

The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
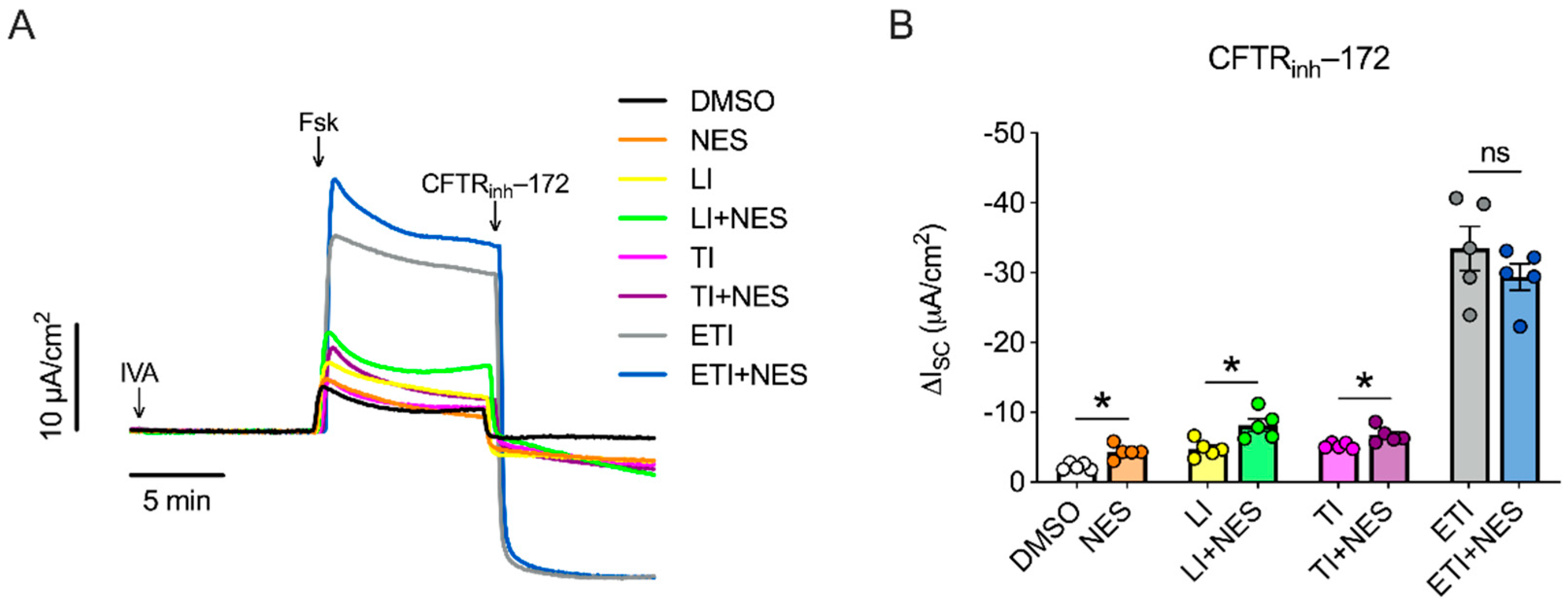
2.1. Nesolicaftor Exerts Differential Effects on Modulator-Corrected CFTR Function in F508del CFTR 16HBEge Cells In Vitro
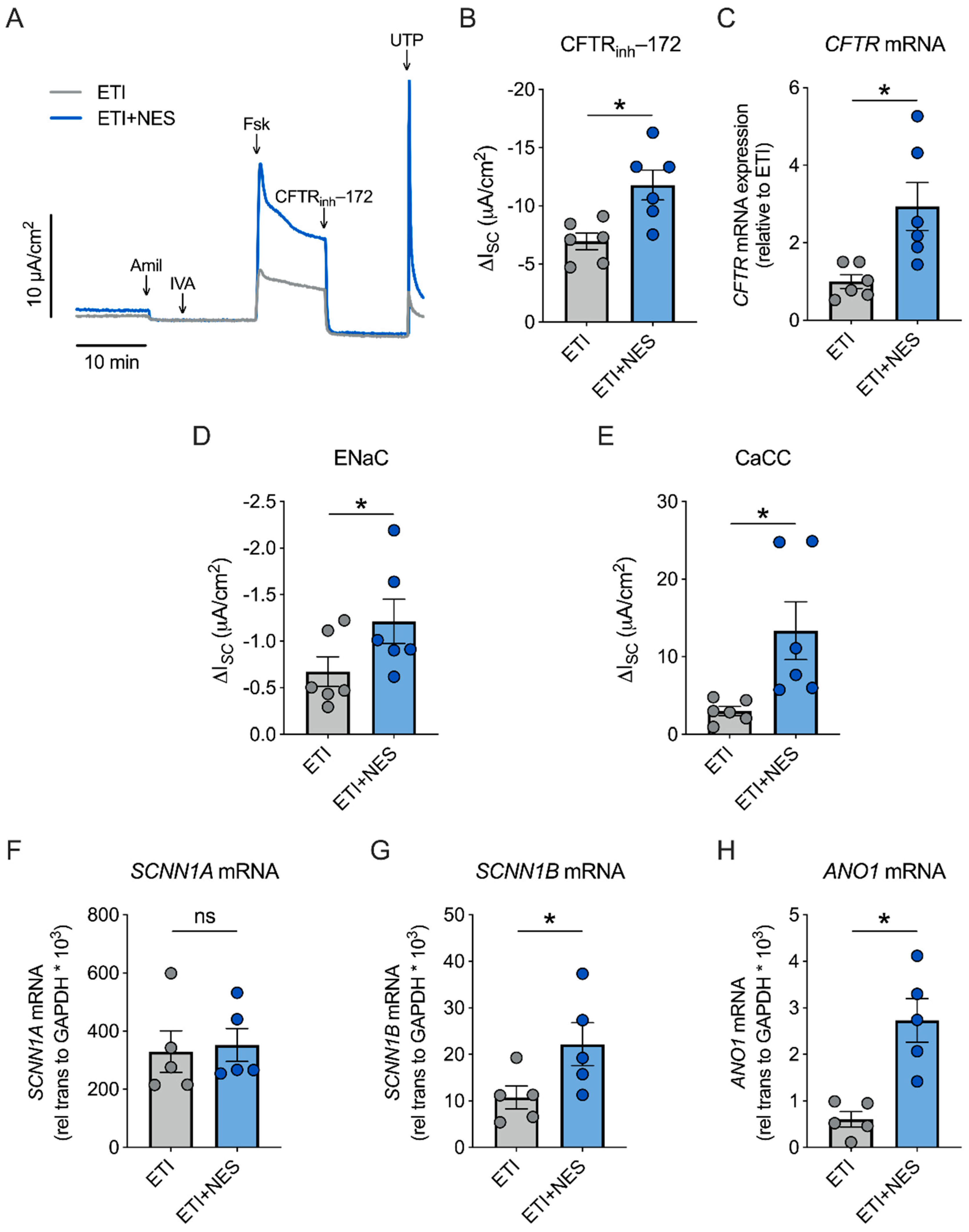
2.2. Nesolicaftor Augments ETI-Corrected F508del CFTR Function in Primary CFBE Cells In Vitro
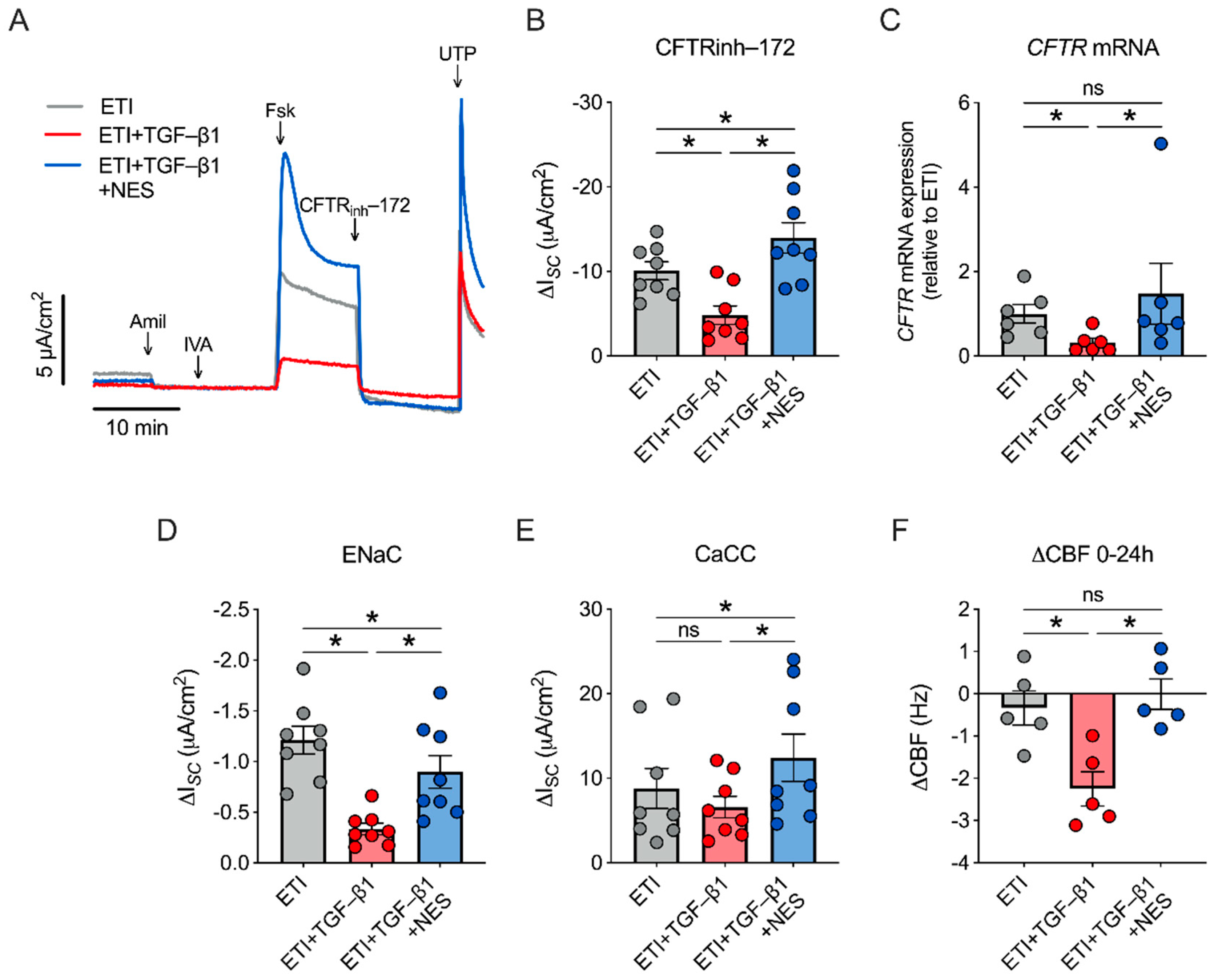
2.3. Nesolicaftor Reverses TGF-β1-Induced Decreases in ETI-Corrected F508del CFTR Function and Ciliary Beating in Primary CFBE Cells In Vitro
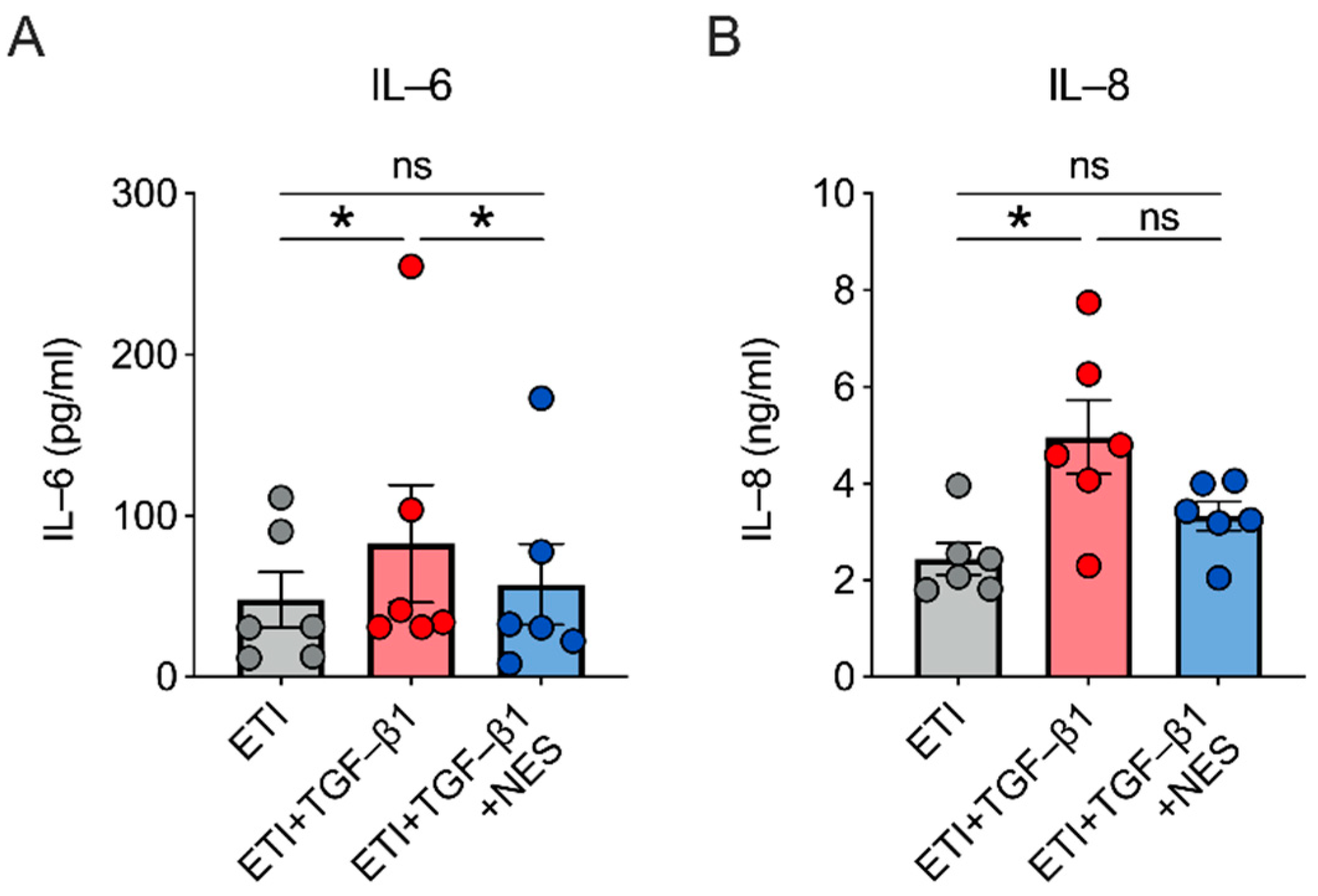
2.4. Nesolicaftor Reverses TGF-β1-Induced Inflammation in ETI-Treated Primary CFBE Cells In Vitro
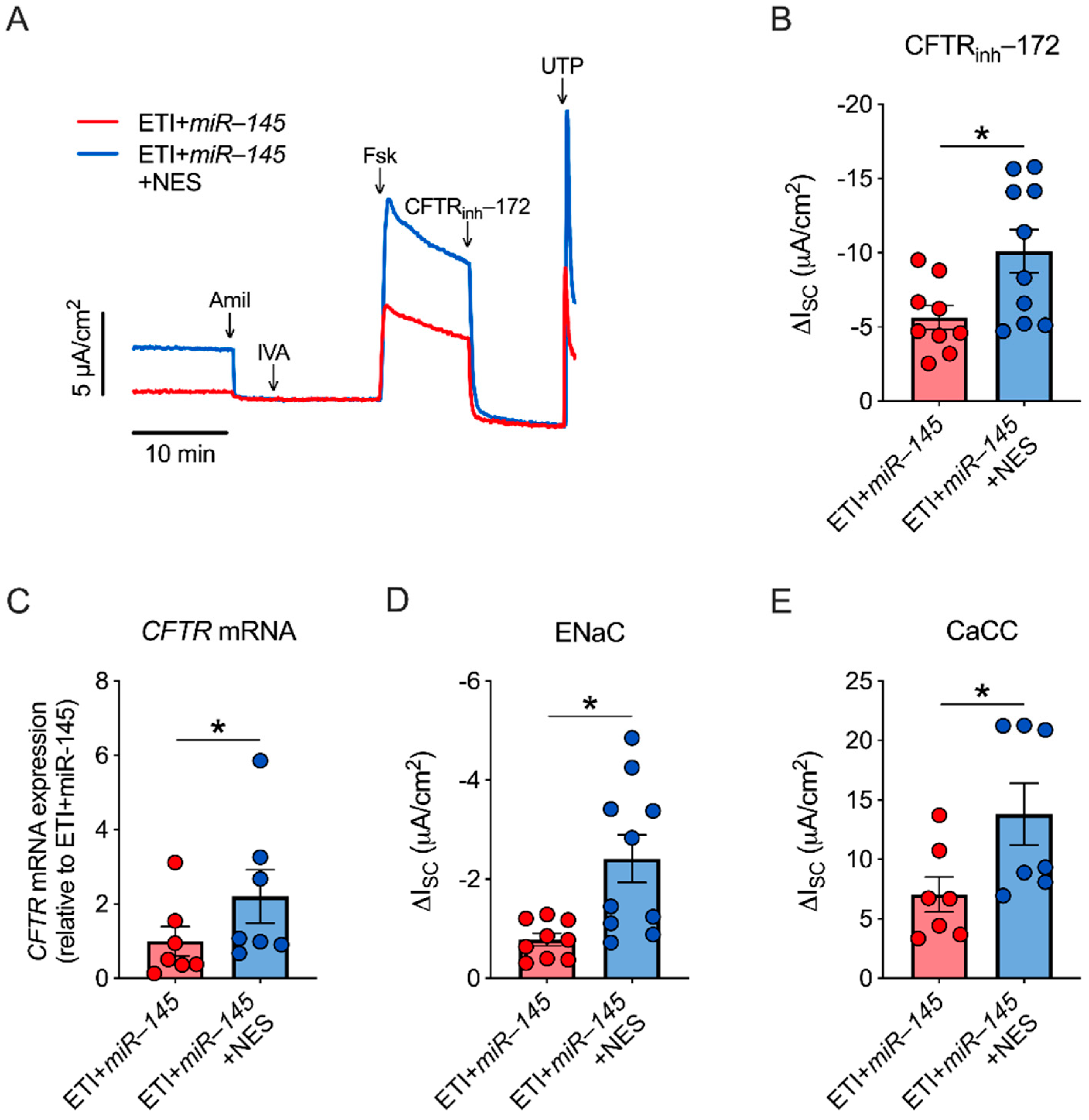
2.5. miR-145 Exposure Mimics TGF-β1 Effects, Which Are Reversed by Nesolicaftor
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Lungs
4.3. Cell Culture
4.4. miR-145 Lentivirus Transduction
4.5. Ussing Chambers
4.6. Ciliary Beat Frequency (CBF)
4.7. Quantitative PCR (qPCR)
4.8. ELISA
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef]
- Manzanares, D.; Gonzalez, C.; Ivonnet, P.; Chen, R.S.; Valencia-Gattas, M.; Conner, G.E.; Larsson, H.P.; Salathe, M. Functional apical large conductance, Ca2+-activated, and voltage-dependent K+ channels are required for maintenance of airway surface liquid volume. J. Biol. Chem. 2011, 286, 19830–19839. [Google Scholar] [CrossRef]
- Perkett, E.A.; Ornatowski, W.; Poschet, J.F.; Deretic, V. Chloroquine normalizes aberrant transforming growth factor beta activity in cystic fibrosis bronchial epithelial cells. Pediatr. Pulmonol. 2006, 41, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, S.M.; Cihil, K.M.; Cornuet, P.K.; Myerburg, M.M.; Swiatecka-Urban, A. Tgf-beta1 inhibits Cftr biogenesis and prevents functional rescue of DeltaF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS ONE 2013, 8, e63167. [Google Scholar] [CrossRef]
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. TGF-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842. [Google Scholar] [CrossRef]
- Fong, C.Y.; Pang, L.; Holland, E.; Knox, A.J. TGF-beta1 stimulates IL-8 release, COX-2 expression, and PGE(2) release in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L201–L207. [Google Scholar] [CrossRef]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Noah, T.L. Transforming growth factor-beta(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1057–1064. [Google Scholar] [CrossRef]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-beta Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef]
- Kim, M.D.; Bengtson, C.D.; Yoshida, M.; Niloy, A.J.; Dennis, J.S.; Baumlin, N.; Salathe, M. Losartan ameliorates TGF-beta1-induced CFTR dysfunction and improves correction by cystic fibrosis modulator therapies. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Kim, M.D.; Baumlin, N.; Yoshida, M.; Polineni, D.; Salathe, S.F.; David, J.K.; Peloquin, C.A.; Wanner, A.; Dennis, J.S.; Sailland, J.; et al. Losartan Rescues Inflammation-related Mucociliary Dysfunction in Relevant Models of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 313–324. [Google Scholar] [CrossRef]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discov. 2018, 23, 111–121. [Google Scholar] [CrossRef]
- Dukovski, D.; Villella, A.; Bastos, C.; King, R.; Finley, D.; Kelly, J.W.; Morimoto, R.I.; Hartl, F.U.; Munoz, B.; Lee, P.S.; et al. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J. Cyst. Fibros. 2020, 19, 733–741. [Google Scholar] [CrossRef]
- Ostareck, D.H.; Ostareck-Lederer, A.; Wilm, M.; Thiele, B.J.; Mann, M.; Hentze, M.W. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell 1997, 89, 597–606. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H.; Hentze, M.W. Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem. Sci. 1998, 23, 409–411. [Google Scholar] [CrossRef]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Venturini, A.; Borrelli, A.; Musante, I.; Scudieri, P.; Capurro, V.; Renda, M.; Pedemonte, N.; Galietta, L.J.V. Comprehensive Analysis of Combinatorial Pharmacological Treatments to Correct Nonsense Mutations in the CFTR Gene. Int. J. Mol. Sci. 2021, 22, 11972. [Google Scholar] [CrossRef] [PubMed]
- Downey, D.G.; Fajac, I.; Flume, P.; O’Carroll, M.; Pressler, T.; Proesmans, M.; Quon, B.; Schwartz, C.; Sutharsen, S.; Jiang, J.; et al. WS11.5 Evaluation of combinations of the CFTR potentiator dirocaftor, corrector posenacaftor and amplifier nesolicaftor in cystic fibrosis subjects with to copies of F508del mutation. In Proceedings of the European Cystic Fibrosis Conference, Lyon, France, 3–6 June 2020. [Google Scholar]
- Valley, H.C.; Bukis, K.M.; Bell, A.; Cheng, Y.; Wong, E.; Jordan, N.J.; Allaire, N.E.; Sivachenko, A.; Liang, F.; Bihler, H.; et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2019, 18, 476–483. [Google Scholar] [CrossRef]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Rehman, T.; Karp, P.H.; Tan, P.; Goodell, B.J.; Pezzulo, A.A.; Thurman, A.L.; Thornell, I.M.; Durfey, S.L.; Duffey, M.E.; Stoltz, D.A.; et al. Inflammatory cytokines TNFalpha and IL-17 enhance the efficacy of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Investig. 2021, 131, e150398. [Google Scholar] [CrossRef]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Martino, M.E.B.; Minges, J.T.; Boyles, S.E.; Guhr Lee, T.N.; Esther, C.R., Jr.; Ribeiro, C.M.P. Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies. Front. Pharmacol. 2021, 12, 628722. [Google Scholar] [CrossRef]
- Flume, P.; Sawicki, G.; Pressler, T.; Schwarz, C.; Fajac, I.; Layish, D.; Bialek, P.; Wilson, S.; Kang, L.; Mclaughlin, B.; et al. WS01.2 Phase 2 initial results evaluating PTI-428, a novel CFTR amplifier, in patients with cystic fibrosis. J. Cyst. Fibros. 2018, 17, S1–S2. [Google Scholar] [CrossRef]
- Makeyev, A.V.; Liebhaber, S.A. The poly(C)-binding proteins: A multiplicity of functions and a search for mechanisms. RNA 2002, 8, 265–278. [Google Scholar] [CrossRef]
- Danahay, H.; Gosling, M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int. J. Mol. Sci. 2020, 21, 2386. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Kumar, C.; Bohl, S.; Klingmueller, U.; Mann, M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell Proteomics. 2009, 8, 443–450. [Google Scholar] [CrossRef]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575, 577–578, 581–572 passim. [Google Scholar] [CrossRef]
- Randell, S.H.; Fulcher, M.L.; O'Neal, W.; Olsen, J.C. Primary epithelial cell models for cystic fibrosis research. Methods Mol. Biol. 2011, 742, 285–310. [Google Scholar] [CrossRef]
- Bengtson, C.D.; Kim, M.D.; Anabtawi, A.; He, J.; Dennis, J.S.; Miller, S.; Yoshida, M.; Baumlin, N.; Salathe, M. Hyperglycaemia in CF adversely affects BK channel function critical for mucus clearance. Eur. Respir. J. 2020, 2000509. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206. [Google Scholar]
- Salathe, M.; Bookman, R.J. Mode of Ca2+ action on ciliary beat frequency in single ovine airway epithelial cells. J. Physiol. 1999, 520, 851–865. [Google Scholar] [CrossRef]
- Sisson, J.H.; Stoner, J.A.; Ammons, B.A.; Wyatt, T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003, 211, 103–111. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bengtson, C.; Silswal, N.; Baumlin, N.; Yoshida, M.; Dennis, J.; Yerrathota, S.; Kim, M.; Salathe, M. The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function. Int. J. Mol. Sci. 2022, 23, 10956. https://doi.org/10.3390/ijms231810956
Bengtson C, Silswal N, Baumlin N, Yoshida M, Dennis J, Yerrathota S, Kim M, Salathe M. The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function. International Journal of Molecular Sciences. 2022; 23(18):10956. https://doi.org/10.3390/ijms231810956
Chicago/Turabian StyleBengtson, Charles, Neerupma Silswal, Nathalie Baumlin, Makoto Yoshida, John Dennis, Sireesha Yerrathota, Michael Kim, and Matthias Salathe. 2022. "The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function" International Journal of Molecular Sciences 23, no. 18: 10956. https://doi.org/10.3390/ijms231810956
APA StyleBengtson, C., Silswal, N., Baumlin, N., Yoshida, M., Dennis, J., Yerrathota, S., Kim, M., & Salathe, M. (2022). The CFTR Amplifier Nesolicaftor Rescues TGF-β1 Inhibition of Modulator-Corrected F508del CFTR Function. International Journal of Molecular Sciences, 23(18), 10956. https://doi.org/10.3390/ijms231810956