
Chloroplast Genome Annotation Tools: Prolegomena to the Identification of Inverted Repeats

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Results
2.1. Comparative Code Analysis
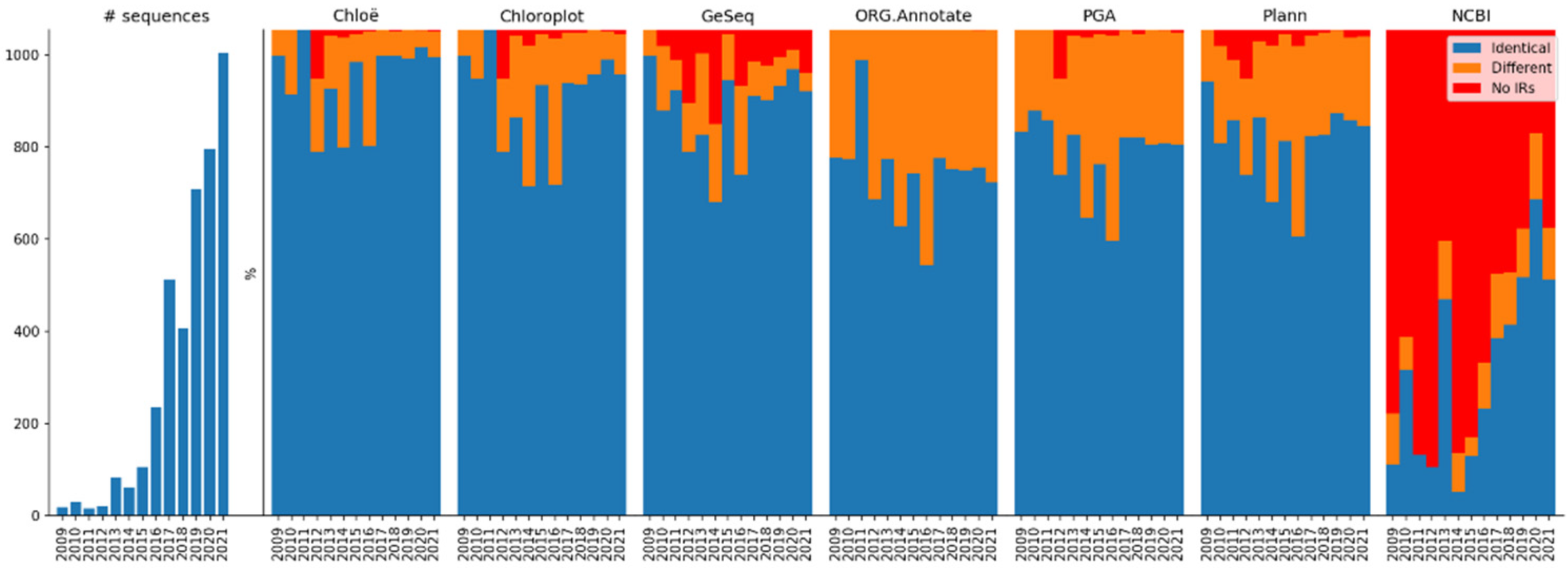
2.2. Comparative Results Analysis: Overview
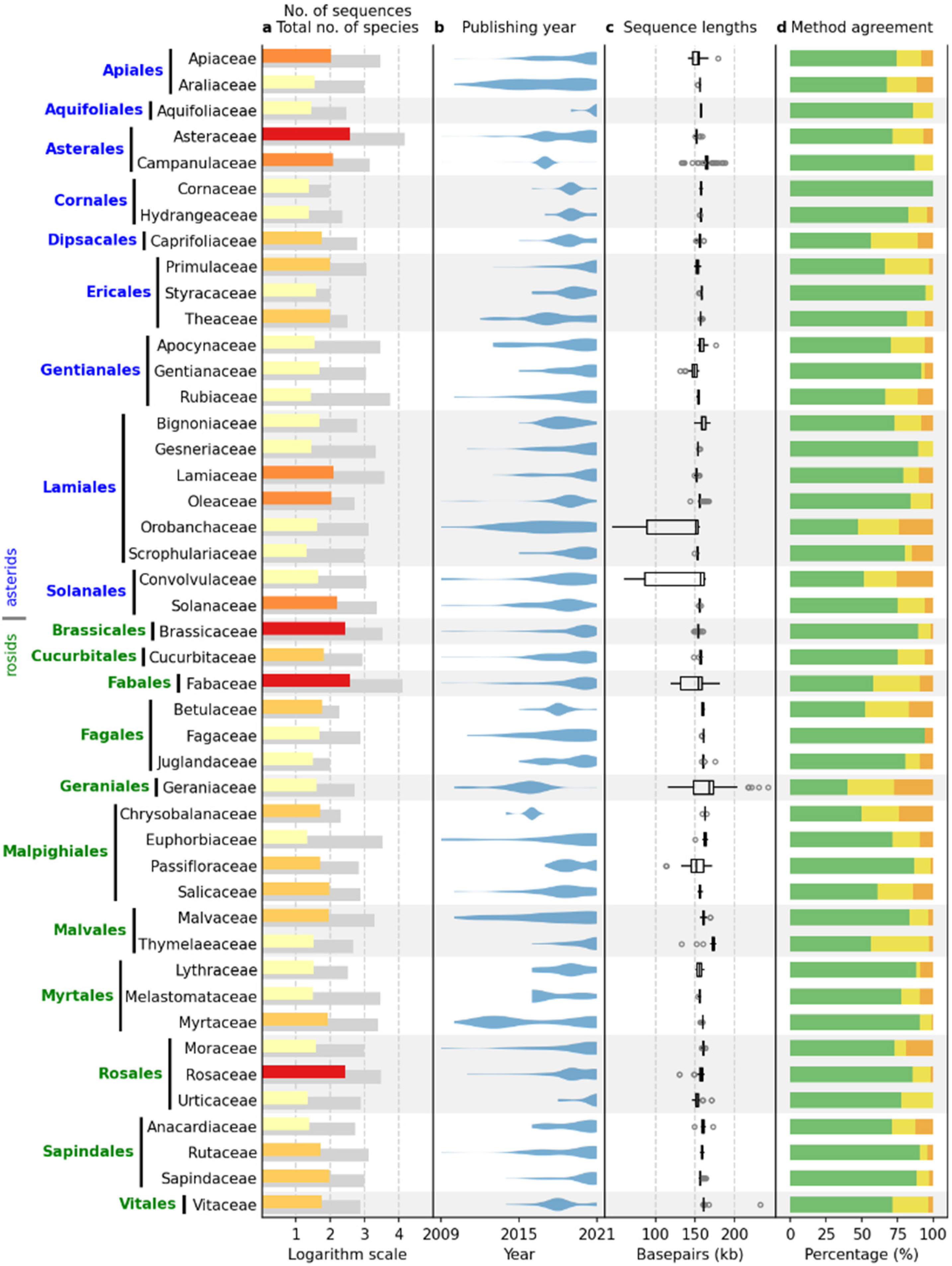
2.3. Comparative Results Analysis: IRL Dataset
2.4. Comparative Results Analysis: IR Dataset
2.4.1. Type of IR Regions Identified
2.4.2. Treatment of Circularity of the Chloroplast Genome
2.4.3. Treatment of Sequences with Ambiguous Characters
2.4.4. Differences between IRa and IRb
2.4.5. Method Agreement
2.4.6. IR-Length Statistics
3. Discussion
3.1. State of the Art
3.2. Further Research
4. Materials and Methods
4.1. Data Acquisition
4.2. Comparative Code Analysis
4.3. Comparative Results Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dagan, T.; Roettger, M.; Stucken, K.; Landan, G.; Koch, R.; Major, P.; Gould, S.B.; Goremykin, V.V.; Rippka, R.; De Marsac, N.T.; et al. Genomes of Stigonematalean Cyanobacteria (Subsection V) and the Evolution of Oxygenic Photosynthesis from Prokaryotes to Plastids. Genome Biol. Evol. 2013, 5, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Baracaldo, P.; Raven, J.A.; Pisani, D.; Knoll, A.H. Early Photosynthetic Eukaryotes Inhabited Low-Salinity Habitats. Proc. Natl. Acad. Sci. USA 2017, 114, E7737–E7745. [Google Scholar] [CrossRef] [PubMed]
- Ruhlman, T.A.; Jansen, R.K. The Plastid Genomes of Flowering Plants. Methods Mol. Biol. 2014, 1132, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-W.; Wing, R.A.; Gruissem, W. The Chloroplast Genome Exists in Multimeric Forms. Proc. Natl. Acad. Sci. USA 1989, 86, 4156–4160. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.J.; Smith, S.B. Moving Pictures and Pulsed-Field Gel Electrophoresis Show Linear DNA Molecules from Chloroplasts and Mitochondria. Curr. Genet. 1990, 17, 421–425. [Google Scholar] [CrossRef]
- Bendich, A.J. Circular Chloroplast Chromosomes: The Grand Illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M.; Chang, Z.; et al. Chloroplast Gene Organization Deduced from Complete Sequence of Liverwort Marchantia Polymorpha Chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The Complete Nucleotide Sequence of the Tobacco Chloroplast Genome: Its Gene Organization and Expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Kuo, L.Y.; Tang, T.Y.; Li, F.W.; Su, H.J.; Chiou, W.L.; Huang, Y.M.; Wang, C.N. Organelle Genome Inheritance in Deparia Ferns (Athyriaceae, Aspleniineae, Polypodiales). Front. Plant Sci. 2018, 9, 486. [Google Scholar] [CrossRef]
- Guillon, J.M.; Raquin, C. Maternal Inheritance of Chloroplasts in the Horsetail Equisetum Variegatum (Schleich.). Curr. Genet. 2000, 37, 53–56. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The Evolution of the Plastid Chromosome in Land Plants: Gene Content, Gene Order, Gene Function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Crosby, K.; Smith, D.R. Does the Mode of Plastid Inheritance Influence Plastid Genome Architecture? PLoS ONE 2012, 7, e46260. [Google Scholar] [CrossRef] [PubMed]
- Tilney-Bassett, R.A.E.; Almouslem, A.B. Variation in Plastid Inheritance between Pelargonium Cultivars and Their Hybrids. Heredity (Edinb) 1989, 63, 145–153. [Google Scholar] [CrossRef]
- Corriveau, J.L.; Coleman, A.W. Rapid Screening Method to Detect Potential Biparental Inheritance of Plastid DNA and Results for Over 200 Angiosperm Species. Am. J. Bot. 1988, 75, 1443. [Google Scholar] [CrossRef]
- Kolodner, R.; Tewari, K.K. Inverted Repeats in Chloroplast DNA from Higher Plants. Proc. Natl. Acad. Sci. USA 1979, 76, 41–45. [Google Scholar] [CrossRef]
- Wu, C.S.; Lai, Y.T.; Lin, C.P.; Wang, Y.N.; Chaw, S.M. Evolution of Reduced and Compact Chloroplast Genomes (CpDNAs) in Gnetophytes: Selection toward a Lower-Cost Strategy. Mol. Phylogenet. Evol. 2009, 52, 115–124. [Google Scholar] [CrossRef]
- Wolf, P.G.; Der, J.P.; Duffy, A.M.; Davidson, J.B.; Grusz, A.L.; Pryer, K.M. The Evolution of Chloroplast Genes and Genomes in Ferns. Plant Mol. Biol. 2011, 76, 251–261. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete Nucleotide Sequence of the Cryptomeria Japonica D. Don. Chloroplast Genome and Comparative Chloroplast Genomics: Diversified Genomic Structure of Coniferous Species. BMC Plant Biol. 2008, 8, 70. [Google Scholar] [CrossRef]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The Complete Chloroplast Genome Sequence of Pelargonium × Hortorum: Organization and Evolution of the Largest and Most Highly Rearranged Chloroplast Genome of Land Plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef]
- Goremykin, V.V.; Hirsch-Ernst, K.I.; Wölfl, S.; Hellwig, F.H. Analysis of the Amborella Trichopoda Chloroplast Genome Sequence Suggests That Amborella Is Not a Basal Angiosperm. Mol. Biol. Evol. 2003, 20, 1499–1505. [Google Scholar] [CrossRef]
- Heinhorst, S.; Cannon, G.C. DNA Replication in Chloroplasts. J. Cell Sci. 1993, 104, 1–9. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA Rearrangements Are More Frequent When a Large Inverted Repeat Sequence Is Lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of Nucleotide Substitution Vary Greatly among Plant Mitochondrial, Chloroplast, and Nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.B.; Palmer, J.D.; Thompson, W.F. Structural Evolution and Flip-Flop Recombination of Chloroplast DNA in the Fern Genus Osmunda. Curr. Genet. 1986, 10, 9054–9058. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the Maintenance of Plant Organelle Genome Stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, G.M.; Downie, S.R. Expansion and Contraction of the Chloroplast Inverted Repeat in Apiaceae Subfamily Apioideae. Syst. Bot. 2000, 25, 648. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and Evolution of the Inverted Repeat-Large Single Copy Junctions in the Chloroplast Genomes of Monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Davis, J.I.; Soreng, R.J. Migration of Endpoints of Two Genes Relative to Boundaries between Regions of the Plastid Genome in the Grass Family (Poaceae). Am. J. Bot. 2010, 97, 874–892. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Chumley, T.W.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Implications of the Plastid Genome Sequence of Typha (Typhaceae, Poales) for Understanding Genome Evolution in Poaceae. J. Mol. Evol. 2010, 70, 149–166. [Google Scholar] [CrossRef]
- Logacheva, M.D.; Schelkunov, M.I.; Penin, A.A. Sequencing and Analysis of Plastid Genome in Mycoheterotrophic Orchid Neottia Nidus-Avis. Genome Biol. Evol. 2011, 3, 1296–1303. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Wu, M.; Palmer, J.D.; Taylor, D.R. Recent Acceleration of Plastid Sequence and Structural Evolution Coincides with Extreme Mitochondrial Divergence in the Angiosperm Genus Silene. Genome Biol. Evol. 2012, 4, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Osorio, B.; Aldrich, J.; Thompson, W.F. Chloroplast DNA Evolution among Legumes: Loss of a Large Inverted Repeat Occurred Prior to Other Sequence Rearrangements. Curr. Genet. 1987, 11, 275–286. [Google Scholar] [CrossRef]
- Cai, Z.; Guisinger, M.; Kim, H.G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive Reorganization of the Plastid Genome of Trifolium Subterraneum (Fabaceae) Is Associated with Numerous Repeated Sequences and Novel DNA Insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The Chloroplast Genome Sequence of Mungbean (Vigna Radiata) Determined by High-Throughput Pyrosequencing: Structural Organization and Phylogenetic Relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of Different Inverted Repeat Copies from the Chloroplast Genomes of Pinaceae and Cupressophytes and Influence of Heterotachy on the Evaluation of Gymnosperm Phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.J.; Wang, T. The Complete Chloroplast Genome Sequence of Cephalotaxus Oliveri (Cephalotaxaceae): Evolutionary Comparison of Cephalotaxus Chloroplast DNAs and Insights into the Loss of Inverted Repeat Copies in Gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef]
- Wu, C.S.; Chaw, S.M. Highly Rearranged and Size-Variable Chloroplast Genomes in Conifers II Clade (Cupressophytes): Evolution towards Shorter Intergenic Spacers. Plant Biotechnol. J. 2014, 12, 344–353. [Google Scholar] [CrossRef]
- Blazier, C.C.; Guisinger, M.M.; Jansen, R.K. Recent Loss of Plastid-Encoded Ndh Genes within Erodium (Geraniaceae). Plant Mol. Biol. 2011, 76, 263–272. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme Reconfiguration of Plastid Genomes in the Angiosperm Family Geraniaceae: Rearrangements, Repeats, and Codon Usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Copetti, D.; Burquez, A.; Bustamante, E.; Charboneau, J.L.M.; Eguiarte, L.E.; Kumar, S.; Lee, H.O.; Lee, J.; McMahon, M.; et al. Exceptional Reduction of the Plastid Genome of Saguaro Cactus (Carnegiea Gigantea): Loss of the Ndh Gene Suite and Inverted Repeat. Am. J. Bot. 2015, 102, 1115–1127. [Google Scholar] [CrossRef]
- Barrett, C.F.; Baker, W.J.; Comer, J.R.; Conran, J.G.; Lahmeyer, S.C.; Leebens-Mack, J.H.; Li, J.; Lim, G.S.; Mayfield-Jones, D.R.; Perez, L.; et al. Plastid Genomes Reveal Support for Deep Phylogenetic Relationships and Extensive Rate Variation among Palms and Other Commelinid Monocots. New Phytol. 2016, 209, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Cauz-Santos, L.A.; da Costa, Z.P.; Callot, C.; Cauet, S.; Zucchi, M.I.; Bergès, H.; van den Berg, C.; Vieira, M.L.C. A Repertory of Rearrangements and the Loss of an Inverted Repeat Region in Passiflora Chloroplast Genomes. Genome Biol. Evol. 2020, 12, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- DePamphilis, C.W.; Palmer, J.D. Loss of Photosynthetic and Chlororespiratory Genes from the Plastid Genome of a Parasitic Flowering Plant. Nature 1990, 348, 337–339. [Google Scholar] [CrossRef]
- Downie, S.R.; Palmer, J.D. Restriction Site Mapping of the Chloroplast DNA Inverted Repeat: A Molecular Phylogeny of the Asteridae. Ann. Missouri Bot. Gard. 1992, 79, 266. [Google Scholar] [CrossRef][Green Version]
- Androsiuk, P.; Jastrzębski, J.P.; Paukszto, Ł.; Makowczenko, K.; Okorski, A.; Pszczółkowska, A.; Chwedorzewska, K.J.; Górecki, R.; Giełwanowska, I. Evolutionary Dynamics of the Chloroplast Genome Sequences of Six Colobanthus Species. Sci. Rep. 2020, 10, 11522. [Google Scholar] [CrossRef]
- Bendich, A.J. Why Do Chloroplasts and Mitochondria Contain so Many Copies of Their Genome? BioEssays 1987, 6, 279–282. [Google Scholar] [CrossRef]
- Twyford, A.D.; Ness, R.W. Strategies for Complete Plastid Genome Sequencing. Mol. Ecol. Resour. 2017, 17, 858–868. [Google Scholar] [CrossRef]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What Can We Do with 1000 Plastid Genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and Participation across 20 Years of Plant Genome Sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef]
- Turudić, A.; Liber, Z.; Grdiša, M.; Jakše, J.; Varga, F.; Šatović, Z. Towards the Well-Tempered Chloroplast DNA Sequences. Plants 2021, 10, 1360. [Google Scholar] [CrossRef]
- Mehl, T.; Gruenstaeudl, M. Airpg: Automatically Accessing the Inverted Repeats of Archived Plastid Genomes. BMC Bioinformatics 2021, 22, 413. [Google Scholar] [CrossRef] [PubMed]
- Lantz, H.; Dominguez Del Angel, V.; Hjerde, E.; Sterck, L.; Capella-Gutierrez, S.; Notredame, C.; Vinnere Pettersson, O.; Amselem, J.; Bouri, L.; Bocs, S.; et al. Ten Steps to Get Started in Genome Assembly and Annotation. F1000Research 2018, 7, 148. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De Novo Assembly of Human Genomes with Massively Parallel Short Read Sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, I. ABySS: A Parallel Assembler for Short Read Sequence Data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- McKain, M.R.; Wilson, M.C. Fast-Plast: A Rapid de Novo Assembly Pipeline for Whole Chloroplast Genomes. Available online: https://github.com/mrmckain/Fast-Plast (accessed on 1 December 2020).
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic Annotation of Organellar Genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Kchouk, M.; Gibrat, J.F.; Elloumi, M. Generations of Sequencing Technologies: From First to Next Generation. Biol. Med. 2017, 9, 1–8. [Google Scholar] [CrossRef]
- Miller, J.R.; Koren, S.; Sutton, G. Genomics Assembly Algorithms for Next-Generation Sequencing Data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Guyeux, C.; Charr, J.C.; Tran, H.T.M.; Furtado, A.; Henry, R.J.; Crouzillat, D.; Guyot, R.; Hamon, P. Evaluation of Chloroplast Genome Annotation Tools and Application to Analysis of the Evolution of Coffee Species. PLoS ONE 2019, 14, e0216347. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, K.; Lucas, S.J. Comparison of Different Annotation Tools for Characterization of the Complete Chloroplast Genome of Corylus Avellana Cv Tombul. BMC Genomics 2019, 20, 874. [Google Scholar] [CrossRef]
- Zhong, X. Assembly, Annotation and Analysis of Chloroplast Genomes. 2020. Available online: https://research-repository.uwa.edu.au/en/publications/assembly-annotation-and-analysis-of-chloroplast-genomes (accessed on 31 December 2021).
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An Online Program for the Versatile Plotting of Organelle Genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A Software Package for Rapid, Accurate, and Flexible Batch Annotation of Plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Huang, D.I.; Cronk, Q.C.B. Plann: A Command-Line Application for Annotating Plastome Sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef]
- Achaz, G.; Boyer, F.; Rocha, E.P.C.; Viari, A.; Coissac, E. Repseek, a Tool to Retrieve Approximate Repeats from Large DNA Sequences. Bioinformatics 2007, 23, 119–121. [Google Scholar] [CrossRef]
- Palmer, J.D.; Nugent, J.M.; Herbon, L.A. Unusual Structure of Geranium Chloroplast DNA: A Triple-Sized Inverted Repeat, Extensive Gene Duplications, Multiple Inversions, and Two Repeat Families. Proc. Natl. Acad. Sci. USA 1987, 84, 769–773. [Google Scholar] [CrossRef]
- Xu, G.; Xu, W. Complete Chloroplast Genomes of Chinese Wild-Growing Vitis Species: Molecular Structures and Comparative and Adaptive Radiation Analysis. Protoplasma 2021, 258, 559–571. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2012; pp. 103–126. [Google Scholar]
- Jung, H.; Ventura, T.; Sook Chung, J.; Kim, W.J.; Nam, B.H.; Kong, H.J.; Kim, Y.O.; Jeon, M.S.; Eyun, S.I. Twelve Quick Steps for Genome Assembly and Annotation in the Classroom. PLoS Comput. Biol. 2020, 16, e1008325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Ali, A. CHLOROPLOT Tool for Organelle Genome Visualization. Available online: https://irscope.shinyapps.io/Chloroplot/ (accessed on 31 December 2021).
- Bellot, S.; Renner, S.S. The Plastomes of Two Species in the Endoparasite Genus Pilostyles (Apodanthaceae) Each Retain Just Five or Six Possibly Functional Genes. Genome Biol. Evol. 2016, 8, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, D.; Senalik, D.; Ames, M.; Zhu, H.; Steffan, S.A.; Harbut, R.; Polashock, J.; Vorsa, N.; Gillespie, E.; Kron, K.; et al. Complete Plastid Genome Sequence of Vaccinium Macrocarpon: Structure, Gene Content, and Rearrangements Revealed by next Generation Sequencing. Tree Genet. Genomes 2013, 9, 489–498. [Google Scholar] [CrossRef]
- Martínez-Alberola, F.; Del Campo, E.M.; Lázaro-Gimeno, D.; Mezquita-Claramonte, S.; Molins, A.; Mateu-Andrés, I.; Pedrola-Monfort, J.; Casano, L.M.; Barreno, E. Balanced Gene Losses, Duplications and Intensive Rearrangements Led to an Unusual Regularly Sized Genome in Arbutus Unedo Chloroplasts. PLoS ONE 2013, 8, e79685. [Google Scholar] [CrossRef] [PubMed]
- Roquet, C.; Coissac, É.; Cruaud, C.; Boleda, M.; Boyer, F.; Alberti, A.; Gielly, L.; Taberlet, P.; Thuiller, W.; Van Es, J.; et al. Understanding the Evolution of Holoparasitic Plants: The Complete Plastid Genome of the Holoparasite Cytinus Hypocistis (Cytinaceae). Ann. Bot. 2016, 118, 885–896. [Google Scholar] [CrossRef]
- Wojciechowski, M.F.; Sanderson, M.J.; Hu, J.M. Evidence on the Monophyly of Astragalus (Fabaceae) and Its Major Subgroups Based on Nuclear Ribosomal DNA ITS and Chloroplast DNA TrnL Intron Data. Syst. Bot. 1999, 24, 409. [Google Scholar] [CrossRef]
- Wojciechowski, M.F.; Sanderson, M.J.; Steele, K.P.; Liston, A. Molecular Phylogeny of the “Temperate Herbaceous Tribes” of Papilionoid Legumes: A Supertree Approach. Adv. Legume Syst. 2000, 9, 277–298. [Google Scholar]
- Zhang, J.; Ruhlman, T.A.; Sabir, J.S.M.; Blazier, J.C.; Weng, M.L.; Park, S.; Jansen, R.K. Coevolution between Nuclear-Encoded DNA Replication, Recombination, and Repair Genes and Plastid Genome Complexity. Genome Biol. Evol. 2016, 8, 622–634. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Zhang, J.; Blazier, J.C.; Sabir, J.S.M.; Jansen, R.K. Recombination-Dependent Replication and Gene Conversion Homogenize Repeat Sequences and Diversify Plastid Genome Structure. Am. J. Bot. 2017, 104, 559–572. [Google Scholar] [CrossRef]
- Jin, D.M.; Wicke, S.; Gan, L.; Yang, J.B.; Jin, J.J.; Yi, T.S. The Loss of the Inverted Repeat in the Putranjivoid Clade of Malpighiales. Front. Plant Sci. 2020, 11, 942. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zeng, X.; Ren, G.; Liu, Z. CGAP: A New Comprehensive Platform for the Comparative Analysis of Chloroplast Genomes. BMC Bioinform. 2013, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an Integrated Web Server for the Annotation, Visualization, Analysis, and GenBank Submission of Completely Sequenced Chloroplast Genome Sequences. BMC Genomics 2012, 13, 715. [Google Scholar] [CrossRef]
- McKain, M.R.; Hartsock, R.H.; Wohl, M.M.; Kellogg, E.A. Verdant: Automated Annotation, Alignment and Phylogenetic Analysis of Whole Chloroplast Genomes. Bioinformatics 2017, 33, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, J.I.; Jeong, Y.S.; Yi, G. AGORA: Organellar Genome Annotation from the Amino Acid and Nucleotide References. Bioinformatics 2018, 34, 2661–2663. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an Integrated Plastome Sequence Annotator and Analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Back, N.; Lang, B. MFannot, Organelle Genome Annotation Websever. Available online: https://megasun.bch.umontreal.ca/apps/mfannot/ (accessed on 31 December 2021).
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A Tool for the Easy Generation of High-Quality Custom Graphical Maps of Plastid and Mitochondrial Genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A Suite of Tools for Generating Physical Maps of Plastid and Mitochondrial Genomes and Visualizing Expression Data Sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method | Description in Publication | Code Analysis |
|---|---|---|
| Chloë | Does not mention IR identification. | Chloë assembles reverse-complement blocks of ~20 bp in size into long sequences allowing up to a few bases (≤10 bp) of mismatches. |
| Chloroplot | The publication contains a description of the main steps. The method can detect nonidentical IR regions allowing small stretches of mismatches. | Chloroplot assembles reverse-complement blocks of size 100 bp into long sequences, sorts the assembled sequences by position, and finds the longest gap between sequences. Assembled sequences downstream of the gap are merged into IRa, and sequences upstream of the gap are merged into IRb. Identified IRa and IRb are checked to ensure that they do not differ in length by more than 1000 bp. In addition, mismatches between the regions are calculated and checked to support the length differences between the regions. If the procedure fails, the same procedure is attempted with blocks of 1000 bp in length. |
| GeSeq | The minimum length for IR annotation is 200 bp. GeSeq only annotates the longest identical IR pair that matches within a submitted sequence. | GeSeq finds leftmost and rightmost exact matches with a size of at least 2000 bp and designates the regions between the match positions as IRa and IRb. |
| ORG.Annotate | - | ORG.Annotate uses repseek to find approximate long matches, scores these matches according to the results of a query of the input sequence in a database containing 45 LSC and 72 SSC regions, and considers the highest scoring matches as IRs. |
| PGA | The IR boundary annotation is performed with a self-blastn search. One parameter can be adjusted to determine IR boundaries: the minimum allowed IR length (default = 1000). | PGA performs self-blasts (-perc_identity 99) on a duplicated sequence to find the maximum inverted match with a length of at least 1000 bp. |
| Plann | Does not mention IR identification. | Plann performs self-blasts (-evalue 1 × 10−200) on an original sequence to find the maximum inverted match with a length between 10,000 and 50,000 bp. |
| Sequence Numbers | Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | NCBI |
|---|---|---|---|---|---|---|---|
| IRs not identified | 109 | 96 | 124 | 4 | 112 | 130 | 131 |
| (77.30%) | (68.09%) | (87.94%) | (2.84%) | (79.43%) | (92.20%) | (92.91%) | |
| IRs identified | 32 | 45 | 17 | 137 | 29 | 11 | 10 |
| (22.70%) | (31.91%) | (12.06%) | (97.16%) | (20.57%) | (7.80%) | (7.09%) | |
| IRs larger than 10 kb | 11 | 5 | 10 | 10 | 10 | 11 | 4 |
| Species (asterids/Ericaceae) with IRs larger than 10 kb | |||||||
| Agapetes malipoensis | + | - | + | + | + | + | - |
| Gaultheria griffithiana | + | - | + | + | + | + | + |
| Rhododendron delavayi | + | - | + | + | + | + | - |
| Rhododendron griersonianum | + | - | + | + | + | + | + |
| Rhododendron kawakamii | + | - | - | + | - | + | + |
| Rhododendron platypodum | + | - | + | + | + | + | + |
| Vaccinium macrocarpon | + | + | + | + | + | + | - |
| Vaccinium oldhamii | + | + | + | + | + | + | - |
| Species (rosids/Geraniaceae/Erodium) with IRs larger than 10 kb | |||||||
| Erodium absinthoides | + | + | + | + | + | + | - |
| Erodium chrysanthum | + | + | + | + | + | + | - |
| Erodium gruinum | + | + | + | - | + | + | - |
| Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | NCBI | |
|---|---|---|---|---|---|---|---|
| Identical IRs | 3720 | 3558 | 3460 | 2783 | 3011 | 3148 | 1819 |
| (93.09%) | (89.04%) | (86.59%) | (69.64%) | (75.35%) | (78.78%) | (45.52%) | |
| Different IRs | 264 | 411 | 265 | 1212 | 965 | 795 | 444 |
| (6.61%) | (10.28%) | (6.63%) | (30.33%) | (24.15%) | (19.90%) | (11.11%) | |
| No IRs | 12 | 27 | 271 | 1 | 20 | 53 | 1733 |
| (0.30%) | (0.68%) | (6.78%) | (0.03%) | (0.50%) | (1.32%) | (43.37%) |
| Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | NCBI | |
|---|---|---|---|---|---|---|---|
| No wrapping | 3434 | 3386 | 3655 | 3995 | 3263 | 3943 | 2248 |
| (86.19%) | (85.31%) | (98.12%) | (100.00%) | (82.07%) | (100.00%) | (99.34%) | |
| Wrapped | 550 | 583 | 70 | 0 | 713 | 0 | 15 |
| (13.81%) | (14.69%) | (1.88%) | (17.93%) | (0.66%) | |||
| Total | 3984 | 3969 | 3725 | 3995 | 3976 | 3943 | 2263 |
| Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | NCBI | |
|---|---|---|---|---|---|---|---|
| With ambiguous characters | |||||||
| Identical IRs | 435 | 375 | 232 | 325 | 338 | 363 | 196 |
| (82.70%) | (71.29%) | (44.11%) | (61.79%) | (64.26%) | (69.01%) | (37.26%) | |
| Different IRs | 86 | 140 | 67 | 201 | 180 | 152 | 72 |
| (16.35%) | (26.61%) | (12.74%) | (38.21%) | (34.22%) | (28.90%) | (13.39%) | |
| No IRs | 5 | 11 | 227 | 0 | 8 | 11 | 258 |
| (0.95%) | (2.09%) | (43.15%) | (0.00%) | (1.52%) | (2.09%) | (49.05%) | |
| Without ambiguous characters | |||||||
| Identical IRs | 3285 | 3183 | 3228 | 2458 | 2673 | 2785 | 1623 |
| (94.67%) | (91.73%) | (93.02%) | (70.84%) | (77.03%) | (80.26%) | (46.77%) | |
| Different IRs | 178 | 271 | 198 | 1011 | 785 | 643 | 372 |
| (5.13%) | (7.81%) | (5.71%) | (29.13%) | (22.62%) | (15.53%) | (10.72%) | |
| No IRs | 7 | 16 | 44 | 1 | 12 | 42 | 1475 |
| (0.20%) | (0.46%) | (1.27%) | (0.03%) | (0.35%) | (1.21%) | (42.51%) | |
| Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | NCBI | |
|---|---|---|---|---|---|---|---|
| IRs differ | 264 | 411 | 265 | 1212 | 965 | 795 | 444 |
| 0 bp | 264 | 177 | 99 | 816 | 522 | 377 | 151 |
| (100.00%) | (43.07%) | (37.36%) | (67.33%) | (54.09%) | (47.42%) | (34.01%) | |
| 1–10 bp | 0 | 122 | 145 | 315 | 331 | 359 | 116 |
| (0.00%) | (29.68%) | (54.71%) | (25.99%) | (34.30%) | (45.16%) | (26.13%) | |
| 11–100 bp | 0 | 75 | 17 | 81 | 111 | 59 | 117 |
| (0.00%) | (18.25%) | (6.42%) | (6.68%) | (11.50%) | (7.42%) | (26.35%) | |
| >100 bp | 0 | 37 | 4 | 0 | 1 | 0 | 60 |
| (0.00%) | (9.00%) | (1.51%) | (0.11%) | (13.51%) |
| Chloë | Chloroplot | GeSeq | ORG.Annotate | PGA | Plann | |
|---|---|---|---|---|---|---|
| Chloë | 63 | 512 | 217 | 10 | 253 | |
| Chloroplot | 270 | 573 | 287 | 61 | 327 | |
| GeSeq | 135 | 12 | 53 | 13 | 85 | |
| ORG.Annotate | 571 | 412 | 754 | 184 | 326 | |
| PGA | 511 | 350 | 833 | 326 | 458 | |
| Plann | 347 | 190 | 527 | 7 | 16 |
| Taxonomic Rank | Asterids (IRL/IR) | Rosids (IRL/IR) | IRL Dataset (Asterids + Rosids) | IR Dataset (Asterids + Rosids) | Total |
|---|---|---|---|---|---|
| Family | 1/64 | 6/82 | 7 | 146 | 151 |
| Genus | 9/569 | 31/750 | 40 | 1319 | 1359 |
| Species/subspecies | 16/1906 | 125/2084 | 141 | 3990 | 4131 |
| Number of sequences | 16/1908 | 125/2088 | 141 | 3996 | 4137 |
| Clade | Family | Genus/Subclade | No. of Sequences | References |
|---|---|---|---|---|
| asterids | Ericaceae | 9 genera 1 | 16 | [77,78] |
| rosids | Apodanthaceae | Pilostyles | 2 | [76] |
| rosids | Cytinaceae | Cytinus | 1 | [79] |
| rosids | Fabaceae | IRLC 2 | 101 | [80,81] |
| rosids | Geraniaceae | Erodium | 10 | [38,39] |
| rosids | Geraniaceae | Monsonia | 3 | [39,82,83] |
| rosids | Lophopyxidaceae | Lophopyxis | 1 | [84] |
| rosids | Putranjivaceae | 2 genera 3 | 7 | [84] |
| Method | Programming Language | Last Code Change | Local | Web Application | Reference |
|---|---|---|---|---|---|
| Chloë | Julia | 08.12.2020. | + | + | [66] |
| Chloroplot | R | 19.08.2021. | + | + | [67] |
| GeSeq 1 | D | 18.12.2020. | - | + | [59] |
| ORG.Annotate | bash | 08.11.2021. | + | - | - |
| PGA | Perl | 29.10.2020. | + | - | [68] |
| Plann | Perl | 26.01.2017. | + | - | [69] |
| airpg | python | 17.09.2021. | + | - | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turudić, A.; Liber, Z.; Grdiša, M.; Jakše, J.; Varga, F.; Šatović, Z. Chloroplast Genome Annotation Tools: Prolegomena to the Identification of Inverted Repeats. Int. J. Mol. Sci. 2022, 23, 10804. https://doi.org/10.3390/ijms231810804
Turudić A, Liber Z, Grdiša M, Jakše J, Varga F, Šatović Z. Chloroplast Genome Annotation Tools: Prolegomena to the Identification of Inverted Repeats. International Journal of Molecular Sciences. 2022; 23(18):10804. https://doi.org/10.3390/ijms231810804
Chicago/Turabian StyleTurudić, Ante, Zlatko Liber, Martina Grdiša, Jernej Jakše, Filip Varga, and Zlatko Šatović. 2022. "Chloroplast Genome Annotation Tools: Prolegomena to the Identification of Inverted Repeats" International Journal of Molecular Sciences 23, no. 18: 10804. https://doi.org/10.3390/ijms231810804
APA StyleTurudić, A., Liber, Z., Grdiša, M., Jakše, J., Varga, F., & Šatović, Z. (2022). Chloroplast Genome Annotation Tools: Prolegomena to the Identification of Inverted Repeats. International Journal of Molecular Sciences, 23(18), 10804. https://doi.org/10.3390/ijms231810804