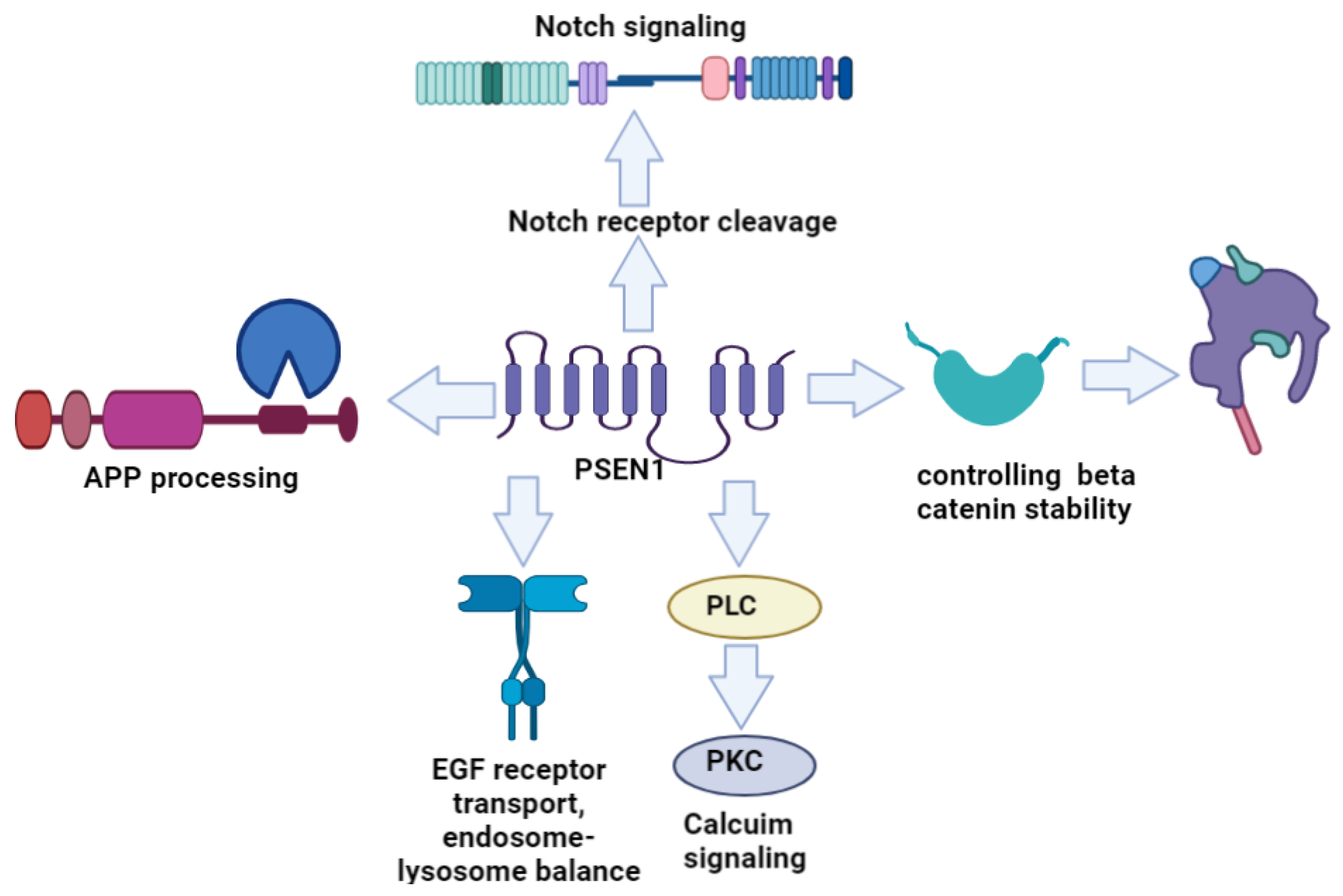{kind=link}
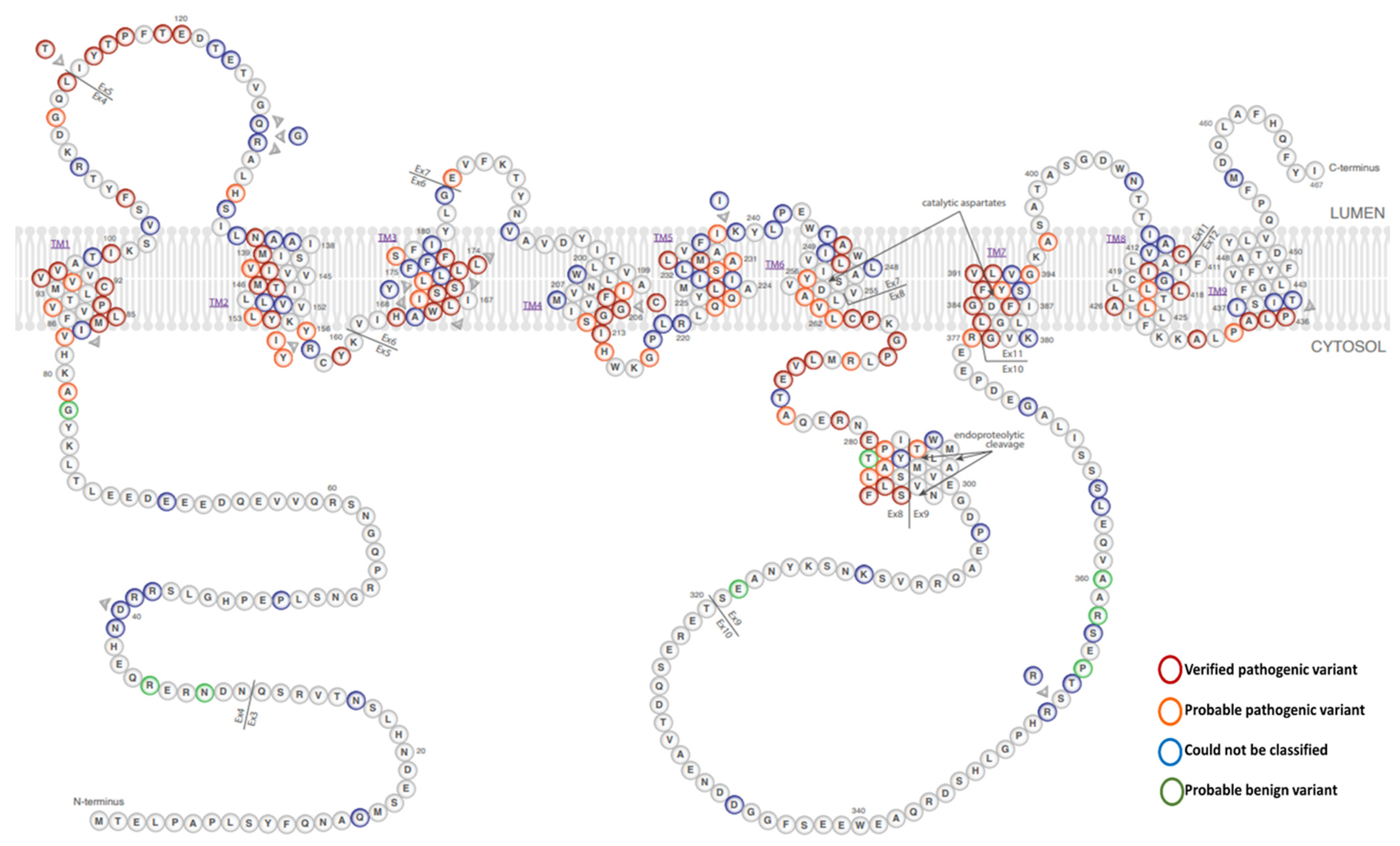{kind=link}
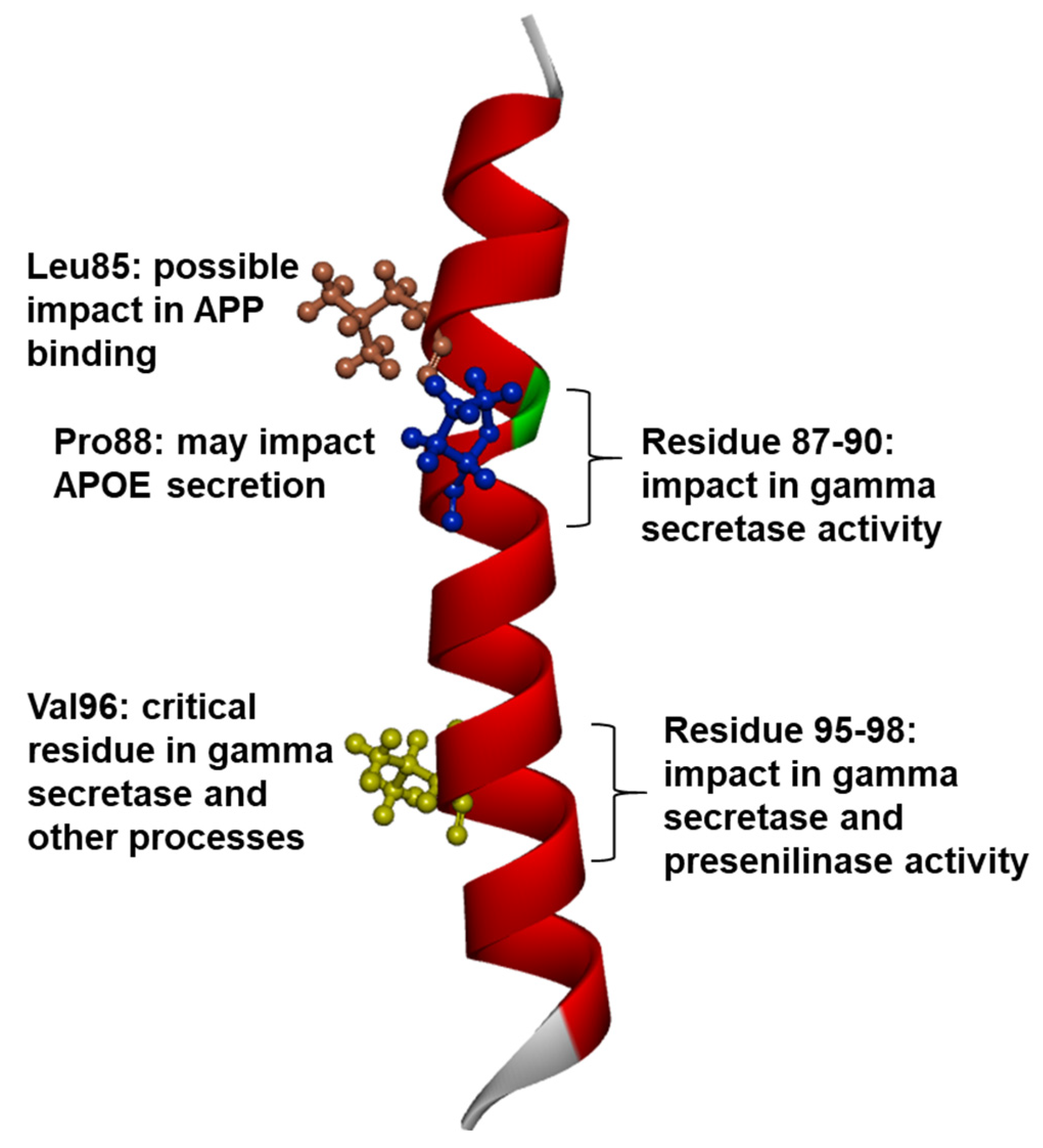{kind=link}
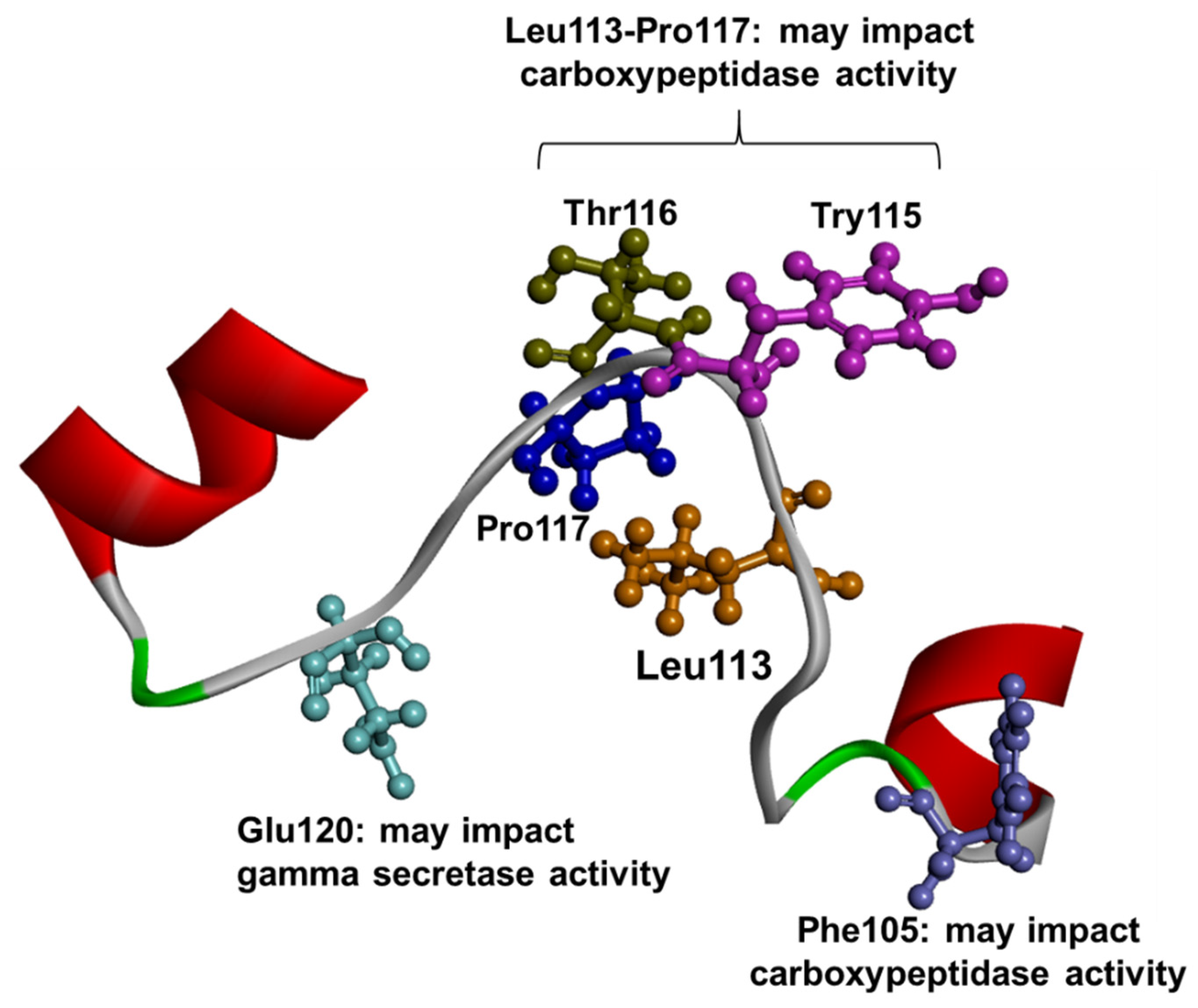{kind=link}
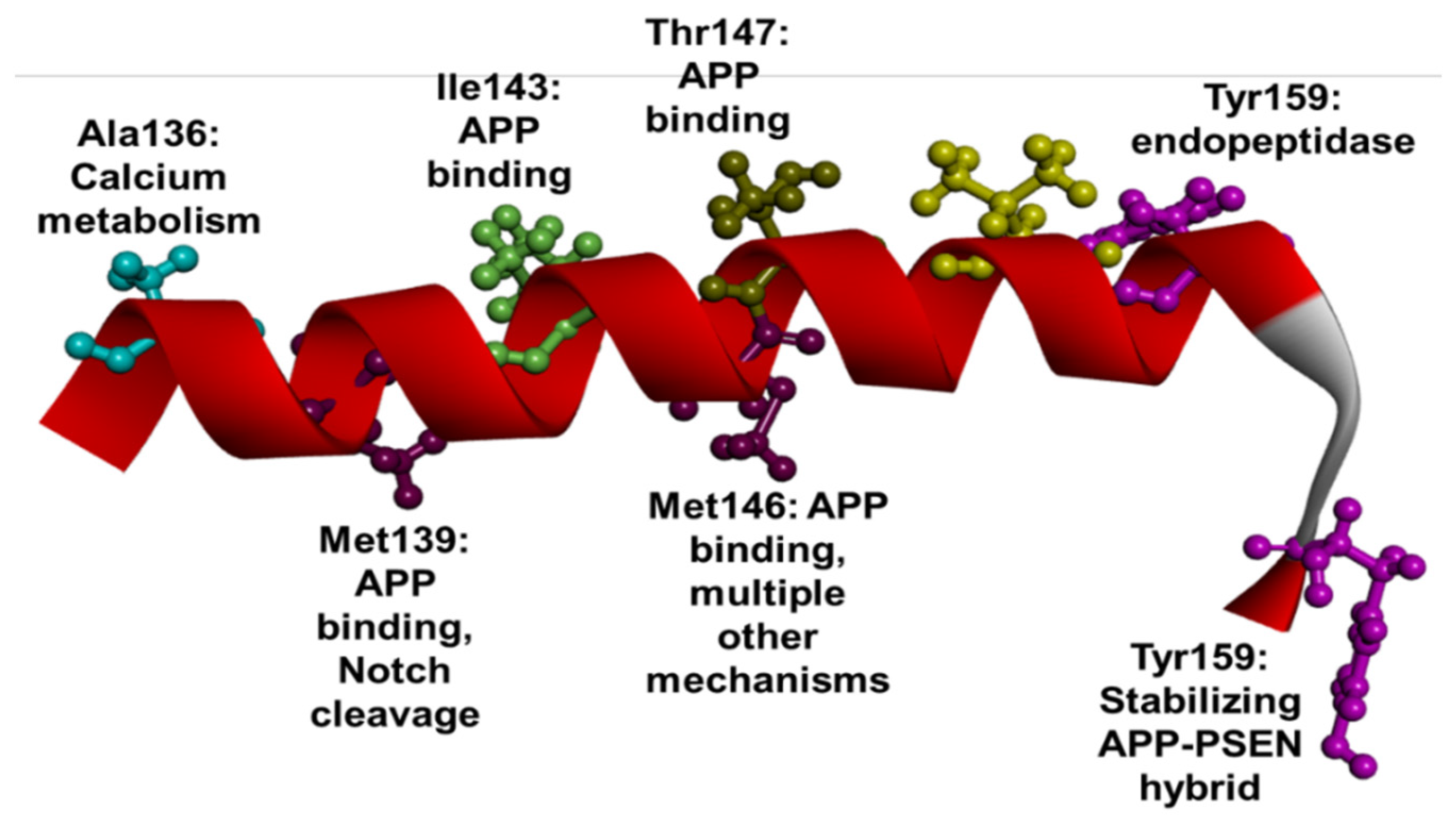{kind=link}
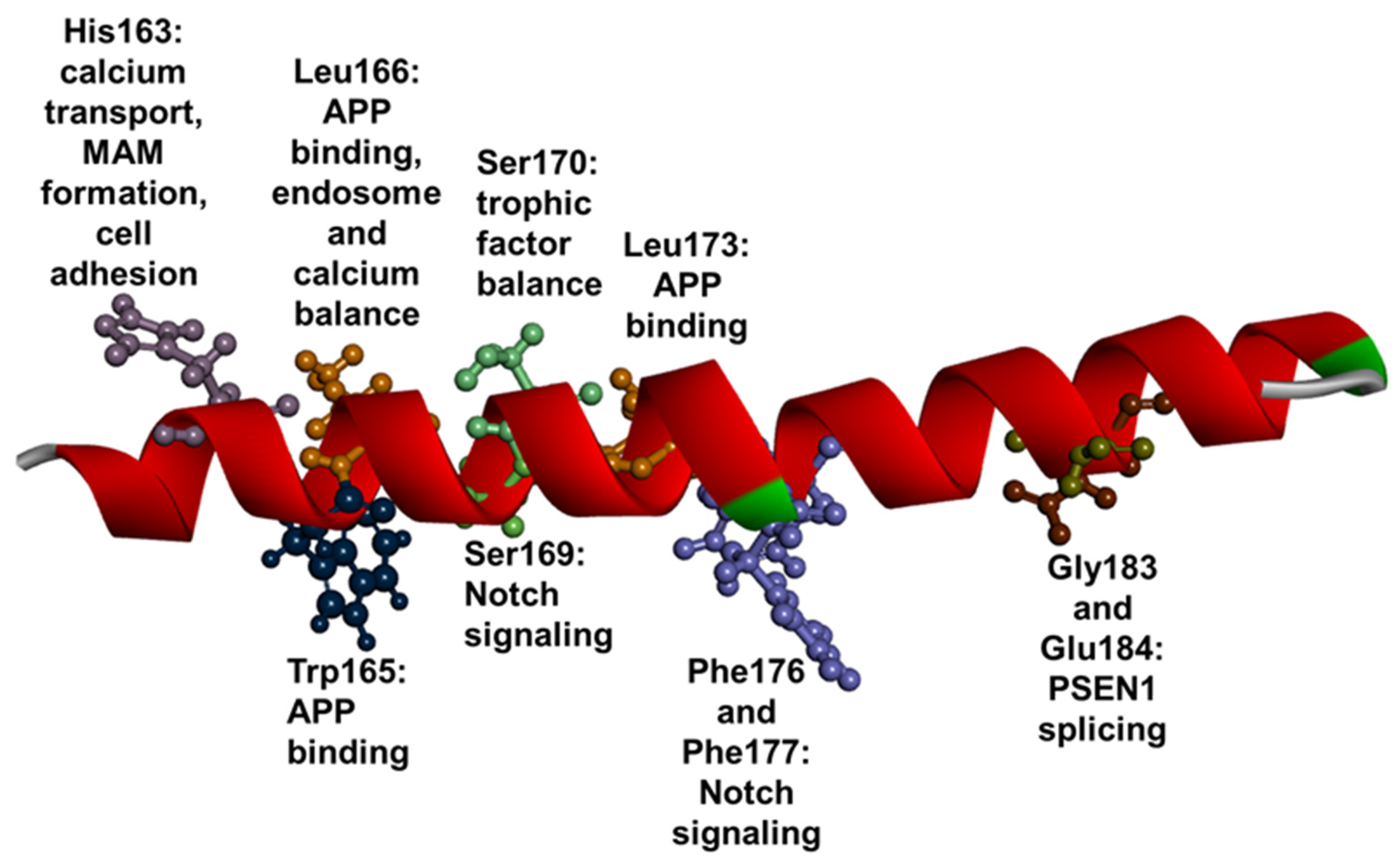{kind=link}
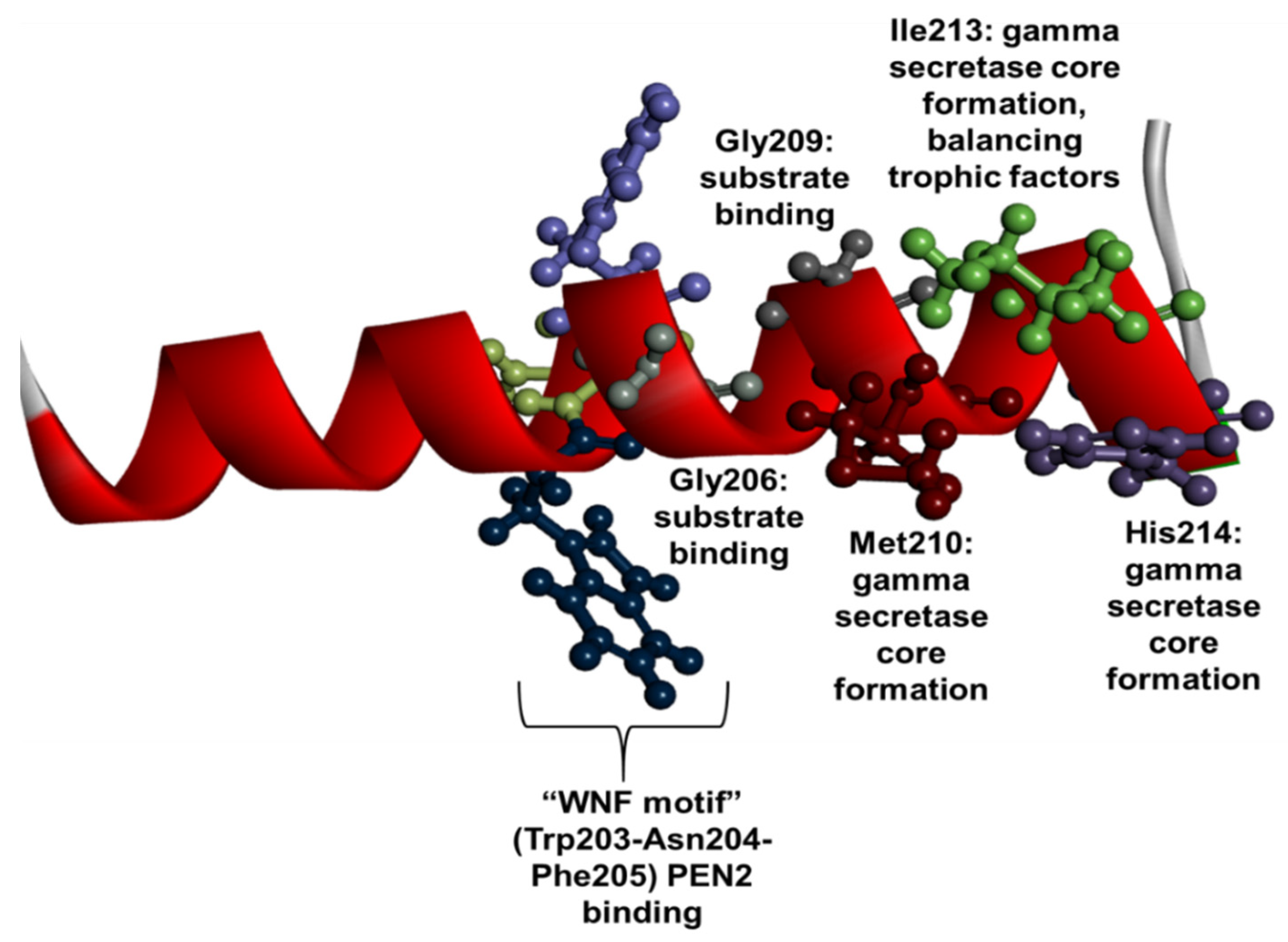{kind=link}
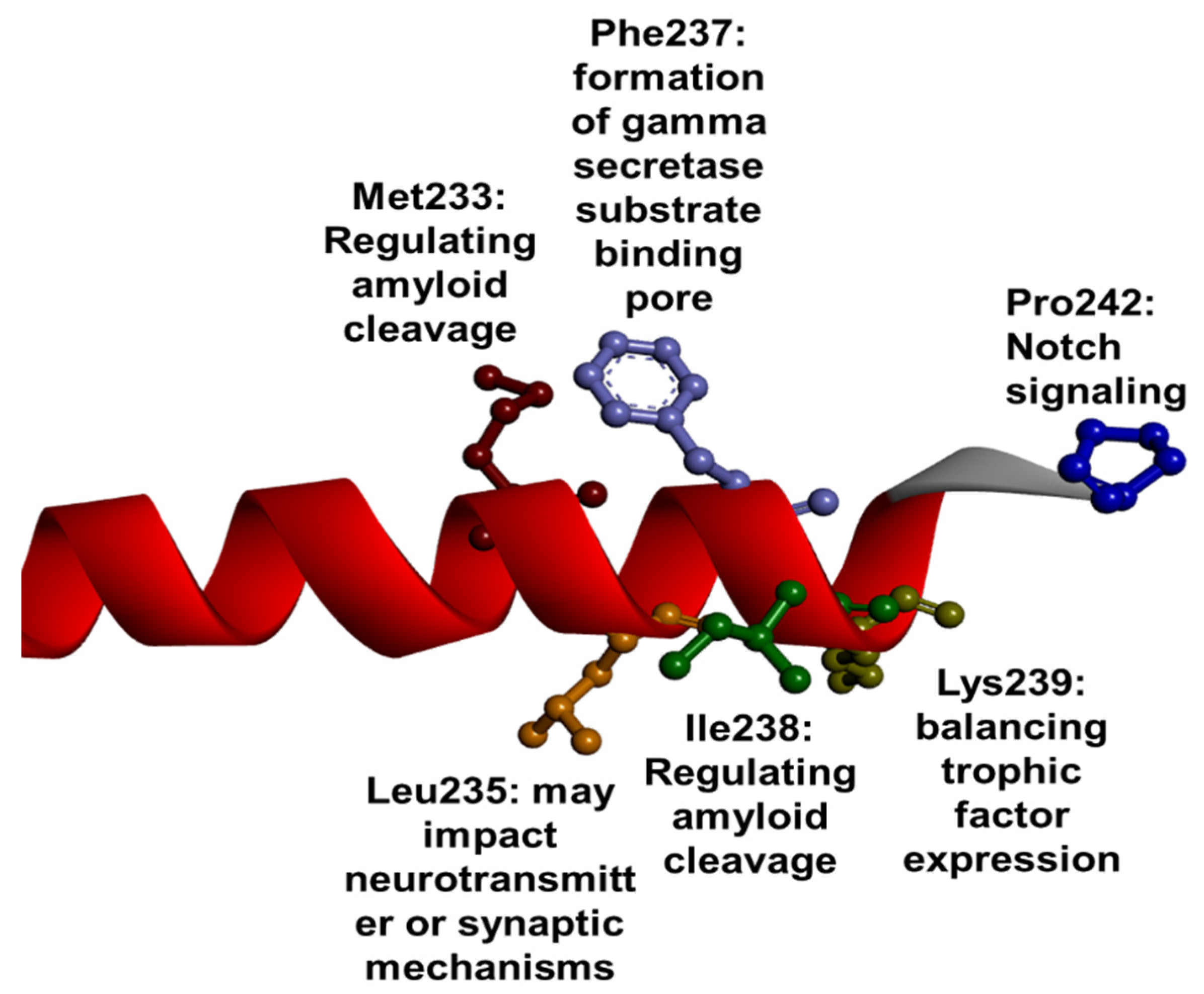{kind=link}
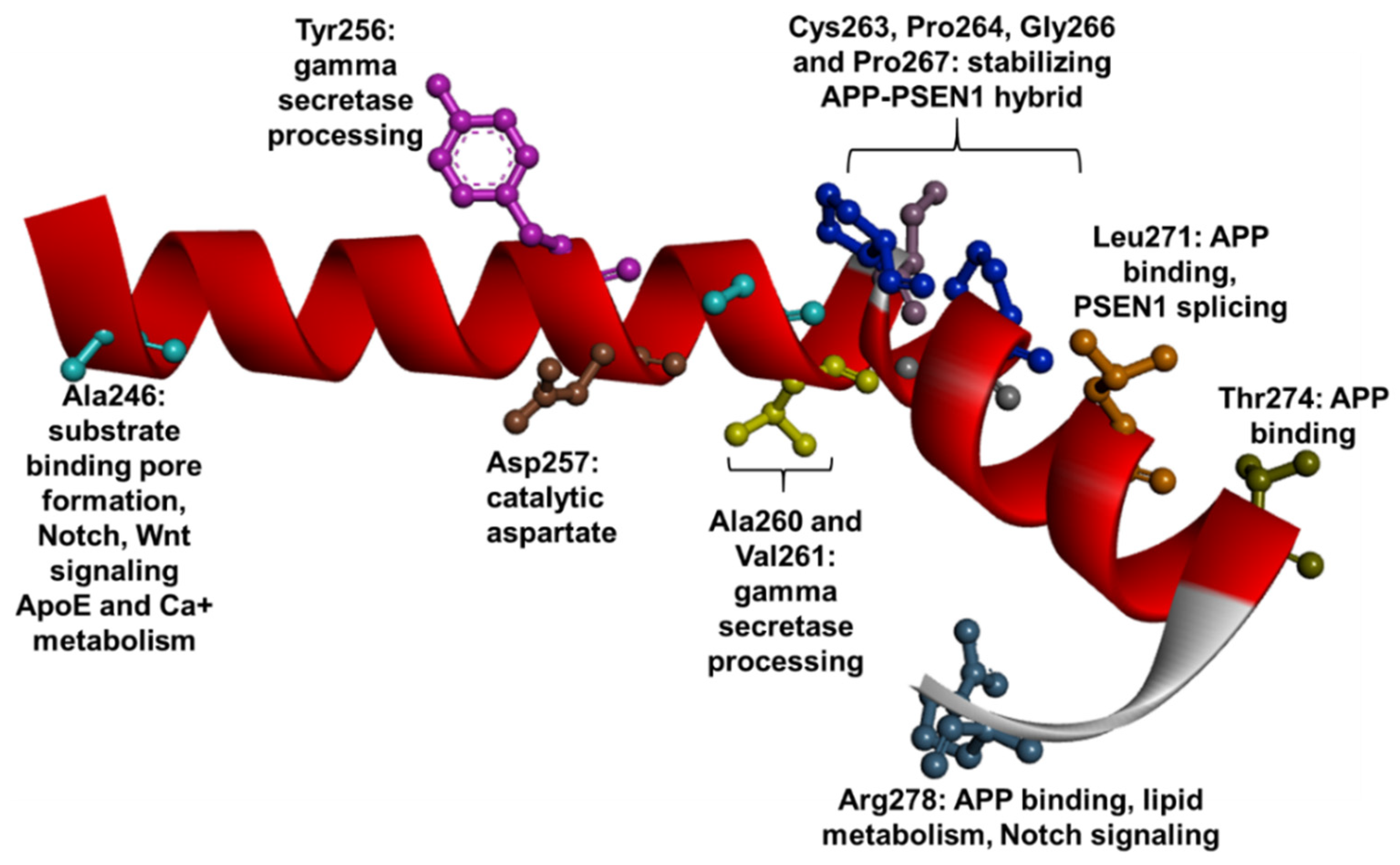{kind=link}
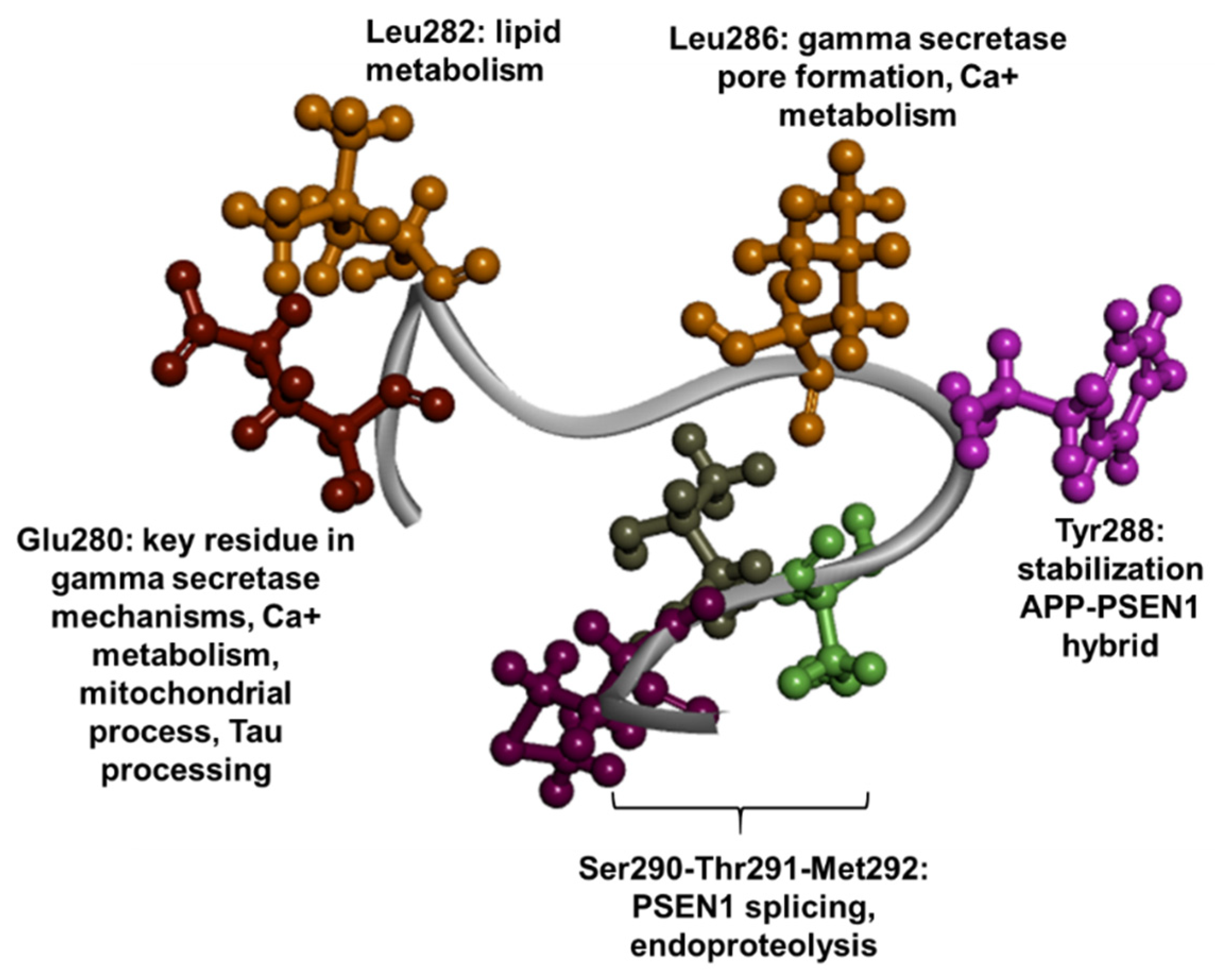{kind=link}
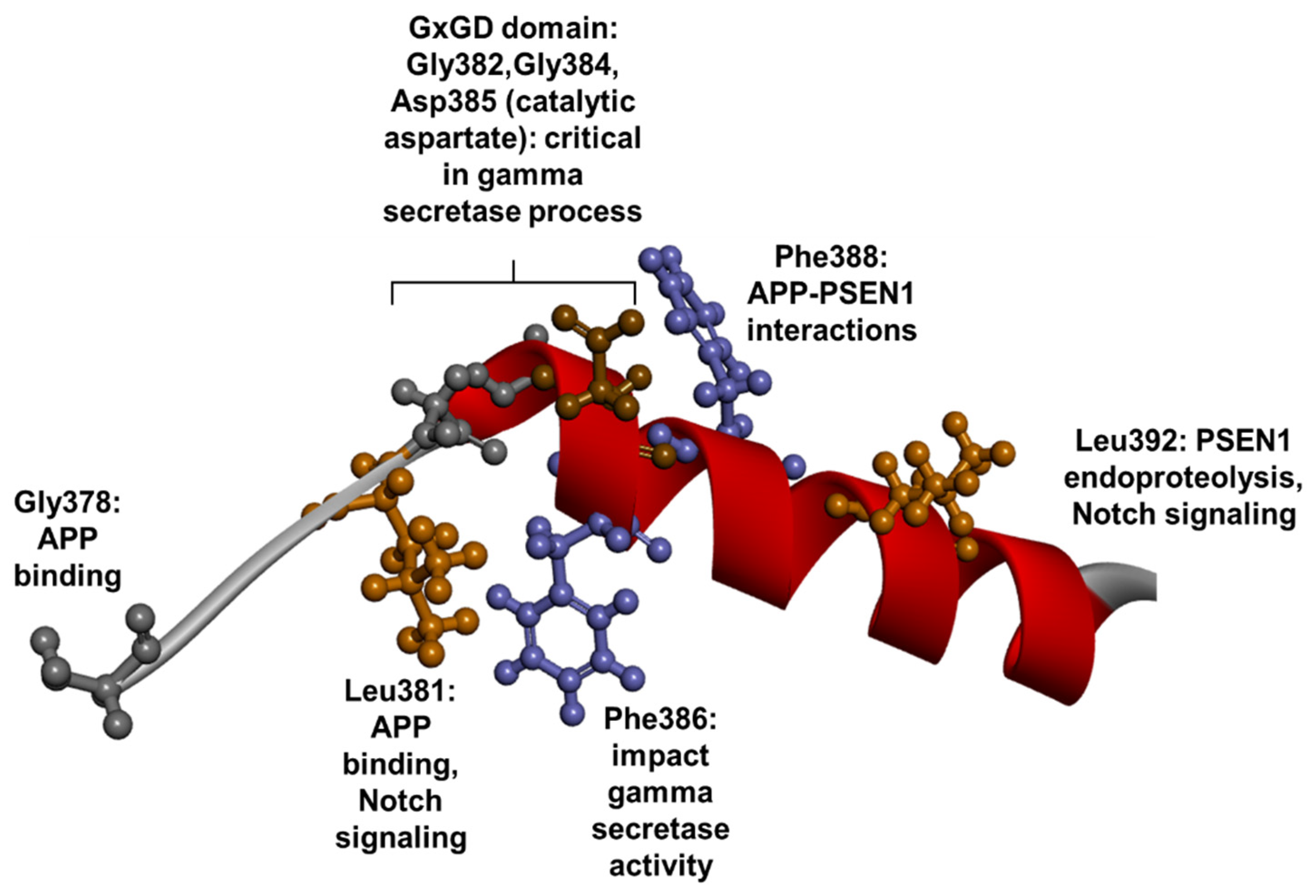{kind=link}
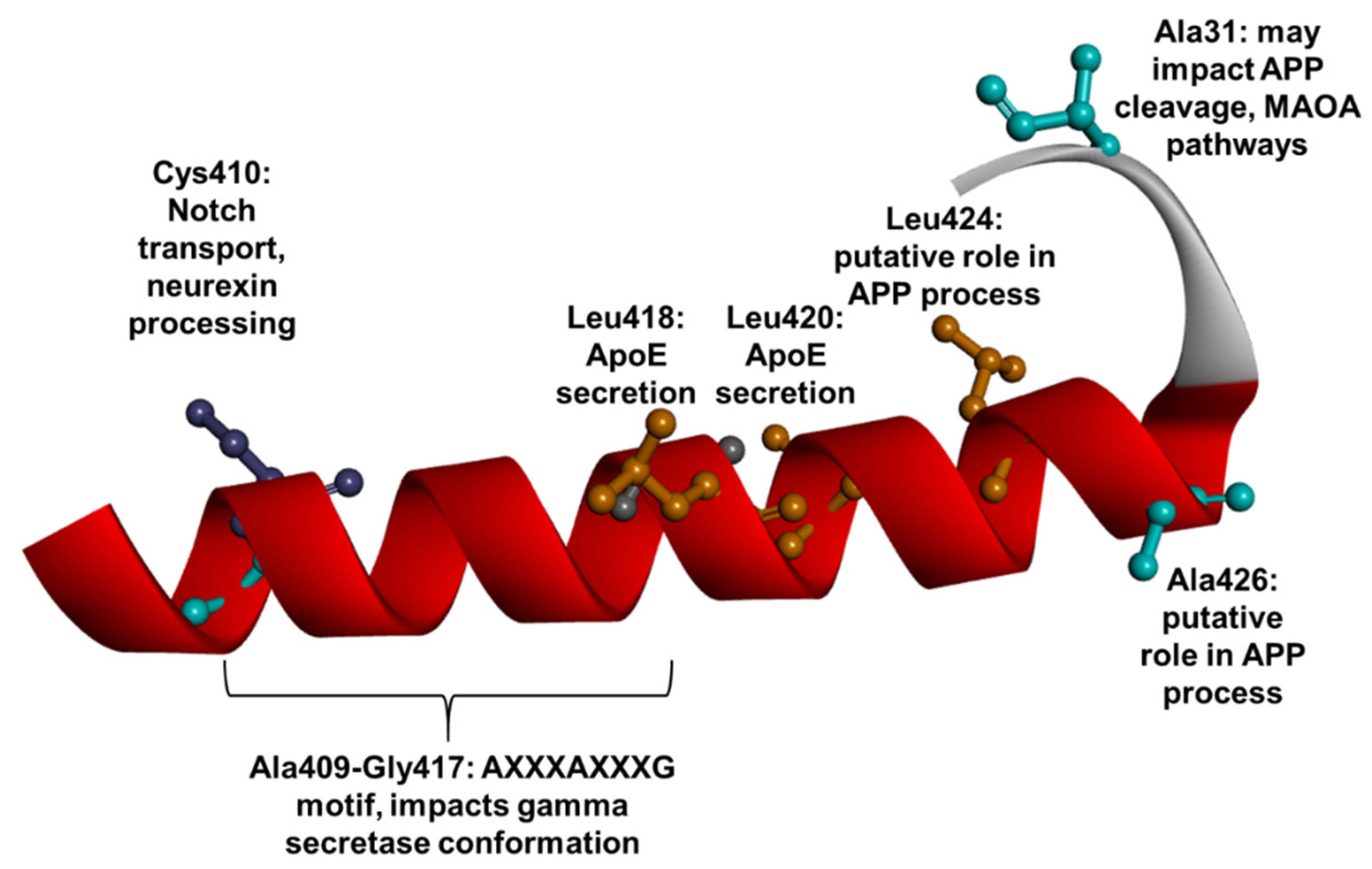{kind=link}
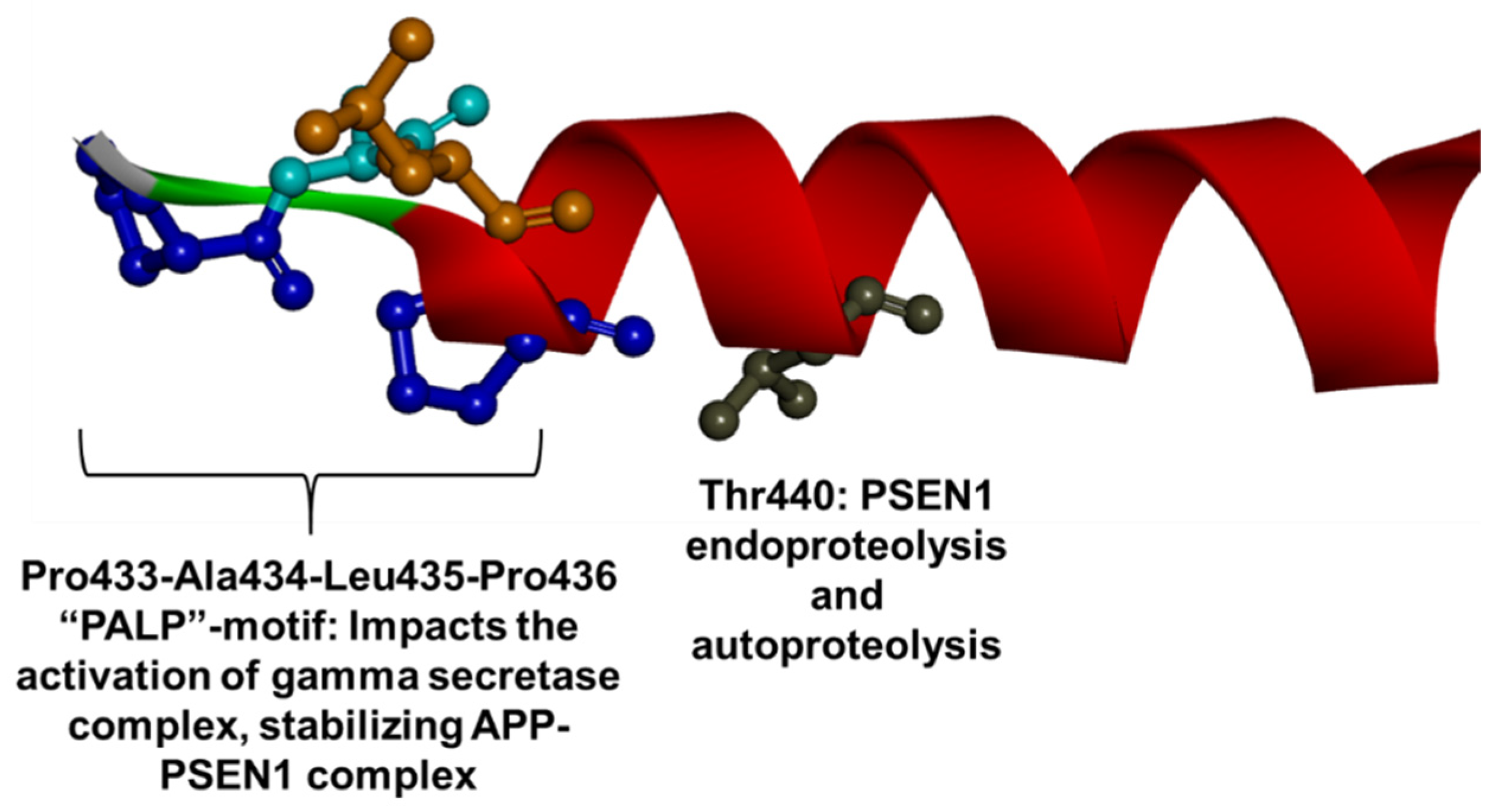{kind=link}
| Criteria | Weighting | Proof |
|---|---|---|
| Pathogenic | Strong |
|
| Moderate |
| |
| Supporting |
| |
| Benign | Supporting |
|
| Strong |
| |
| Protective (?) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene. Int. J. Mol. Sci. 2022, 23, 10970. https://doi.org/10.3390/ijms231810970
Bagaria J, Bagyinszky E, An SSA. Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene. International Journal of Molecular Sciences. 2022; 23(18):10970. https://doi.org/10.3390/ijms231810970
Chicago/Turabian StyleBagaria, Jaya, Eva Bagyinszky, and Seong Soo A. An. 2022. "Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene" International Journal of Molecular Sciences 23, no. 18: 10970. https://doi.org/10.3390/ijms231810970
APA StyleBagaria, J., Bagyinszky, E., & An, S. S. A. (2022). Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene. International Journal of Molecular Sciences, 23(18), 10970. https://doi.org/10.3390/ijms231810970