

Fluorine Atoms on C6H5-Corrole Affect the Interaction with Mpro and PLpro Proteases of SARS-CoV-2: Molecular Docking and 2D-QSAR Approaches

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
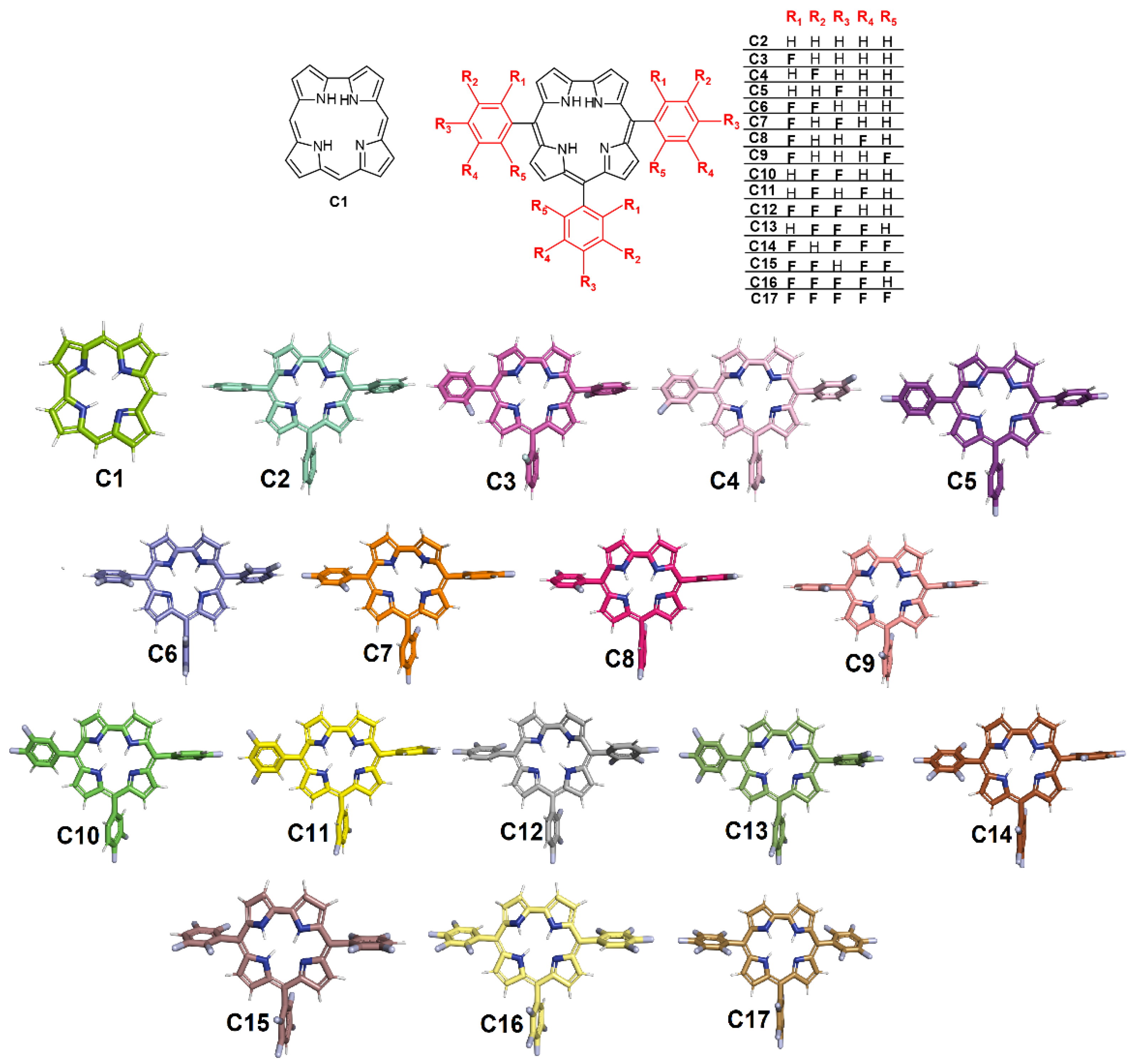
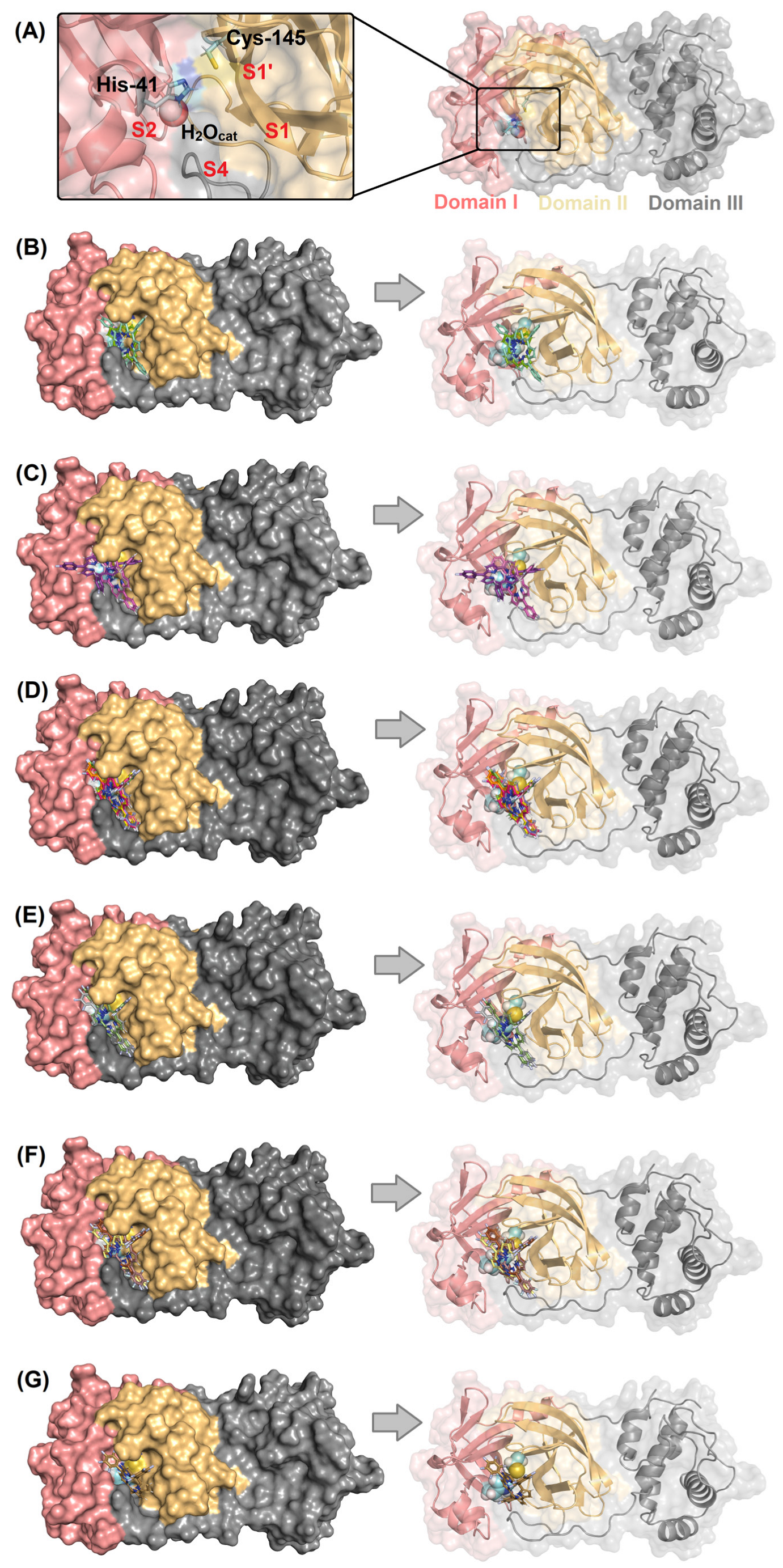
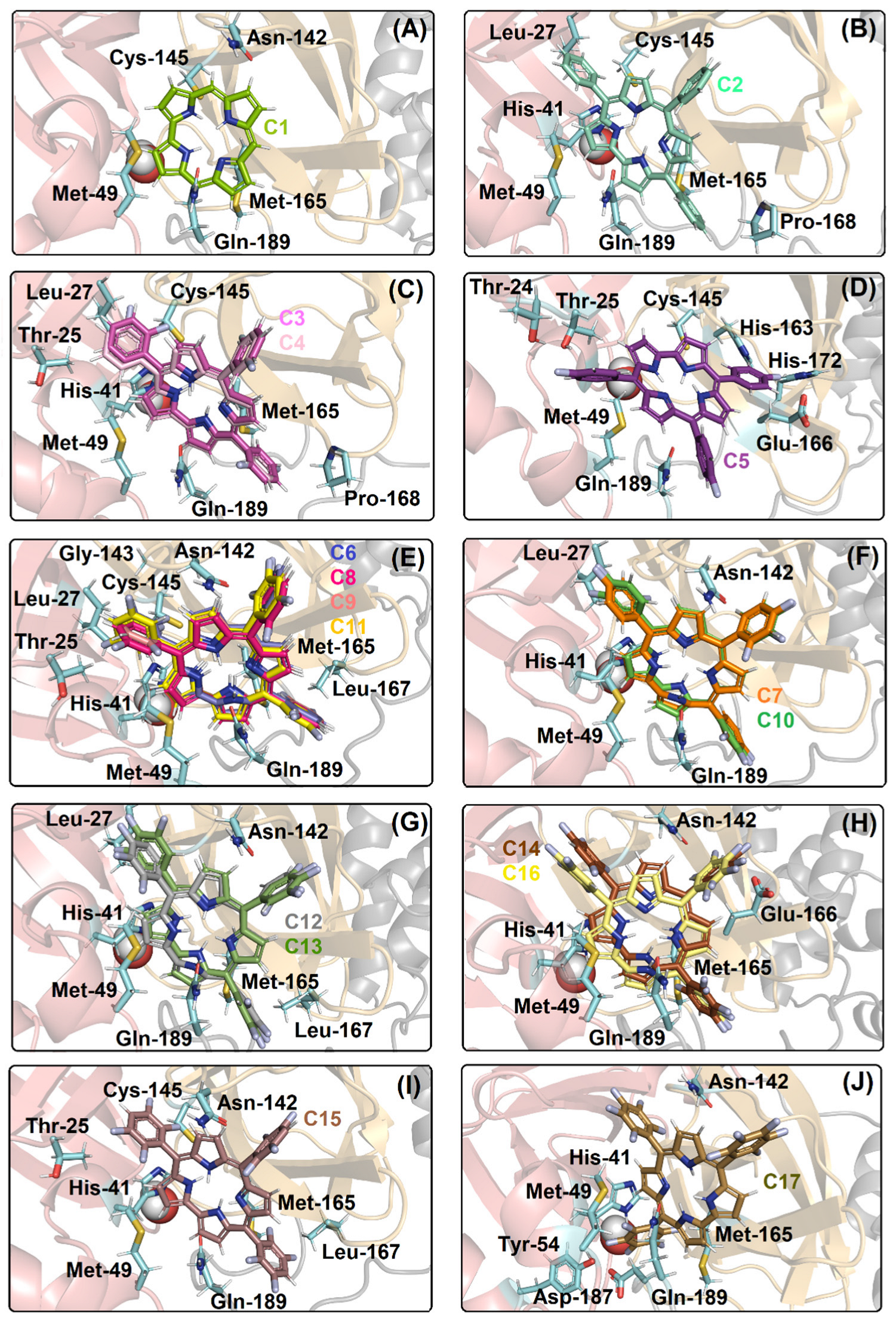
2.1. Molecular Docking Evaluation for Mpro Enzyme
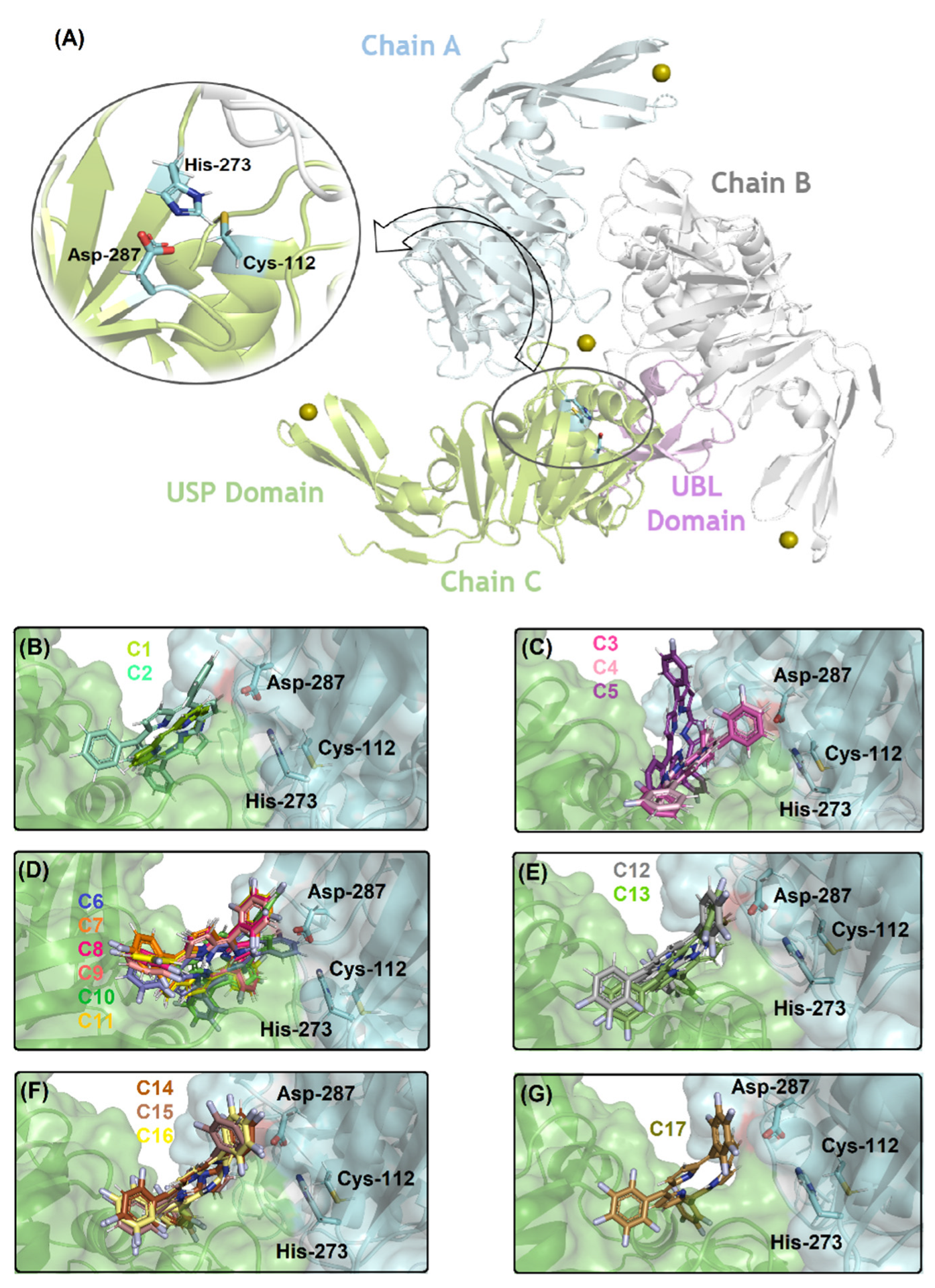
2.2. Molecular Docking Evaluation for PLpro Enzyme
2.3. 2D Quantitative Structure–Activity Relationship (2D-QSAR) Model
3. Discussion
4. Materials and Methods
4.1. In Silico Calculations Procedure
4.2. The 2D-QSAR Procedure
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dos Santos, W.G. Natural history of COVID-19 and current knowledge on treatment therapeutic options. Biomed. Pharmacother. 2020, 129, 110493. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 28 August 2022).
- Liu, X.-H.; Zhang, X.; Lu, Z.-H.; Zhu, Y.-S.; Wang, T. Potential molecular targets of nonstructural proteins for the development of antiviral drugs against SARS-CoV-2 infection. Biomed. Phamacother. 2021, 133, 111035. [Google Scholar] [CrossRef] [PubMed]
- Chaves, O.A.; Fintelman-Rodrigues, N.; Wang, X.; Sacramento, C.Q.; Temerozo, J.R.; Ferreira, A.C.; Mattos, M.; Pereira-Dutra, F.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Commercially Available Flavonols Are Better SARS-CoV-2 Inhibitors than Isoflavone and Flavones. Viruses 2022, 14, 1458. [Google Scholar] [CrossRef]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Dalmat, Y.-M. SARS-CoV-2 histoire de mutants. Option/Bio 2021, 32, 10. [Google Scholar] [CrossRef]
- Sharma, T.; Abohashrh, M.; Baig, M.H.; Dong, J.-J.; Alam, M.M.; Ahmad, I.; Irfan, S. Screening of drug databank against WT and mutant main protease of SARS-CoV-2: Towards finding potential compound for repurposing against COVID-19. Saudi J. Biol. Sci. 2021, 28, 3152–3159. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus (COVID-19)|Drugs. Available online: https://www.fda.gov/drugs/emergency-preparedness-drugs/coronaviruscovid-19-drugs (accessed on 15 August 2022).
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of COVID-19-Final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Rangsinth, P.; Sillapachaiyaporn, C.; Nilkhet, S.; Tencomnao, T.; Ung, A.T.; Chuchawankul, S. Mushroom-derived bioactive compounds potentially serve as the inhibitors of SARS-CoV-2 main protease: An in silico approach. J. Tradit. Complement. Med. 2021, 11, 158–172. [Google Scholar] [CrossRef]
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Fintelman-Rodrigues, N.; Sacramento, C.Q.; Lima, C.R.; da Silva, F.S.; Ferreira, A.C.; Mattos, M.; de Freitas, C.S.; Soares, V.C.; Dias, S.S.G.; Temerozo, J.R.; et al. Atazanavir, alone or in combination with ritonavir, inhibits SARS-CoV-2 replication and proinflammatory cytokine production. Antimicrob. Agents Chemother. 2020, 64, e00825-20. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLPro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Chaves, O.A.; Sacramento, C.Q.; Ferreira, A.C.; Mattos, M.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Vazquez, L.; Pinto, D.P.; Silveira, G.P.E.; Fonseca, L.B.; et al. Atazanavir is a competitive inhibitor of SARS-CoV-2 Mpro, impairing variants replication in vitro and in vivo. Pharmaceuticals 2022, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Chaves, O.A.; Sacramento, C.Q.; Fintelman-Rodruigues, N.; Temerozo, J.R.; Pereira-Dutra, F.; Mizurini, D.M.; Monteiro, R.Q.; Vazquez, L.; Bozza, P.T.; Castro-Faria-Neto, H.C.; et al. Apixaban, an orally available anticoagulant, inhibits SARS-CoV-2 replication and its major protease in a non-competitive way. J. Mol. Cell Biol. 2022, 6, mjac039. [Google Scholar] [CrossRef] [PubMed]
- Santos-Filho, O.A. Identification of potential inhibitors of severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) main protease from non-natural and natural sources: A molecular docking study. J. Braz. Chem. Soc. 2020, 31, 2638–2643. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Rajpoot, S.; Alagumuthu, M.; Baig, M.S. Dual targeting of 3CLpro and PLpro of SARS-CoV-2: A novel structure-base design approach to treat COVID-19. Curr. Res. Struct. Biol. 2021, 3, 9–18. [Google Scholar] [CrossRef]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 proteases for COVID-19 antiviral development. Front. Chem. 2022, 9, 819165. [Google Scholar] [CrossRef]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef]
- Pfizer’s Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk of Hospitalization or Death by 89% in Interim Analysis of Phase 2/3 Epic-HR Study. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate (accessed on 15 August 2022).
- Ahmad, B.; Batool, M.; Ain, Q.U.; Kim, M.S.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef]
- Acunha, T.V.; Chaves, O.A.; Iglesias, B.A. Fluorescent pyrene moiety in fluorinated C6F5-corroles increases the interaction with HSA and CT-DNA. J. Porphyr. Phthalocyanines 2021, 25, 75–94. [Google Scholar] [CrossRef]
- Ding, T.; Alemán, E.A.; Modarelli, D.A.; Ziegler, C.J. Photophysical properties of a series of free-base corroles. J. Phys. Chem. A 2005, 109, 7411–7417. [Google Scholar] [CrossRef] [PubMed]
- Ventura, B.; Esposti, A.D.; Koszarna, B.; Gryko, D.T.; Flamigni, L. Photophysical characterization of free-base corroles, promising chromophores for light energy conversion and singlet oxygen generation. New J. Chem. 2005, 29, 1559–1566. [Google Scholar] [CrossRef]
- Acunha, T.V.; Matiuzzi, B.R.; Silva, J.A.; Galindo, D.D.M.; Chaves, O.A.; Rocha, V.N.; Piquini, P.C.; Kohler, M.H.; Boni, L.; Iglesias, B.A. Unveiling the photophysical, biomolecule binding and photo-oxidative capacity of novel Ru(II)-polypyridyl corroles: A multipronged approach. J. Mol. Liq. 2021, 340, 117223. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, H.-J.; Huang, H.; Wang, H.-H.; Wu, S.; Liu, H.-Y.; Zhang, H.-T. The photodynamic activity and toxicity evaluation of 5,10,15-tris(ethoxylcarbonyl)corrole phosphorus(V) in vivo and in vitro. Eur. J. Med. Chem. 2019, 163, 779–786. [Google Scholar] [CrossRef]
- Bornhuetter, T.; Shamali, N.; Saltsman, I.; Mahammed, A.; Gross, Z.; Daeschlein, G.; Roeder, B. Singlet oxygen luminescence kinetics under PDI relevant conditions of pathogenic dermatophytes and molds. J. Photochem. Photobiol. B 2018, 178, 606–613. [Google Scholar] [CrossRef]
- Goslinskia, T.; Piskorz, J. Fluorinated porphyrinoids and their biomedical applications. J. Photochem. Photobiol. C 2011, 12, 304–321. [Google Scholar] [CrossRef]
- Teo, R.D.; Hwang, J.Y.; Termini, J.; Gross, Z.; Gray, H.B. Fighting cancer with corroles. Chem. Rev. 2017, 117, 2711–2729. [Google Scholar] [CrossRef]
- Aviezer, D.; Cotton, S.; David, M.; Segev, A.; Khaselev, N.; Galili, N.; Gross, Z.; Yayon, A. Porphyrin analogues as novel antagonists of fibroblast growth factor and vascular endothelial growth factor receptor binding that inhibit endothelial cell proliferation, tumor progression, and metastasis. Cancer Res. 2000, 60, 2973–2980. [Google Scholar]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef]
- Gu, C.; Wu, Y.; Guo, H.; Zhu, Y.; Xu, W.; Wang, Y.; Zhou, Y.; Sun, Z.; Cai, X.; Li, Y.; et al. Protoporphyrin IX and verteporfin potently inhibit SARS-CoV-2 infection in vitro and in a mouse model expressing human ACE2. Sci. Bull. 2021, 66, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Kipshidze, N.; Yeo, N.; Kipshidze, N. Photodynamic therapy for COVID-19. Nat. Photonics 2020, 14, 651–652. [Google Scholar] [CrossRef]
- Conrado, P.C.V.; Sakita, K.M.; Arita, G.S.; Galinari, C.B.; Gonçalves, R.S.; Lopes, L.D.G.; Lonardoni, M.V.C.; Teixeira, J.J.V.; Bonfim-Mendonça, P.S.; Kioshima, E.S. A systematic review of photodynamic therapy as an antiviral treatment: Potential guidance for dealing with SARS-CoV-2. Photodiagnosis Photodyn. Ther. 2021, 34, 102221. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Chaves, O.A.; Barros, L.S.; Oliveira, M.C.C.; Sant’Anna, C.M.R.; Ferreira, A.B.B.; Silva, F.A.; Cesarin-Sobrinho, D.; Netto-Ferreira, J.C. Biological interactions of fluorinated chalcones: Stimulation of tyrosinase activity and binding to bovine serum albumin. J. Fluor. Chem. 2017, 199, 30–38. [Google Scholar] [CrossRef]
- Chaves, O.A.; Oliveira, C.H.C.S.; Ferreira, R.C.; Melos, J.L.R.; Rodrigues-Santos, C.E.; Echevarria, A.; Cesarin-Sobrinho, D. Investigation of interaction between human plasmatic albumin and potential fluorinated anti-trypanosomal drugs. J. Fluor. Chem. 2017, 199, 103–112. [Google Scholar] [CrossRef]
- Spessard, G.O. ACD Labs/LogP dB 3.5 and ChemSketch 3.5. J. Chem. Inf. Comput. Sci. 1998, 19, 1250–1253. [Google Scholar] [CrossRef]
- de Oliveira, D.B.; Gaudio, A.C. BuildQSAR: A new computer program for QSAR analysis. Mol. Inform. 2001, 19, 599–601. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef]
- Gaudio, A.C.; Zandonade, E. Proposição, validação e análise dos modelos que correlacionam estrutura química e atividade biológica. Quim. Nova 2001, 24, 658–671. [Google Scholar] [CrossRef]
- Muhammad, I.A.; Muangchoo, K.; Muhammad, A.; Ajingi, Y.S.; Muhammad, I.Y.; Umar, I.D.; Muhammad, A.B. A Computational study to identify potential inhibitors of SARS-CoV-2 main protease (Mpro) from Eucalyptus active compounds. Computation 2020, 8, 79. [Google Scholar] [CrossRef]
- Debia, N.P.; Rodríguez, J.J.P.; da Silveira, C.H.; Chaves, O.A.; Iglesias, B.A.; Rodembusch, F.S.; Lüdtke, D.S. Synthesis and photophysics of benzazole based triazoles with amino acid-derived pendant units. Multiparametric optical sensors for BSA and CT-DNA in solution. J. Mol. Liq. 2020, 309, 113092. [Google Scholar] [CrossRef]
- Bombaça, A.C.S.; Silva, L.A.; Chaves, O.A.; da Silva, L.S.; Barbosa, J.M.C.; da Silva, A.M.; Ferreira, A.B.B.; Menna-Barreto, R.F.S. Novel N,N-di-alkylnaphthoimidazolium derivative of β-lapachone impaired Trypanosoma cruzi mitochondrial electron transport system. Biomed. Pharmacother. 2021, 135, 111186. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CL pro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Comm. 2020, 11, 3202. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Worrall, L.J.; Vuckovic, M.; Rosell, F.I.; Gentile, F.; Ton, A.-T.; Caveney, N.A.; Ban, F.; Cherkasov, A.; Paetzel, M.; et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020, 11, 5877. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Guzman, C.A.; Ruiz-Pernía, J.J.; Tuñon, I. Unraveling the SARS-CoV-2 main protease mechanism using multiscale methods. ACS Catal. 2020, 21, 12544–12554. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Harris, R.C.; Tsai, C.-C.; Ellis, C.R.; Shen, J. Proton-coupled conformational allostery modulates the inhibitor selectivity for β-secretase. J. Phys. Chem. Lett. 2017, 8, 4832–4837. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.A.; Verma, N.; Harris, R.C.; Liu, R.; Shen, J. Assessment of proton-coupled conformational dynamics of SARS and MERS coronavirus papain-like proteases: Implication for designing broad-spectrum antiviral inhibitors. J. Chem. Phys. 2020, 153, 115101. [Google Scholar] [CrossRef]
- Lechuga, G.C.; Souza-Silva, F.; Sacramento, C.Q.; Trugilho, M.R.O.; Valente, R.H.; Napoleão-Pêgo, P.; Dias, S.S.G.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Carels, N.; et al. SARS-CoV-2 proteins bind to hemoglobin and its metabolites. Int. J. Mol. Sci. 2021, 22, 9035. [Google Scholar] [CrossRef]
- Gubarev, Y.A.; Lebedeva, N.S.; Yurina, E.S.; Syrbu, S.S.; Kiselev, A.N.; Lebedev, M.A. Possible therapeutic targets and promising drugs based on unsymmetrical hetaryl-substituted porphyrins to combat SARS-CoV-2. J. Pharm. Anal. 2021, 11, 691–698. [Google Scholar] [CrossRef]
- Koifman, O.I.; Lebedeva, N.S.; Gubarev, Y.A.; Koifman, M.O. Modeling the binding of protoporphyrin IX, verteporfin, and chlorin e6 to SARS-CoV-2 proteins. Chem. Heterocycl. Compd. 2021, 57, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction–Chemistry at Your Fingertips. Available online: https://www.wavefun.com/ (accessed on 15 August 2022).
- Cambridge Crystallographic Data Center. Available online: http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ (accessed on 15 August 2022).
- PyMol Delano. Available online: https://pymol.org/2/ (accessed on 15 August 2022).
- Osterberg, T.; Norinder, U. Prediction of drug transport processes using simple parameters and PLS statistics. The use of ACD/logP and ACD/ChemSketch descriptors. Eur. J. Pharm. Sci. 2001, 12, 327–337. [Google Scholar] [CrossRef]
- Rodrigues-Santos, C.E.; Leonor, L.; Bortoluzzi, A.J.; Canto-Cavalheiro, M.M.; Machado, G.C.; Echevarria, A. Synthesis, antileishmanial activity and structure–activity relationship of 1-N-X-phenyl-3-N′-Y-phenyl-benzamidines. Eur. J. Med. Chem. 2013, 67, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, N.S. Physical Organic Chemistry, 2nd ed.; Prentice Hall: Hoboken, NJ, USA, 1995. [Google Scholar]
- SwissADEME. Available online: www.swissademe.ch (accessed on 15 August 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mpro | PLpro |
|---|---|---|
| C1 | 25.76 | 25.25 |
| C2 | 35.68 | 32.44 |
| C3 | 35.98 | 30.41 |
| C4 | 36.44 | 31.76 |
| C5 | 24.65 | 30.38 |
| C6 | 34.98 | 31.29 |
| C7 | 22.43 | 30.13 |
| C8 | 36.24 | 31.99 |
| C9 | 34.50 | 31.84 |
| C10 | 24.92 | 34.69 |
| C11 | 35.64 | 32.39 |
| C12 | 25.47 | 31.18 |
| C13 | 26.37 | 40.65 |
| C14 | 21.76 | 37.95 |
| C15 | 36.57 | 35.07 |
| C16 | 24.81 | 38.97 |
| C17 | 22.84 | 39.29 |
| Compound | Amino Acid Residue | Interaction Type | Distance (Å) |
|---|---|---|---|
| Met-49 | Van der Waals | 1.70 | |
| Asn-142 | Van der Waals | 2.90 | |
| C1 | Cys-145 | Hydrogen bonding | 3.20 |
| Met-165 | Van der Waals | 3.20 | |
| Gln-189 | Van der Waals | 3.60 | |
| Leu-27 | Van der Waals | 2.90 | |
| His-41 | Van der Waals | 3.00 | |
| Met-49 | Van der Waals | 1.90 | |
| C2 | Cys-145 | Van der Waals | 2.60 |
| Met-165 | Van der Waals | 2.30 | |
| Pro-168 | Van der Waals | 3.00 | |
| Gln-189 | Van der Waals | 2.10 | |
| Thr-25 | Van der Waals | 1.20 | |
| Leu-27 | Van der Waals | 2.70 | |
| His-41 | Van der Waals | 2.70 | |
| C3, C4 | Met-49 | Van der Waals | 2.40 |
| Cys-145 | Van der Waals | 3.00 | |
| Met-165 | Van der Waals | 2.60 | |
| Pro-168 | Van der Waals | 2.80 | |
| Gln-1896 | Hydrogen bonding | 3.70 | |
| Thr-24 | Hydrogen bonding | 2.10 | |
| Thr-25 | Van der Waals | 1.50 | |
| Met-49 | Van der Waals | 2.50 | |
| C5 | Cys-145 | Van der Waals | 3.70 |
| His-163 | Van der Waals | 1.40 | |
| Glu-166 | Van der Waals | 2.40 | |
| His-172 | Van der Waals | 1.60 | |
| Gln-189 | Van der Waals | 2.40 | |
| Thr-25 | Van der Waals | 2.60 | |
| Leu-27 | Van der Waals | 1.80 | |
| His-41 | Van der Waals | 2.70 | |
| Met-49 | Van der Waals | 1.80 | |
| C6, C8, C9, C11 | Asn-142 | Van der Waals | 1.90 |
| Gly-143 | Van der Waals | 2.60 | |
| Cys-145 | Hydrogen bonding | 2.20 | |
| Met-165 | Van der Waals | 2.60 | |
| Leu-167 | Van der Waals | 2.70 | |
| Gln-189 | Hydrogen bonding | 2.00 | |
| Leu-27 | Van der Waals | 2.10 | |
| His-41 | Van der Waals | 1.50 | |
| C7, C10 | Met-49 | Van der Waals | 1.70 |
| Asn-142 | Van der Waals | 3.70 | |
| Gln-189 | Hydrogen bonding | 2.00 | |
| Leu-27 | Van der Waals | 2.20 | |
| His-41 | Van der Waals | 2.40 | |
| Met-49 | Van der Waals | 1.60 | |
| C12, C13 | Asn-142 | Van der Waals | 2.00 |
| Met-165 | Van der Waals | 1,20 | |
| Leu-167 | Van der Waals | 2.80 | |
| Gln-189 | Hydrogen bonding | 2.10 | |
| His-41 | Van der Waals | 3.10 | |
| Met-49 | Van der Waals | 2.50 | |
| C14, C16 | Asn-142 | Van der Waals | 2.50 |
| Met-165 | Van der Waals | 1.10 | |
| Glu-166 | Van der Waals | 3.40 | |
| Gln-189 | Hydrogen bonding | 2.10 | |
| Thr-25 | Van der Waals | 2.80 | |
| His-41 | Van der Waals | 2.90 | |
| Met-49 | Van der Waals | 2.30 | |
| C15 | Asn-142 | Van der Waals | 3.70 |
| Cys-145 | Hydrogen bonding | 2.30 | |
| Met-165 | Van der Waals | 2.70 | |
| Leu-167 | Van der Waals | 2.20 | |
| Glu-189 | Van der Waals | 3.00 | |
| His-41 | Van der Waals | 2.40 | |
| Met-49 | Van der Waals | 2.70 | |
| Tyr-54 | Van der Waals | 1.70 | |
| C17 | Asn-142 | Van der Waals | 2.40 |
| Met-165 | Van der Waals | 1.20 | |
| Asp-187 | Van der Waals | 1.30 | |
| Gln-189 | Hydrogen bonding | 2.00 | |
| H2Ocat | Van der Waals | 2.50 |
| Compound | Amino Acid Residue/Chain | Interaction Type | Distance (Å) |
|---|---|---|---|
| Lys-106/Chain A | Van der Waals | 3.40 | |
| Asp-287/Chain A | Van der Waals | 3.10 | |
| C1 | Leu-290/Chain A | Van der Waals | 3.20 |
| Pro-249/Chain C | Van der Waals | 1.80 | |
| Asn-258/Chain C | Van der Waals | 3.70 | |
| Tyr-265/Chain C | Van der Waals | 1.70 | |
| Trp-107/Chain A | Van der Waals | 1.80 | |
| Asp-287/Chain A | Van der Waals | 3.50 | |
| C2 | Leu-290/Chain A | Van der Waals | 0.60 |
| Pro-249/Chain C | Van der Waals | 3.20 | |
| Tyr-265/Chain C | Van der Waals | 3.20 | |
| Pro-300/Chain C | Van der Waals | 1.80 | |
| Lys-106/Chain A | Van der Waals | 3.00 | |
| Trp-107/Chain A | Van der Waals | 1.80 | |
| Lys-278/Chain A | Van der Waals | 2.00 | |
| C3, C4 | Asp-287/Chain A | Van der Waals | 3.70 |
| Leu-290/Chain A | Van der Waals | 3.20 | |
| Pro-249/Chain C | Van der Waals | 1.70 | |
| Tyr-265/Chain C | Van der Waals | 2.30 | |
| Asn–268/Chain C | Hydrogen bonding | 2.10 | |
| Tyr-269/Chain C | Van der Waals | 3.50 | |
| Lys-106/Chain A | Van der Waals | 3.10 | |
| Trp-107/Chain A | Van der Waals | 2.10 | |
| C5 | Leu-290/Chain A | Van der Waals | 2.60 |
| Leu-163/Chain C | Van der Waals | 3.30 | |
| Pro-249/Chain C | Van der Waals | 2.50 | |
| Tyr-265/Chain C | Van der Waals | 3.00 | |
| Trp-107/Chain A | Van der Waals | 2.50 | |
| Asp-287/Chain A | Van der Waals | 2.90 | |
| Leu-290/Chain A | Van der Waals | 2.80 | |
| C6 | Pro-249/Chain C | Van der Waals | 1.70 |
| Tyr-265/Chain C | Van der Waals | 2.90 | |
| Asn-268/Chain C | Hydrogen bonding | 2.90 | |
| Tyr-269/Chain C | Van der Waals | 2.20 | |
| Pro-300/Chain C | Van der Waals | 2.80 | |
| Trp-107/Chain A | Van der Waals | 3.70 | |
| Lys-275/Chain A | Hydrogen bonding | 1.10 | |
| Asp-287/Chain A | Van der Waals | 2.20 | |
| C7, C8, C9, C10, C11 | Leu-290/Chain A | Van der Waals | 1.20 |
| Pro-249/Chain C | Van der Waals | 1.30 | |
| Gln-251/Chain C | Hydrogen bonding | 3.30 | |
| Tyr-265/Chain C | Van der Waals | 2.60 | |
| Asn-268/Chain C | Hydrogen bonding | 2.90 | |
| Trp-107/Chain A | Van der Waals | 1.90 | |
| Lys-275/Chain A | Hydrogen bonding | 2.50 | |
| Asp-287/Chain A | Van der Waals | 3.10 | |
| C12, C13 | Leu-290/Chain A | Van der Waals | 2.00 |
| Pro-249/Chain C | Van der Waals | 1.70 | |
| Tyr-265/Chain C | Van der Waals | 3.20 | |
| Asn-268/Chain C | Hydrogen bonding | 3.30 | |
| Pro-300/Chain C | Van der Waals | 2.90 | |
| Trp-107/Chain A | Van der Waals | 1.90 | |
| Lys-275/Chain A | Hydrogen bonding | 1.70 | |
| Asp-287/Chain A | Van der Waals | 2.30 | |
| C14, C15, C16 | Leu-290/Chain A | Van der Waals | 1.40 |
| Pro-249/Chain C | Van der Waals | 1.60 | |
| Tyr-265/Chain C | Van der Waals | 3.30 | |
| Asn-268/Chain C | Hydrogen bonding | 3.70 | |
| Pro-300/Chain C | Van der Waals | 3.40 | |
| Trp-107/Chain A | Van der Waals | 3.10 | |
| Lys-275/Chain A | Hydrogen bonding | 2.60 | |
| Asp-287/Chain A | Van der Waals | 3.40 | |
| Leu-290/Chain A | Van der Waals | 1.20 | |
| C17 | Asp-165/Chain C | Van der Waals | 1.90 |
| Pro-249/Chain C | Van der Waals | 1.50 | |
| Tyr-265/Chain C | Van der Waals | 2.90 | |
| Asn-268/Chain C | Hydrogen bonding | 3.70 | |
| Pro-300/Chain C | Van der Waals | 2.70 |

| Compound | MR | MV | POLZ | ST | δC1 | δC2 |
|---|---|---|---|---|---|---|
| C1 | 90.28 | 214.0 | 35.8 | 78.2 | 83.0 | 136.3 |
| C2 | 164.0 | 409.8 | 65.0 | 63.4 | 105.1 | 135.0 |
| C3 | 164.0 | 422.5 | 65.0 | 60.4 | 105.1 | 139.3 |
| C4 | 164.0 | 422.5 | 65.0 | 60.4 | 105.1 | 135.0 |
| C5 | 164.0 | 422.5 | 65.0 | 60.4 | 101.8 | 131.5 |
| C6 | 164.0 | 435.1 | 65.0 | 57.7 | 105.1 | 139.3 |
| C7 | 164.0 | 435.1 | 65.0 | 57.7 | 101.8 | 135.8 |
| C8 | 164.0 | 435.1 | 65.0 | 57.7 | 105.1 | 139.3 |
| C9 | 164.0 | 435.1 | 65.0 | 57.7 | 105.1 | 143.6 |
| C10 | 164.0 | 435.1 | 65.0 | 57.7 | 101.8 | 131.5 |
| C11 | 164.0 | 435.1 | 65.0 | 57.7 | 105.1 | 135.0 |
| C12 | 164.0 | 447.7 | 65.0 | 55.2 | 101.8 | 135.8 |
| C13 | 164.0 | 447.7 | 65.0 | 55.2 | 101.8 | 131.5 |
| C14 | 164.0 | 460.3 | 65.0 | 53.0 | 101.8 | 135.8 |
| C15 | 164.0 | 460.3 | 65.0 | 53.0 | 101.1 | 143.6 |
| C16 | 164.0 | 460.3 | 65.0 | 53.0 | 101.8 | 135.8 |
| C17 | 164.0 | 473.0 | 65.0 | 51.0 | 101.8 | 140.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaves, O.A.; Rodrigues-Santos, C.E.; Echevarria, Á.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Castro-Faria-Neto, H.C.; Souza, T.M.L.e. Fluorine Atoms on C6H5-Corrole Affect the Interaction with Mpro and PLpro Proteases of SARS-CoV-2: Molecular Docking and 2D-QSAR Approaches. Int. J. Mol. Sci. 2022, 23, 10936. https://doi.org/10.3390/ijms231810936
Chaves OA, Rodrigues-Santos CE, Echevarria Á, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, Castro-Faria-Neto HC, Souza TMLe. Fluorine Atoms on C6H5-Corrole Affect the Interaction with Mpro and PLpro Proteases of SARS-CoV-2: Molecular Docking and 2D-QSAR Approaches. International Journal of Molecular Sciences. 2022; 23(18):10936. https://doi.org/10.3390/ijms231810936
Chicago/Turabian StyleChaves, Otávio Augusto, Cláudio Eduardo Rodrigues-Santos, Áurea Echevarria, Carolina Q. Sacramento, Natalia Fintelman-Rodrigues, Jairo R. Temerozo, Hugo Caire Castro-Faria-Neto, and Thiago Moreno Lopes e Souza. 2022. "Fluorine Atoms on C6H5-Corrole Affect the Interaction with Mpro and PLpro Proteases of SARS-CoV-2: Molecular Docking and 2D-QSAR Approaches" International Journal of Molecular Sciences 23, no. 18: 10936. https://doi.org/10.3390/ijms231810936
APA StyleChaves, O. A., Rodrigues-Santos, C. E., Echevarria, Á., Sacramento, C. Q., Fintelman-Rodrigues, N., Temerozo, J. R., Castro-Faria-Neto, H. C., & Souza, T. M. L. e. (2022). Fluorine Atoms on C6H5-Corrole Affect the Interaction with Mpro and PLpro Proteases of SARS-CoV-2: Molecular Docking and 2D-QSAR Approaches. International Journal of Molecular Sciences, 23(18), 10936. https://doi.org/10.3390/ijms231810936