
Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors
 , , , ,
, , , ,  , ,
, , 

Abstract
1. Introduction
2. Results and Discussion
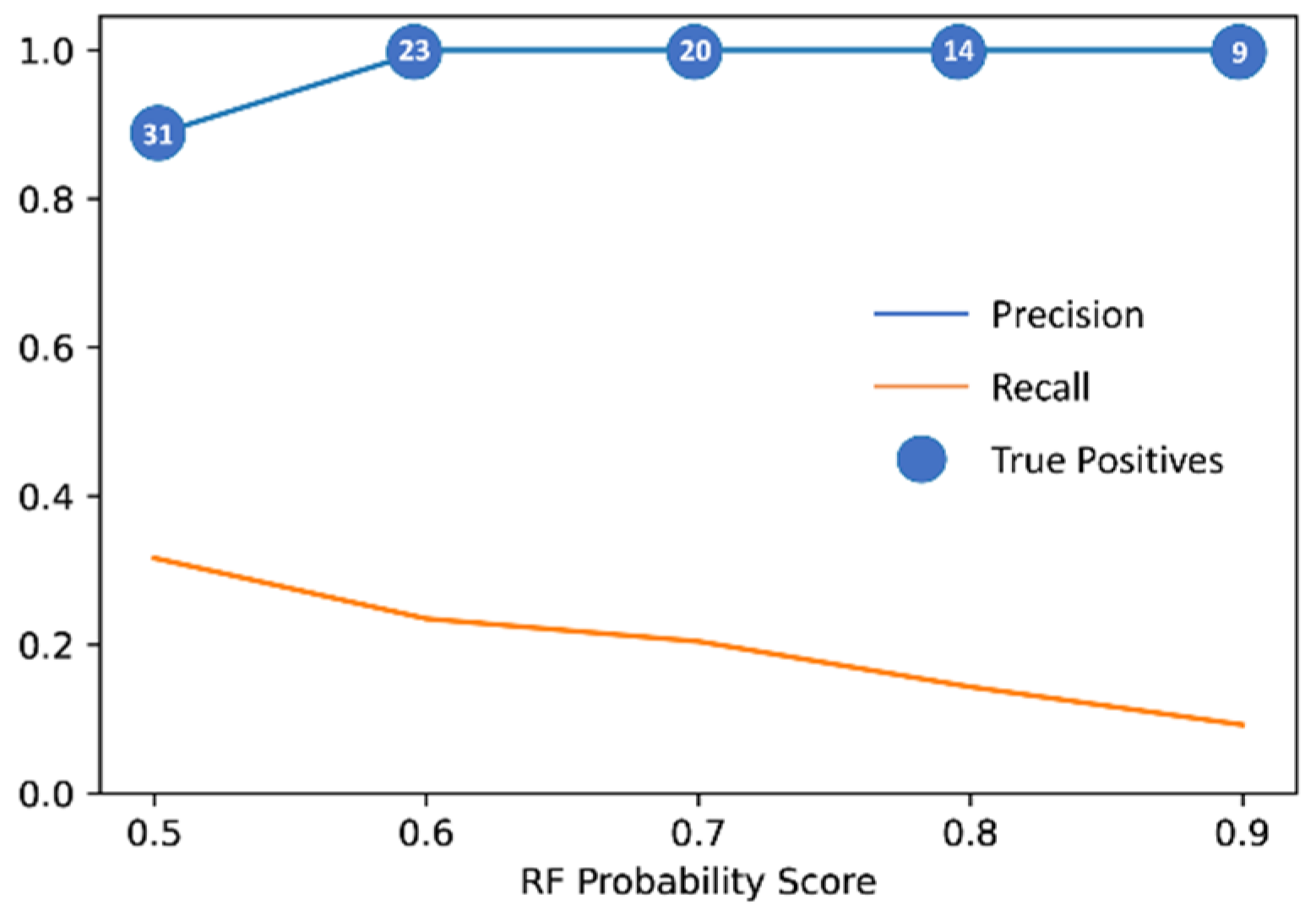
2.1. Machine Learning Model Generation, Optimization and Evaluation
2.2. Virtual Screening
2.3. Antiproliferative Assays
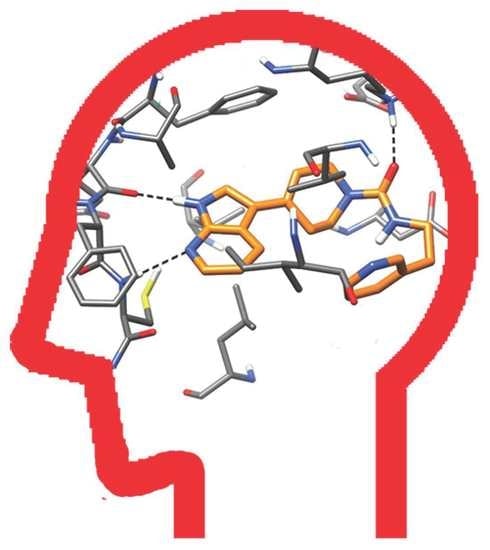
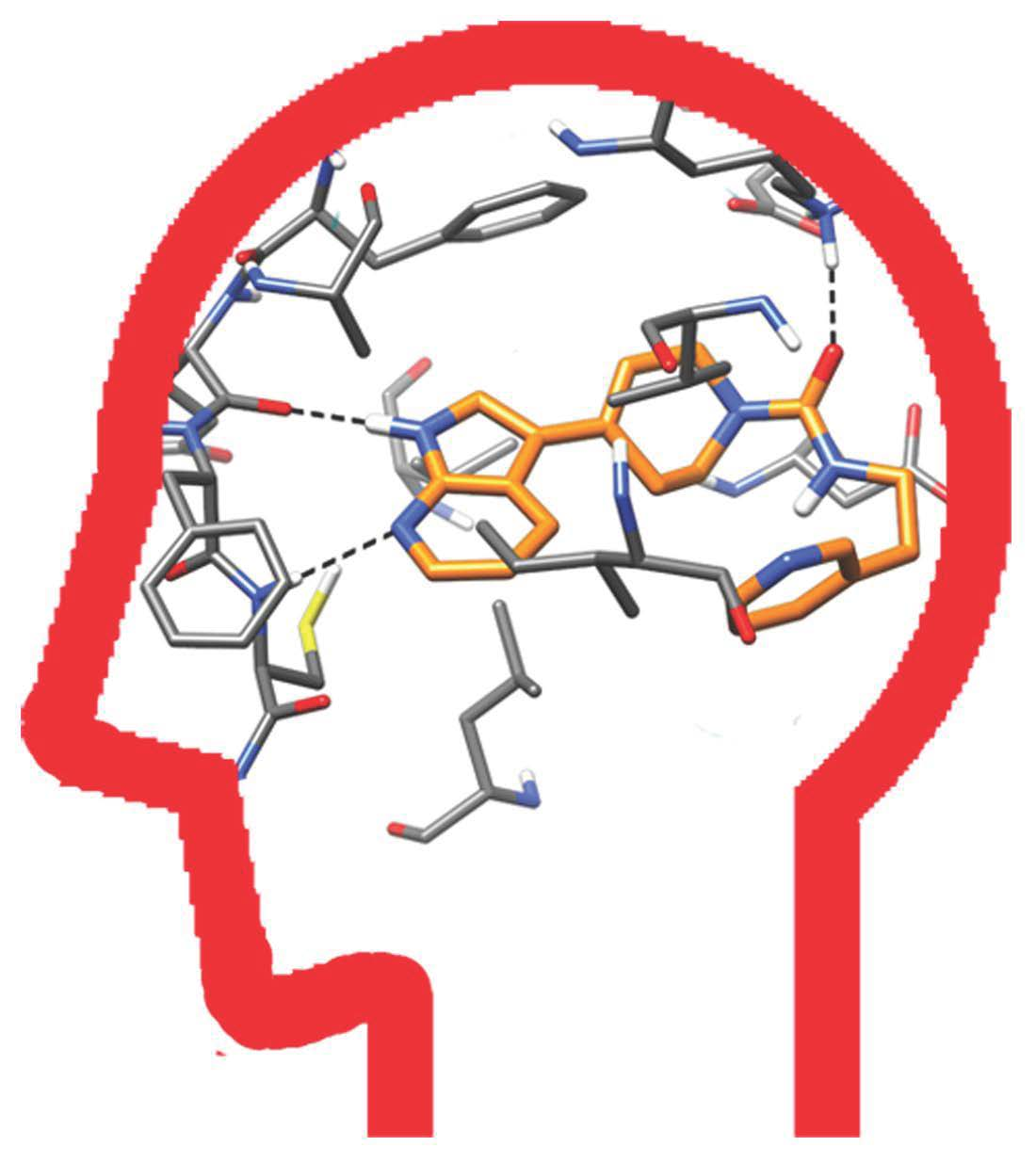
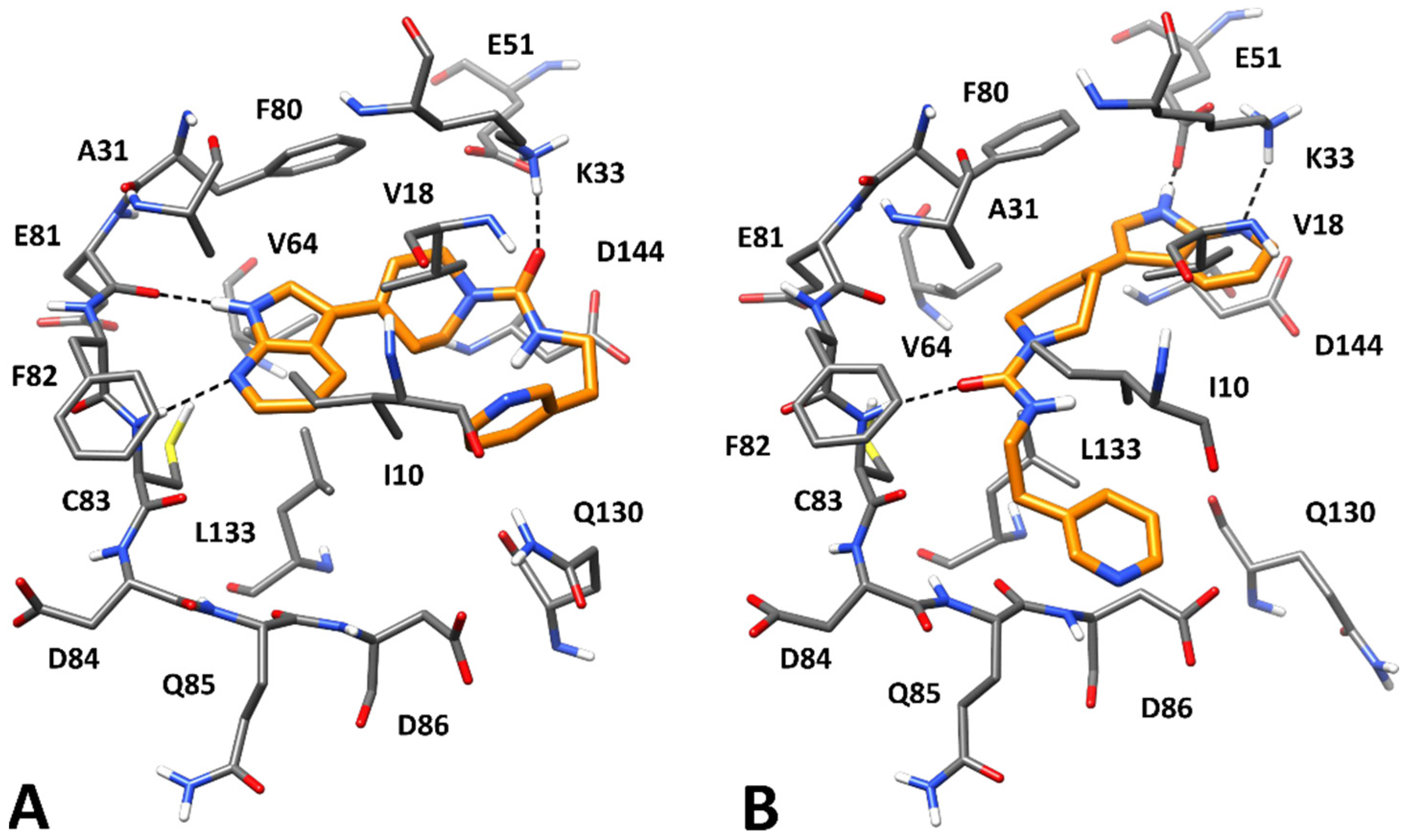
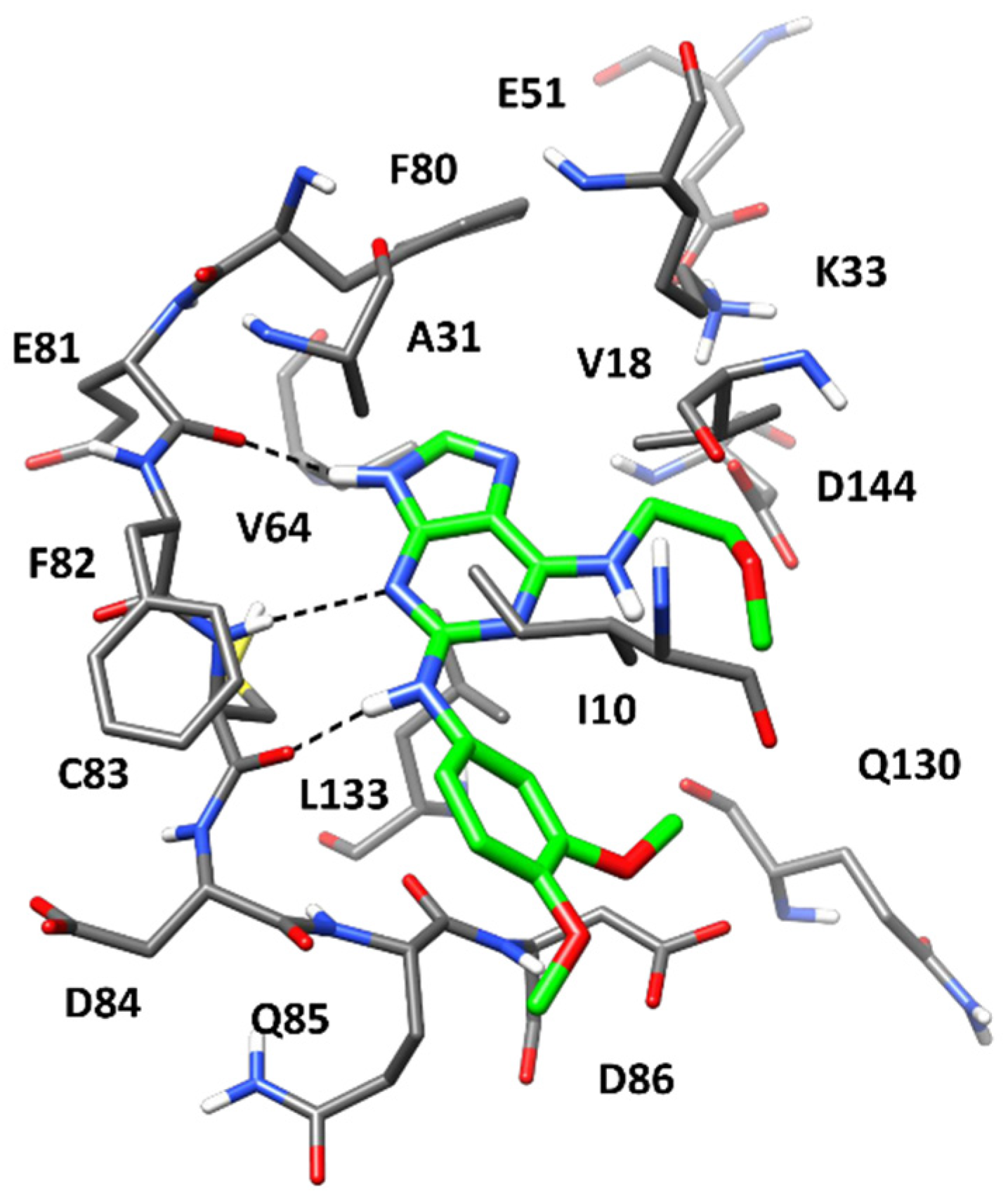
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. Machine Learning Data Sets
3.2. Molecular Representations
3.3. Machine Learning Methods
3.4. Model Building and Evaluation
3.5. Model Evaluation
3.6. Consensus Approach
3.7. Database Generation and Machine Learning Screening
3.8. In Vitro Cdk5 Inhibition Activity
3.9. Molecular Docking Calculations
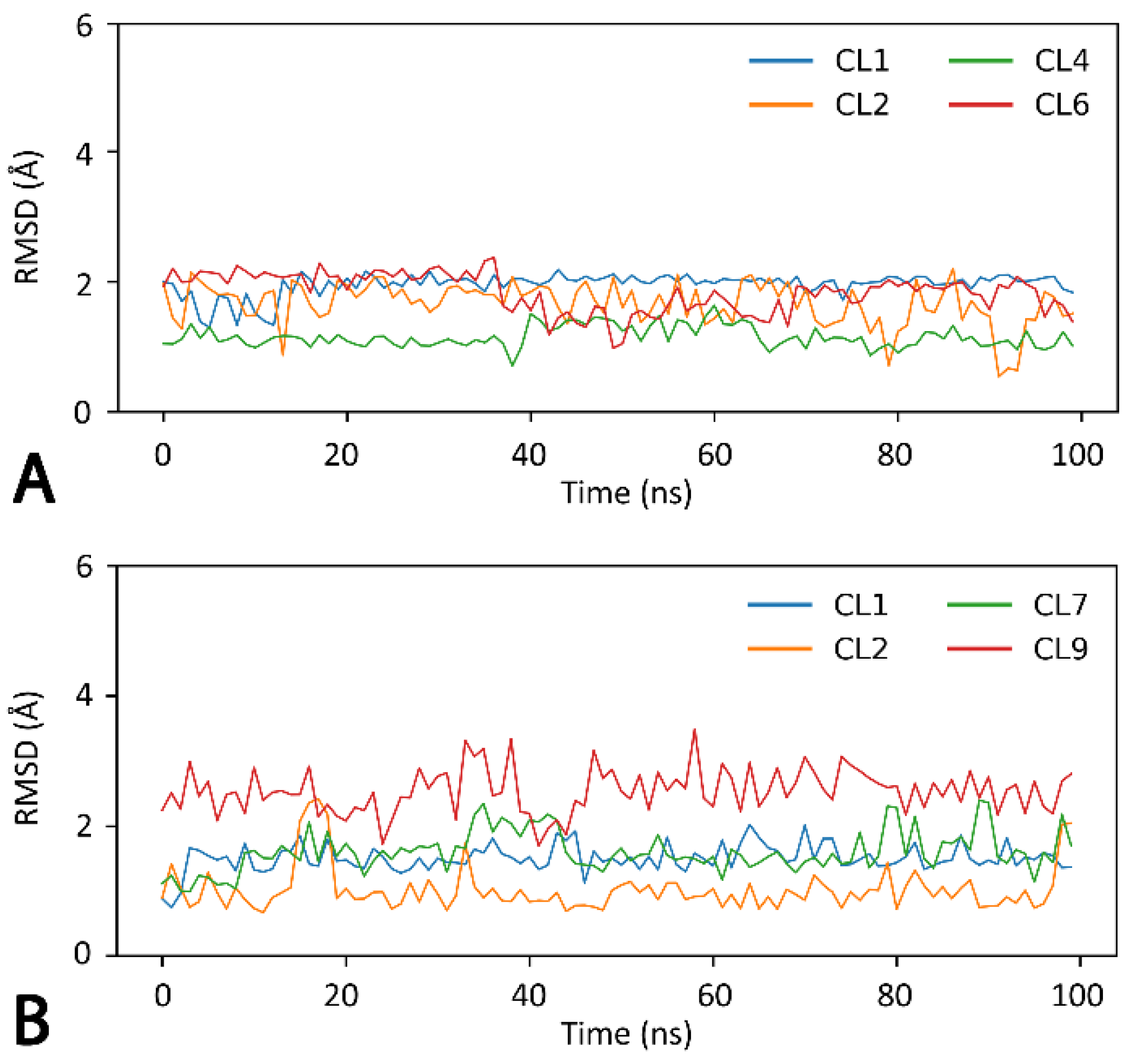
3.10. Molecular Dynamics (MD) Simulations
3.11. Binding Energy Evaluation
3.12. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Takahashi, T.; Caviness, V.S.; Harlow, E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 1993, 119, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.; Agostinho, P. Cdk5: Multitasking between physiological and pathological conditions. Prog. Neurobiol. 2011, 94, 49–63. [Google Scholar] [CrossRef]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.H.; Musacchio, A. Structure and regulation of the CDK5-p25(nck5a) complex. Mol. Cell 2001, 8, 657–669. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.D.; Crocker, S.J.; Jackson-Lewis, V.; Jordan-Sciutto, K.L.; Hayley, S.; Mount, M.P.; O’Hare, M.J.; Callaghan, S.; Slack, R.S.; Przedborski, S.; et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 13650–13655. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, N.P.S.; Al-Sarraj, S.T.; Anderson, V.; Kibble, M.; Leigh, N.; Miller, C.C.J. Cyclin-dependent kinase-5 is associated with lipofuscin in motor neurones in amyotrophic lateral sclerosis. Neurosci. Lett. 1998, 245, 45–48. [Google Scholar] [CrossRef]
- Patrick, C.; Crews, L.; Desplats, P.; Dumaop, W.; Rockenstein, E.; Achim, C.L.; Everall, I.P.; Masliah, E. Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am. J. Pathol. 2011, 178, 1646–1661. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; White, M.G.; Akay, C.; Chodroff, R.A.; Robinson, J.; Lindl, K.A.; Dichter, M.A.; Qian, Y.; Mao, Z.; Kolson, D.L.; et al. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J. Neurochem. 2007, 103, 439–455. [Google Scholar] [CrossRef]
- Binukumar, B.K.; Zheng, Y.-L.; Shukla, V.; Amin, N.D.; Grant, P.; Pant, H.C. TFP5, a peptide derived from p35, a Cdk5 neuronal activator, rescues cortical neurons from glucose toxicity. J. Alzheimers. Dis. 2014, 39, 899–909. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Eggers, J.P.; Grandgenett, P.M.; Collisson, E.C.; Lewallen, M.E.; Tremayne, J.; Singh, P.K.; Swanson, B.J.; Andersen, J.M.; Caffrey, T.C.; High, R.R.; et al. Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras. Clin. Cancer Res. 2011, 17, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Zhang, J.; Xiong, M.; Wang, X.; Luo, X.; Han, L.; Meng, Y.; Zhang, Y.; Liao, W.; Liu, S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5-AP-1 axis. Cell Death Dis. 2016, 7, e2415. [Google Scholar] [CrossRef] [PubMed]
- Strock, C.J.; Park, J.-I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Li, L.; Zhang, J.; Lei, Y.; Wang, L.; Liu, D.-X.; Feng, J.; Hou, P.; Yao, R.; Zhang, Y.; et al. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013, 3, 2932. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Z.; Mao, W.; Ahmed, A.A.; Yang, H.; Zhou, J.; Jennings, N.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Miranda, R.; et al. CDK5 Regulates Paclitaxel Sensitivity in Ovarian Cancer Cells by Modulating AKT Activation, p21Cip1- and p27Kip1-Mediated G1 Cell Cycle Arrest and Apoptosis. PLoS ONE 2015, 10, e0131833. [Google Scholar] [CrossRef]
- Catania, A.; Urban, S.; Yan, E.; Hao, C.; Barron, G.; Allalunis-Turner, J. Expression and localization of cyclin-dependent kinase 5 in apoptotic human glioma cells. Neuro. Oncol. 2001, 3, 89–98. [Google Scholar] [CrossRef]
- Levacque, Z.; Rosales, J.L.; Lee, K.-Y. Level of cdk5 expression predicts the survival of relapsed multiple myeloma patients. Cell Cycle 2012, 11, 4093–4095. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Jia, Y.; Liu, T.; Wang, M.; Lv, W.; Zhang, R.; Shi, J.; Liu, L. CDK5 neutralizes the tumor suppressing effect of BIN1 via mediating phosphorylation of c-MYC at Ser-62 site in NSCLC. Cancer Cell Int. 2019, 19, 226. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Liu, L.; Jin, Y.; Fu, S.-B.; Rosales, J.L. Cdk5 mediates vimentin Ser56 phosphorylation during GTP-induced secretion by neutrophils. J. Cell. Physiol. 2012, 227, 739–750. [Google Scholar] [CrossRef]
- Xie, W.; Liu, C.; Wu, D.; Li, Z.; Li, C.; Zhang, Y. Phosphorylation of kinase insert domain receptor by cyclin-dependent kinase 5 at serine 229 is associated with invasive behavior and poor prognosis in prolactin pituitary adenomas. Oncotarget 2016, 7, 50883–50894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liebl, J.; Weitensteiner, S.B.; Vereb, G.; Takács, L.; Fürst, R.; Vollmar, A.M.; Zahler, S. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J. Biol. Chem. 2010, 285, 35932–35943. [Google Scholar] [CrossRef] [PubMed]
- Courapied, S.; Sellier, H.; de Carné Trécesson, S.; Vigneron, A.; Bernard, A.-C.; Gamelin, E.; Barré, B.; Coqueret, O. The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J. Biol. Chem. 2010, 285, 26765–26778. [Google Scholar] [CrossRef] [PubMed]
- Lenjisa, J.L.; Tadesse, S.; Khair, N.Z.; Kumarasiri, M.; Yu, M.; Albrecht, H.; Milne, R.; Wang, S. CDK5 in oncology: Recent advances and future prospects. Future Med. Chem. 2017, 9, 1939–1962. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.J.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; Sisk, C.M.C.; Patton, N. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef]
- Tian, H.; Jiang, X.; Tao, P. PASSer: Prediction of Allosteric Sites Server. Mach. Learn. Sci. Technol. 2021, 2, 35015. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef]
- Russo Spena, C.; De Stefano, L.; Poli, G.; Granchi, C.; El Boustani, M.; Ecca, F.; Grassi, G.; Grassi, M.; Canzonieri, V.; Giordano, A.; et al. Virtual screening identifies a PIN1 inhibitor with possible antiovarian cancer effects. J. Cell. Physiol. 2019, 234, 15708–15716. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Di Stefano, M.; Martinelli, E.; Macchia, M.; Martinelli, A.; Poli, G.; Tuccinardi, T. VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions. Int. J. Mol. Sci. 2022, 23, 2105. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, S.; Sharma, M.C. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp. Mol. Pathol. 2007, 82, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.H.; Malojcic, G.; Clugston, S.L.; Williams, B.; Coeffet-Le Gal, M.; Pan-Zhou, X.R.; Venkatachalan, S.; Harmange, J.C.; Ledeboer, M. Discovery and Optimization of Highly Selective Inhibitors of CDK5. J. Med. Chem. 2022, 65, 3575–3596. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612-20. [Google Scholar] [CrossRef]
- QUACPAC 2.1.3.0: OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 1 June 2022).
- Metz, J.T.; Johnson, E.F.; Soni, N.B.; Merta, P.J.; Kifle, L.; Hajduk, P.J. Navigating the kinome. Nat. Chem. Biol. 2011, 7, 200–202. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 1 June 2022).
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Carracedo-Reboredo, P.; Liñares-Blanco, J.; Rodríguez-Fernández, N.; Cedrón, F.; Novoa, F.J.; Carballal, A.; Maojo, V.; Pazos, A.; Fernandez-Lozano, C. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Ralaivola, L.; Swamidass, S.J.; Saigo, H.; Baldi, P. Graph kernels for chemical informatics. Neural Netw. 2005, 18, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z. Introduction to machine learning: K-nearest neighbors. Ann. Transl. Med. 2016, 4, 218. [Google Scholar] [CrossRef]
- Cova, T.F.G.G.; Pais, A.A.C.C. Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns. Front. Chem. 2019, 7, 809. [Google Scholar] [CrossRef]
- D’Ascenzio, M.; Secci, D.; Carradori, S.; Zara, S.; Guglielmi, P.; Cirilli, R.; Pierini, M.; Poli, G.; Tuccinardi, T.; Angeli, A.; et al. 1,3-Dipolar Cycloaddition, HPLC Enantioseparation, and Docking Studies of Saccharin/Isoxazole and Saccharin/Isoxazoline Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors. J. Med. Chem. 2020, 63, 2470–2488. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Poli, G.; Lapillo, M.; Jha, V.; Mouawad, N.; Caligiuri, I.; Macchia, M.; Minutolo, F.; Rizzolio, F.; Tuccinardi, T.; Granchi, C. Computationally driven discovery of phenyl(piperazin-1-yl)methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 589–596. [Google Scholar] [CrossRef]
- Poli, G.; Lapillo, M.; Granchi, C.; Caciolla, J.; Mouawad, N.; Caligiuri, I.; Rizzolio, F.; Langer, T.; Minutolo, F.; Tuccinardi, T. Binding investigation and preliminary optimisation of the 3-amino-1,2,4-triazin-5(2 H )-one core for the development of new Fyn inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 956–961. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | MCC | Precision | Recall |
|---|---|---|---|
| Consensus | 0.43 | 0.88 | 0.31 |
| RF | 0.42 | 0.87 | 0.30 |
| SVM | 0.42 | 0.65 | 0.52 |
| MLP | 0.41 | 0.59 | 0.61 |
| KNN | 0.33 | 0.58 | 0.46 |
| Compound ID | Structure | IC50 (μM) |
|---|---|---|
| CPD1 |  | 3.43 ± 0.01 |
| CPD2 |  | >30.0 |
| CPD3 |  | >30.0 |
| CPD4 |  | 1.27 ± 0.07 |
| IC50 (Mean ± SD, nM) | ||||
|---|---|---|---|---|
| Compound | HCT116 | MDA-MB-231 | OVCAR3 | A2780 |
| CPD1 | 183 ± 27 | 1474 ± 44 | 12.0 ± 1.1 | 93.7 ± 21.6 |
| CPD4 | 1766 ± 230 | 2127 ± 349 | 2118 ± 63 | 678 ± 123 |
| Cisplatin | 2218 ± 6 | >10,000 | 681 ± 50 | 275 ± 39 |
| CPD1 | VDW | ELE | EPB | ENPOLAR | ΔPBSA |
|---|---|---|---|---|---|
| CL1 | −47.2 | −32.4 | 48.0 | −4.3 | −35.9 |
| CL2 | −47.0 | −40.2 | 58.3 | −4.2 | −33.1 |
| CL6 | −46.8 | −23.6 | 48.6 | −4.3 | −26.0 |
| CL4 | −44.6 | −23.6 | 49.2 | −4.2 | −23.2 |
| CPD4 | VDW | ELE | EPB | ENPOLAR | ΔPBSA |
| CL7 | −45.6 | −24.8 | 40.2 | −4.2 | −34.4 |
| CL1 | −44.7 | −24.1 | 43.8 | −4.4 | −29.4 |
| CL2 | −44.9 | −23.2 | 48.2 | −4.3 | −24.3 |
| CL9 | −37.2 | −22.6 | 44.4 | −4.1 | −19.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stefano, M.; Galati, S.; Ortore, G.; Caligiuri, I.; Rizzolio, F.; Ceni, C.; Bertini, S.; Bononi, G.; Granchi, C.; Macchia, M.; et al. Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors. Int. J. Mol. Sci. 2022, 23, 10653. https://doi.org/10.3390/ijms231810653
Di Stefano M, Galati S, Ortore G, Caligiuri I, Rizzolio F, Ceni C, Bertini S, Bononi G, Granchi C, Macchia M, et al. Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors. International Journal of Molecular Sciences. 2022; 23(18):10653. https://doi.org/10.3390/ijms231810653
Chicago/Turabian StyleDi Stefano, Miriana, Salvatore Galati, Gabriella Ortore, Isabella Caligiuri, Flavio Rizzolio, Costanza Ceni, Simone Bertini, Giulia Bononi, Carlotta Granchi, Marco Macchia, and et al. 2022. "Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors" International Journal of Molecular Sciences 23, no. 18: 10653. https://doi.org/10.3390/ijms231810653
APA StyleDi Stefano, M., Galati, S., Ortore, G., Caligiuri, I., Rizzolio, F., Ceni, C., Bertini, S., Bononi, G., Granchi, C., Macchia, M., Poli, G., & Tuccinardi, T. (2022). Machine Learning-Based Virtual Screening for the Identification of Cdk5 Inhibitors. International Journal of Molecular Sciences, 23(18), 10653. https://doi.org/10.3390/ijms231810653