
Comparative Analysis of Chloroplast Genome in Saccharum spp. and Related Members of ‘Saccharum Complex’


Abstract
1. Introduction
2. Results
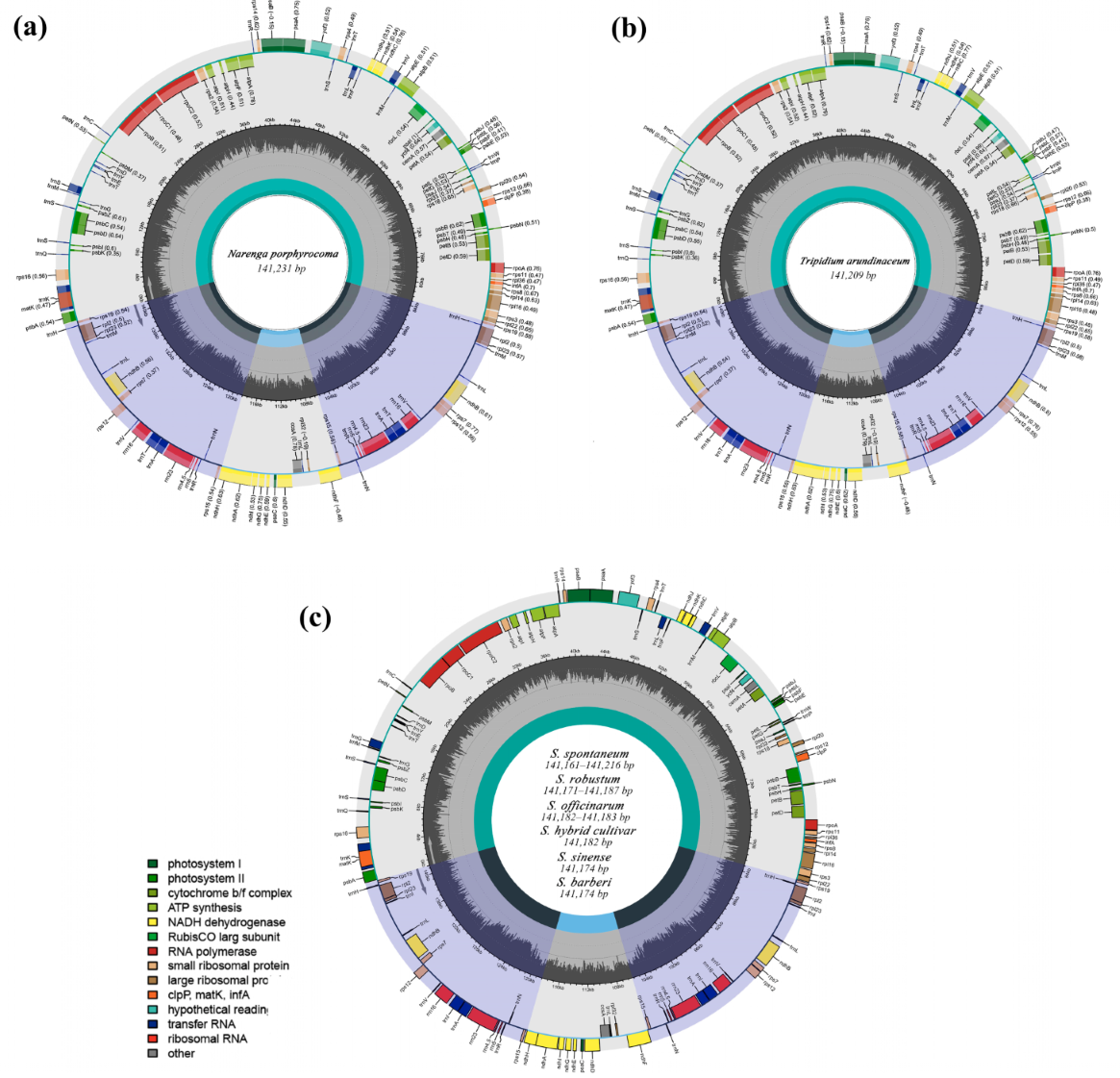
2.1. Plastid Genome Structure and Organization
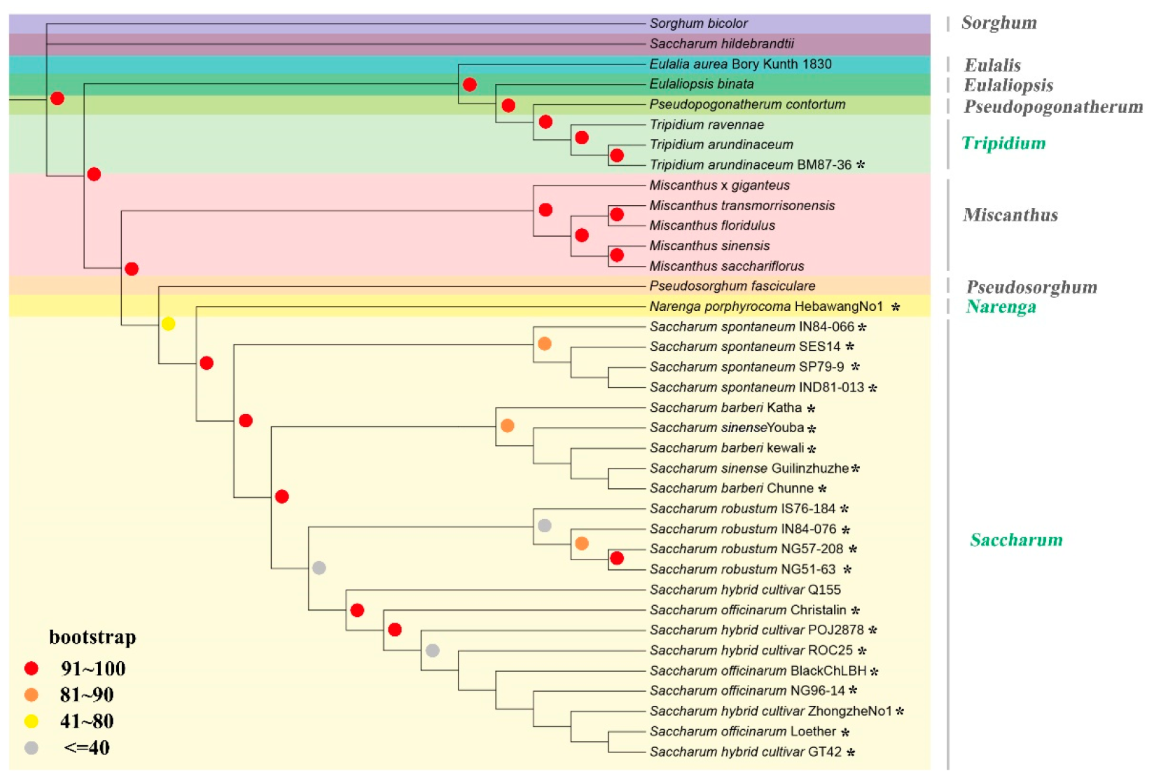
2.2. Phylogenetic Analysis of Saccharinae Species
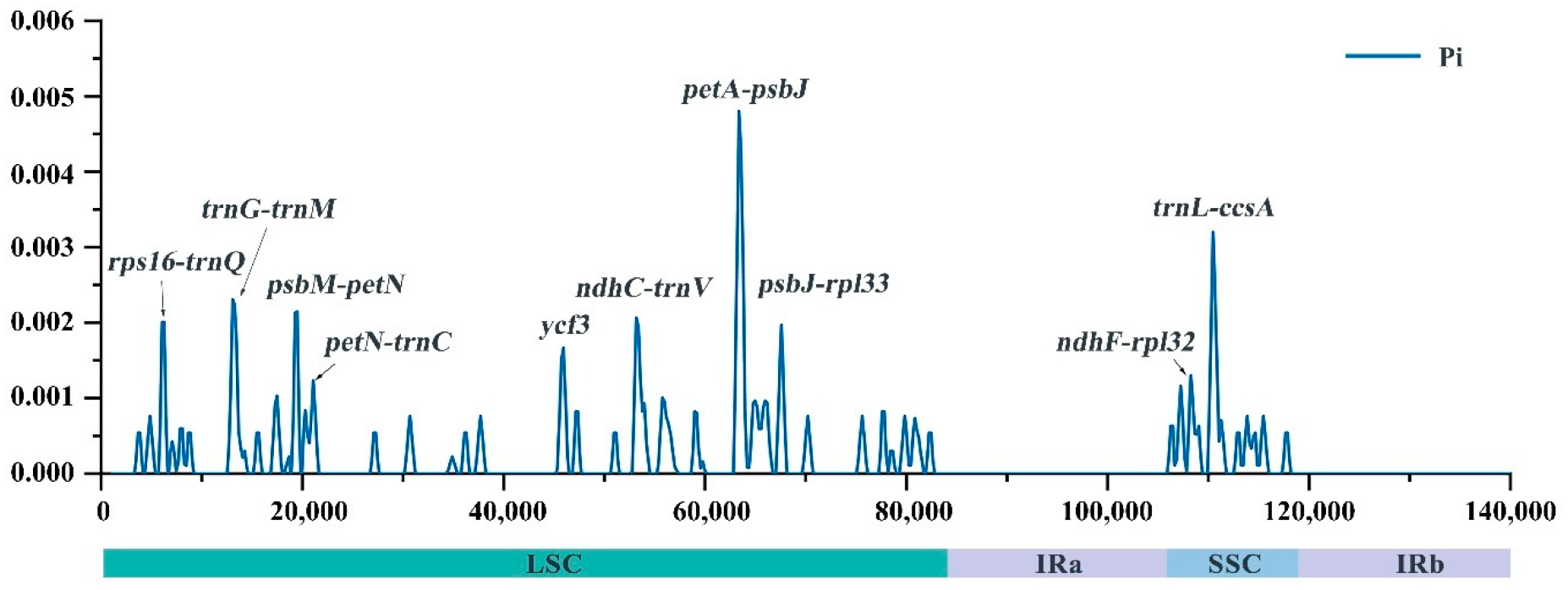
2.3. Repeat Analysis and Genetic Diversity Assessment
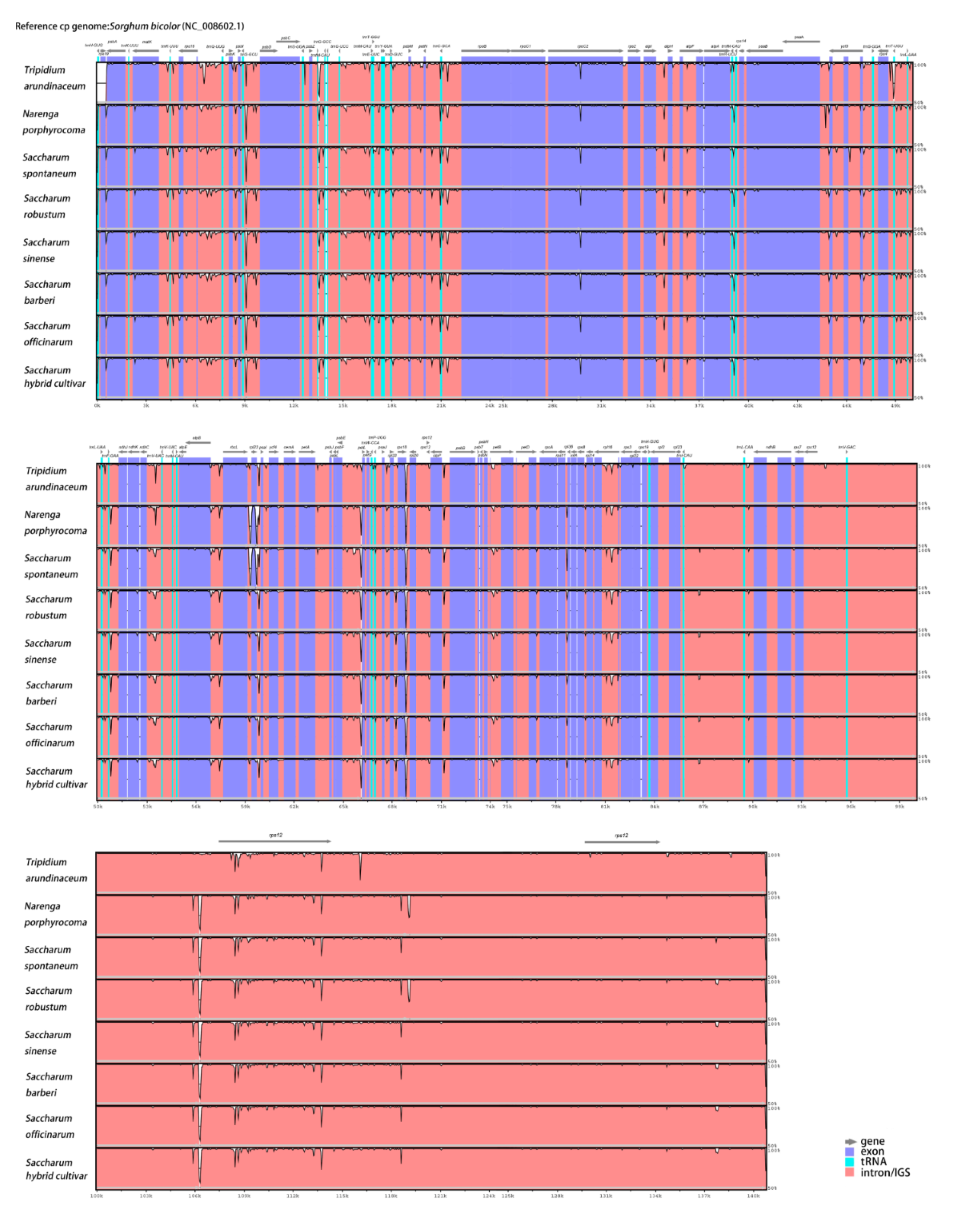
2.4. Comparison of Chloroplast Genome Sequences
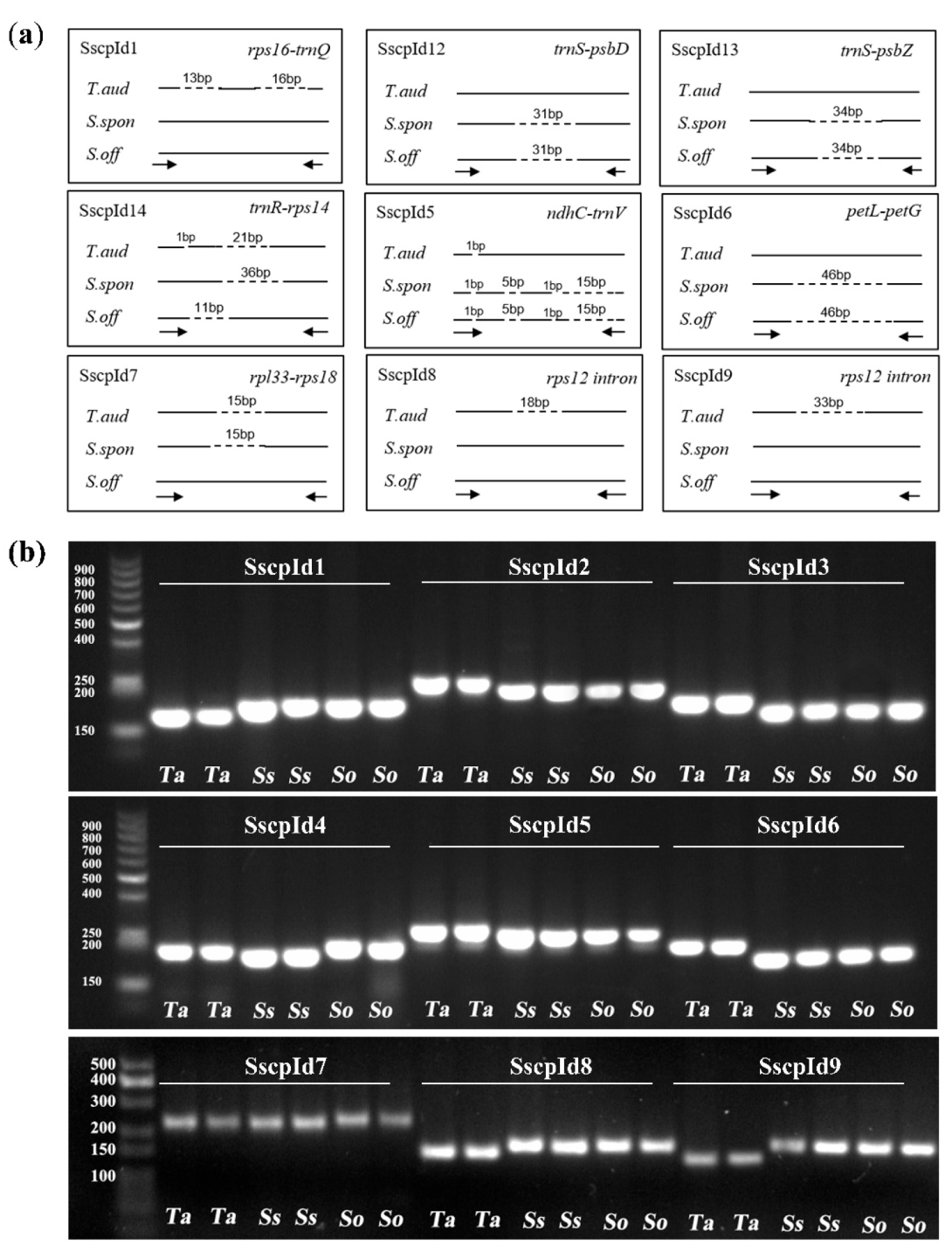
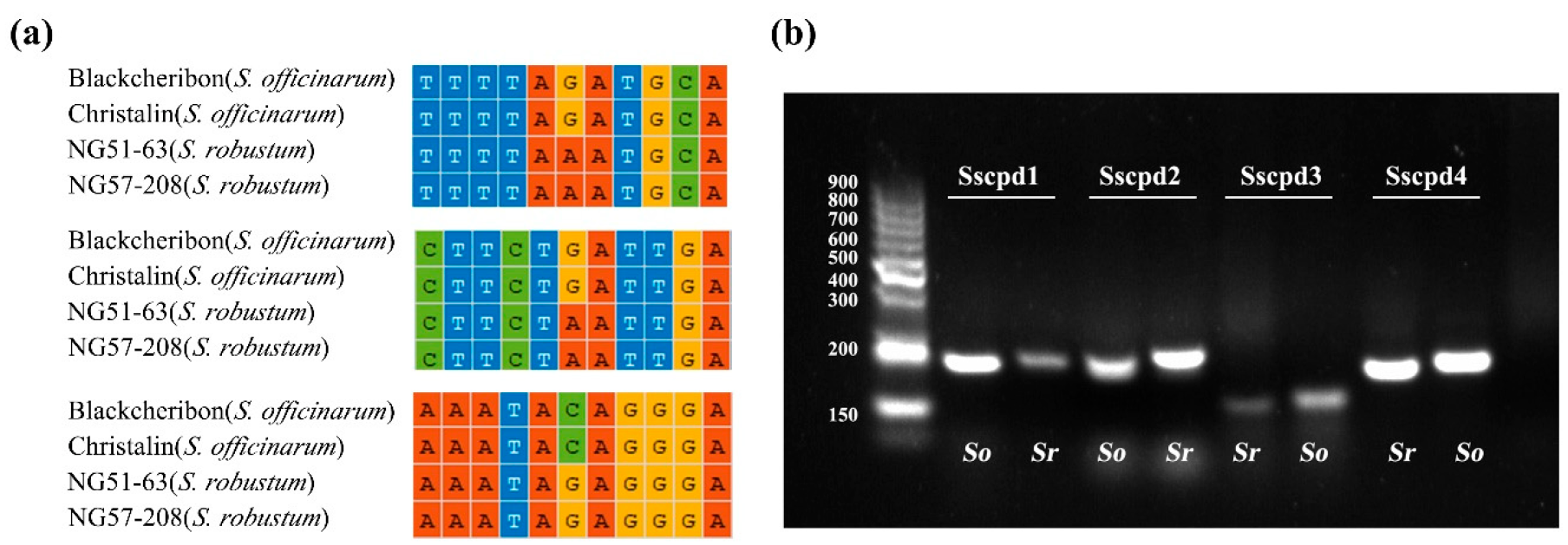
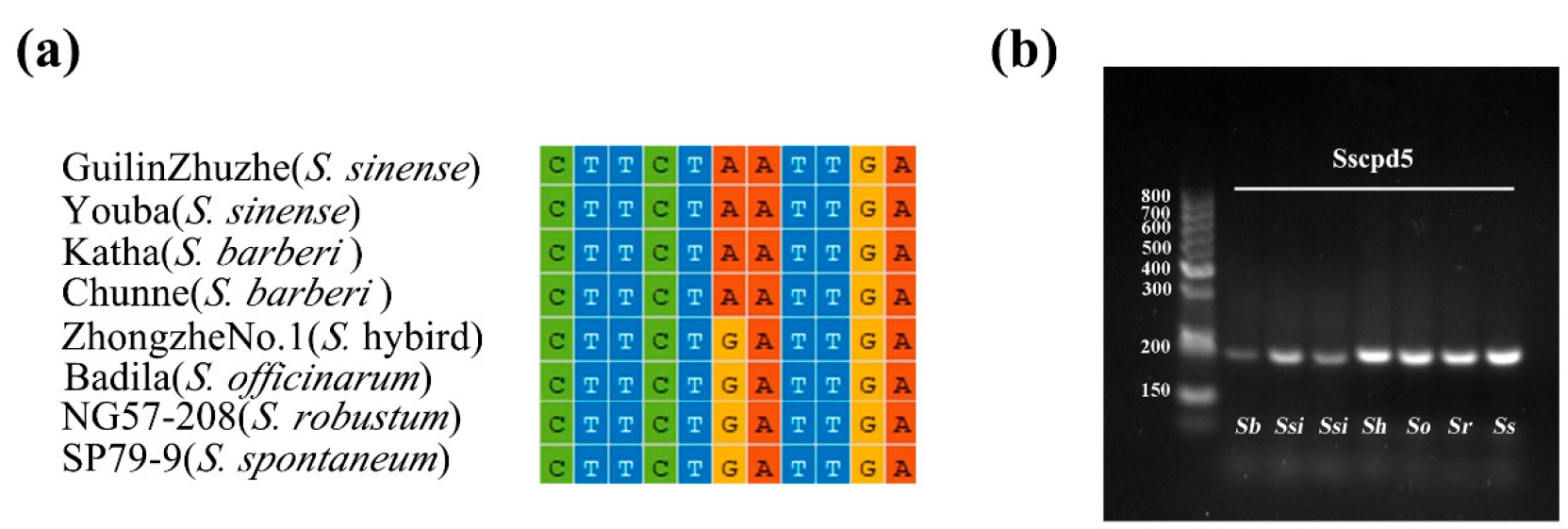
2.5. Development of Markers to Discriminate Saccharum and Sugarcane-Relative Species
3. Discussion
3.1. Differences in Gene and Structure of Chloroplast Genomes among Saccharinae Species
3.2. Plastid View on the Evolutionary Relationships within Saccharinae
3.3. Development of Markers at the Inter- and Intra-Species Level for Saccharum
4. Materials and Methods
4.1. DNA Extraction, Library Construction, and Sequencing
4.2. Plastome Assemblies and Annotation
4.3. Phylogenetic Analyses
4.4. Plastome Polymorphism
4.5. Comparative Genome Alignment
4.6. Development and Validation of InDel and dCAPS Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- James, G.; Tate, B. Sugarcane; Wiley Online Library: Hoboken, NJ, USA, 2004. [Google Scholar]
- Yu, F.; Chai, J.; Li, X.; Yu, Z.; Yang, R.; Ding, X.; Wang, Q.; Wu, J.; Yang, X.; Deng, Z. Chromosomal Characterization of Tripidium arundinaceum Revealed by Oligo-FISH. Int. J. Mol. Sci. 2021, 22, 8539. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Wang, Q.; Qiu, Y.; Qin, Y.; Li, X.; Wu, Q.; He, W.; Guo, Y.; Zhang, W.; Chen, J.; et al. Production, Identification and Characterization of Erianthus rockii × Narenga porphyrocoma Intergeneric Hybrids as a New Germplasm for Sugarcane Breeding and Genetic Research. Sugar Tech 2020, 22, 389–395. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, R.; Ou, H.; Gui, Y.; Wei, J.; Zhou, H.; Tan, H.; Li, Y. Comprehensive transcriptome analysis reveals genes in response to water deficit in the leaves of Saccharum narenga (Nees ex Steud.) hack. BMC Plant Biol. 2018, 18, 250. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, X.; Zeng, Q.; Zheng, Z.; Wu, C.; Yang, R.; Feng, X.; Wu, Z.; Fan, L.; Huang, Z. Sugarcane Breeding, Germplasm Development and Related Molecular Research in China. Sugar Tech 2022, 24, 73–85. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, H.; Pan, Y.; Lu, X. Genetic variability among the chloroplast genomes of sugarcane (Saccharum spp.) and its wild progenitor species Saccharum spontaneum L. Genet. Mol. Res. 2014, 13, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- C.P.W.G.; Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794-12797. [Google Scholar]
- Dong, W.; Xu, C.; Li, W.; Xie, X.; Lu, Y.; Liu, Y.; Jin, X.; Suo, Z. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 2017, 8, 1148. [Google Scholar] [CrossRef]
- Wu, L.; Nie, L.; Xu, Z.; Li, P.; Wang, Y.; He, C.; Song, J.; Yao, H. Comparative and phylogenetic analysis of the complete chloroplast genomes of three Paeonia Section Moutan Species (Paeoniaceae). Front. Genet. 2020, 11, 980. [Google Scholar] [CrossRef]
- Celiński, K.; Kijak, H.; Wojnicka-Półtorak, A.; Buczkowska-Chmielewska, K.; Sokołowska, J.; Chudzińska, E. Effectiveness of the DNA barcoding approach for closely related conifers discrimination: A case study of the Pinus mugo complex. C. R. Biol. 2017, 340, 339–348. [Google Scholar] [CrossRef]
- Loeuille, B.; Thode, V.; Siniscalchi, C.; Andrade, S.; Rossi, M.; Pirani, J.R. Extremely low nucleotide diversity among thirty-six new chloroplast genome sequences from Aldama (Heliantheae, Asteraceae) and comparative chloroplast genomics analyses with closely related genera. PeerJ 2021, 9, e10886. [Google Scholar] [CrossRef]
- Sokołowska, J.; Fuchs, H.; Celiński, K. New insight into taxonomy of European mountain pines, Pinus mugo complex, based on complete chloroplast genomes sequencing. Plants 2021, 10, 1331. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Yoon, Y.-M.; Park, J.-C.; Park, J.-H.; Kim, K.-H.; Kim, Y.-K.; Son, J.-H.; Park, T.-I. De Novo Transcriptome Assembly and SNP Discovery for the Development of dCAPS Markers in Oat. Agronomy 2022, 12, 184. [Google Scholar] [CrossRef]
- Nishijima, R.; Yoshida, K.; Sakaguchi, K.; Yoshimura, S.I.; Sato, K.; Takumi, S. RNA Sequencing-Based Bulked Segregant Analysis Facilitates Efficient D-genome Marker Development for a Specific Chromosomal Region of Synthetic Hexaploid Wheat. Int. J. Mol. Sci. 2018, 19, 3749. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, B.; Li, Z.; Guo, T.; Zhao, J.; Guan, Z.; Liu, K. Identification and characterization of a new dwarf locus DS-4 encoding an Aux/IAA7 protein in Brassica napus. Theor. Appl. Genet. 2019, 132, 1435–1449. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Tsudzuki, T.; Takahashi, S.; Shimada, H.; Kadowaki, K.-i. Complete Nucleotide Sequence of the Sugarcane (Saccharum officinarum) Chloroplast Genome: A Comparative Analysis of Four Monocot Chloroplast Genomes. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2004, 11, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Calsa Júnior, T.; Carraro, D.M.; Benatti, M.R.; Barbosa, A.C.; Kitajima, J.P.; Carrer, H. Structural features and transcript-editing analysis of sugarcane (Saccharum officinarum L.) chloroplast genome. Curr. Genet. 2004, 46, 366–373. [Google Scholar] [CrossRef]
- Vidigal, P.M.; Coelho, A.S.; Novaes, E.; Barbosa, M.H.; Peternelli, L.A. Complete Chloroplast Genome Sequence and Annotation of the Saccharum Hybrid Cultivar RB867515. Genome Announc. 2016, 4, e01157-16. [Google Scholar] [CrossRef]
- Evans, D.L.; Joshi, S.V. Complete chloroplast genomes of Saccharum spontaneum, Saccharum officinarum and Miscanthus floridulus (Panicoideae: Andropogoneae) reveal the plastid view on sugarcane origins. Syst. Biodivers. 2016, 14, 548–571. [Google Scholar] [CrossRef]
- Xu, F.; He, L.; Gao, S.; Su, Y.; Li, F.; Xu, L. Comparative Analysis of two Sugarcane Ancestors Saccharum officinarum and S. spontaneum based on Complete Chloroplast Genome Sequences and Photosynthetic Ability in Cold Stress. Int. J. Mol. Sci. 2019, 20, 3828. [Google Scholar] [CrossRef]
- Lloyd Evans, D.; Joshi, S.V.; Wang, J. Whole chloroplast genome and gene locus phylogenies reveal the taxonomic placement and relationship of Tripidium (Panicoideae: Andropogoneae) to sugarcane. BMC Evol. Biol. 2019, 19, 33. [Google Scholar] [CrossRef]
- Tsuruta, S.I.; Ebina, M.; Kobayashi, M.; Takahashi, W. Complete Chloroplast Genomes of Erianthus arundinaceus and Miscanthus sinensis: Comparative Genomics and Evolution of the Saccharum Complex. PLoS ONE 2017, 12, e0169992. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.L.; Hughes, B. Complete Chloroplast Genomes of Saccharum giganteum, Saccharum longisetosum, Cleistachne sorghoides, Sarga timorense, Narenga porphyrocoma and Tripsacum dactyloides. Comparisons with ITS phylogeny and Placement within Saccharum. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lloyd Evans, D.; Hlongwane, T.T.; Joshi, S.V.; Riano Pachon, D.M. The sugarcane mitochondrial genome: Assembly, phylogenetics and transcriptomics. PeerJ 2019, 7, e7558. [Google Scholar] [CrossRef] [PubMed]
- Welker, C.A.D.; McKain, M.R.; Vorontsova, M.S.; Peichoto, M.C.; Kellogg, E.A. Plastome phylogenomics of sugarcane and relatives confirms the segregation of the genus Tripidium (Poaceae: Andropogoneae). Taxon 2019, 68, 246–267. [Google Scholar] [CrossRef]
- Lloyd Evans, D.; Hughes, B.; Joshi, V. Comparative whole plastome and low copy number phylogenetics of the core Saccharinae and Sorghinae. bioRxiv 2022. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Pompidor, N.; Charron, C.; Hervouet, C.; Bocs, S.; Droc, G.; Rivallan, R.; Manez, A.; Mitros, T.; Swaminathan, K.; Glaszmann, J.-C.; et al. Three founding ancestral genomes involved in the origin of sugarcane. Ann. Bot. 2021, 127, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Welker, C.A.; Souza-Chies, T.T.; Longhi-Wagner, H.M.; Peichoto, M.C.; McKain, M.R.; Kellogg, E.A. Phylogenetic analysis of Saccharum sl (Poaceae; Andropogoneae), with emphasis on the circumscription of the South American species. Am. J. Bot. 2015, 102, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Elkonin, L.; Kibalnik, O.; Zavalishina, A.; Gerashchenkov, G.; Rozhnova, N. Genetic function of cytoplasm in plants with special emphasis on sorghum. In Chloroplasts and Cytoplasm. Structure and Function; Nova Science Publishers: New York, NY, USA, 2018; pp. 97–155. [Google Scholar]
- Zhang, Y.; Fang, Z.; Wang, Q.; Liu, Y.; Yang, L.; Zhuang, M.; Sun, P. Chloroplast subspecies-specific SNP detection and its maternal inheritance in Brassica oleracea L. by using a dCAPS marker. J. Hered. 2012, 103, 606–611. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Guo, Y.; Huang, F.; Wang, Q.; Chai, J.; Yu, F.; Wu, J.; Zhang, M.; Deng, Z. Authenticity Identification of Saccharum officinarum and Saccharum spontaneum Germplasm Materials. Agronomy 2022, 12, 819. [Google Scholar] [CrossRef]
- D’Hont, A.; Rao, P.S.; Feldmann, P.; Grivet, L.; Islam-Faridi, N.; Taylor, P.; Glaszmann, J.C. Identification and characterisation of sugarcane intergeneric hybrids, Saccharum officinarum × Erianthus arundinaceus, with molecular markers and DNA in situ hybridisation. Theor. Appl. Genet. 1995, 91, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Pachakkil, B.; Terajima, Y.; Ohmido, N.; Ebina, M.; Irei, S.; Hayashi, H.; Takagi, H. Cytogenetic and agronomic characterization of intergeneric hybrids between Saccharum spp. hybrid and Erianthus arundinaceus. Sci. Rep. 2019, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, J.; Wang, P.; Lin, Y.; Fu, C.; Deng, Z.; Wang, Q.; Li, Q.; Chen, R.; Zhang, M. Characterization of Chromosome Inheritance of the Intergeneric BC2 and BC3 Progeny between Saccharum spp. and Erianthus arundinaceus. PLoS ONE 2015, 10, e0133722. [Google Scholar] [CrossRef]
- Wood, B. Restriction fragment length polymorphism distinguishes members of Saccharum sinense and S. barberi. Plant Physiol. 1987, 83, 15. [Google Scholar]
- Zhang, H.; Li, F.; He, L.; Zhong, H.; Yang, Q.; He, S. Identification of sugarcane interspecies hybrids with RAPDs. Afr. J. Biotechnol. 2008, 7, 1072–1074. [Google Scholar]
- Manigbas, N.L.; Villegas, L.C. Microsatellite markers in hybridity tests to identify true hybrids of sugarcane. Philipp. J. Crop Sci. 2004, 29, 23–32. [Google Scholar]
- Todd, J.; Pan, Y.-B.; Boykin, D. Fidelity of Sugarcane Crosses Assessed with SSR Markers. Agronomy 2020, 10, 386. [Google Scholar] [CrossRef]
- Ahmad, A.; Wang, J.-D.; Pan, Y.-B.; Sharif, R.; Gao, S.-J. Development and Use of Simple Sequence Repeats (SSRs) Markers for Sugarcane Breeding and Genetic Studies. Agronomy 2018, 8, 260. [Google Scholar] [CrossRef]
- Santos, J.; Barbosa, G.; Neto, C.; Almeida, C. Efficiency of biparental crossing in sugarcane analyzed by SSR markers. Crop Breed. Appl. Biotechnol. 2014, 14, 102–107. [Google Scholar] [CrossRef]
- Xavier, M.A.; Pinto, L.R.; Fávero, T.M.; Perecin, D.; Carlini-Garcia, L.A.; Landell, M.G. Paternity identification in sugarcane polycrosses by using microsatellite markers. Genet. Mol. Res. 2014, 13, 2268–2277. [Google Scholar] [CrossRef]
- Liu, X.-H.; Fang, F.-X.; Zhang, R.-H.; Song, H.-Z.; Yang, R.-Z.; Gao, Y.-J.; Ou, H.-P.; Lei, J.-C.; Luo, T.; Duan, W.-X.; et al. Identification of Progenies from Sugarcane × Narenga porphyrocoma (Hance) Bor. by SSR Marker. Southwest China J. Agric. Sci. 2012, 25, 38–43. [Google Scholar]
- Shavrukov, Y.N. CAPS markers in plant biology. Russ. J. Genet. Appl. Res. 2016, 6, 279–287. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.M.J.; Harris, N.; Gibson, M.; Lyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M.A.; et al. Apollo: A sequence annotation editor. Genome Biol. 2022, 3, research0082.1. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An Online Program for the Versatile Plotting of Organelle Genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.; Wang, G. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Non-Saccharum | S. spontaneum | S. robustum | S. sinense | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | BM87-36 | HBW1 | SES14 | SP79-9 | IN84-066 | IND81-013 | NG51-63 | NG57-208 | IS76-184 | IN84-076 | Guilinzhuzhe | Youba |
| Length/bp | 141,209 | 141,231 | 141,162 | 141,216 | 141,191 | 141,161 | 141,171 | 141,171 | 141,187 | 141,187 | 141,174 | 141,174 |
| LSC/bp | 83,167 | 83,107 | 83,039 | 83,093 | 83,068 | 83,038 | 83,036 | 83,036 | 83,056 | 83,056 | 83,043 | 83,043 |
| SSC/bp | 12,513 | 12,533 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | 12,538 | 12,538 | 12,538 | 12,538 |
| IR/bp | 22,763 | 22,794 | 22,789 | 22,789 | 22,789 | 22,789 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 |
| Species | S. barberi | S. officinarum | S. hybrid | |||||||||
| Accession | Katha | Kewali | Chunne | Loether | Christalin | BlackChLBH | NG96-14 | POJ2878 | ROC25 | GT42 | ZhongzheNo1 | |
| Length/bp | 141,174 | 141,174 | 141,174 | 141,183 | 141,182 | 141,182 | 141,183 | 141,182 | 141,182 | 141,182 | 141,182 | |
| LSC/bp | 83,043 | 83,043 | 83,043 | 83,048 | 83,047 | 83,047 | 83,048 | 83,047 | 83,047 | 83,047 | 83,047 | |
| SSC/bp | 12,538 | 12,538 | 12,538 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | 12,542 | |
| IR/bp | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | 22,795 | |
| Primer | Forward Reverse | Region | PCR Product Size (bp) | ||
|---|---|---|---|---|---|
| T. aru | S. spon | S. off | |||
| SscpId1 | GGATCGACTCTTTCCCAACA CCCAAAAACGACCAATCTTT | rps16-trnQ | 130 | 159 | 159 |
| SscpId2 | TGACAGAGGCAAGAAATAACGA GCCCGCTGAAGAGAAAATC | trnS-psbD | 235 | 204 | 204 |
| SscpId3 | AAGACCGGAGCTATCAACCA TGAAAGTGGACTTGCATCTGA | trnS-psbZ | 157 | 123 | 123 |
| SscpId4 | CCATTAGACAATGGACGCTTCT TGCTCCATATTCCTTCTTTATGA | trnR-rps14 | 188 | 172 | 198 |
| SscpId5 | AACCTAATGAAAATCGGATGATTA GCGAATTCCTATTTTGTTTGAA | ndhC-trnV | 249 | 228 | 228 |
| SscpId6 | AGGGACTCATGTTCCGTCTG GCATGAAGGGGTTAAATTCC | petL-petG | 191 | 145 | 145 |
| SscpId7 | TCGTGTGTTCGATTTTTCCA TTTTGATCCAGAACCAGAAGAA | rpl33-rps18 | 235 | 235 | 250 |
| SscpId8 | TTTGGCAAAGAAAAATAGAGTACG ATTCCTCAAAACGAGGCTCA | rps12 intron | 155 | 173 | 173 |
| SscpId9 | TTTGGCATTAGTTCATGAGGA AAGGAAATGGCTATCAAGAACG | rps12 intron | 143 | 176 | 176 |
| Primer | Forward Reverse | Region | Digestion Enzyme | PCR Product Size (bp)/ Target SNP | |
|---|---|---|---|---|---|
| S. rob | S. off | ||||
| Sscpd1 | CGTACTCCTAATCGAATTTGTATTTT CAGTGGTAAAAGTGTGATTCGTTC | trnG-trnM | TaqI | 226/A | 199/G |
| Sscpd2 | CGTACTCCTAATCGAATTTGTATTCTA TGGTAAAAGTGTGATTCGTTCTATT | trnG-trnM | XbaI | 224/A | 197/G |
| Sscpd3 | AACAAAAGAATAAATCCAGGGATTCCA CGGGTCCTTACTCCCCTTTA | rbcL-psaI | TaqI | 158/C | 174/G |
| Sscpd4 | TCTTCTTTATTCTTCGAATTGATTGC TGCGTATTTGATTCCATTATCGT | petA-psbJ | TaqI | 216/A | 201/G |
| Primer | Forward Reverse | Region | Digestion Enzyme | PCR Product Size (bp)/ Target SNP | |
|---|---|---|---|---|---|
| S. sin/ S. bar | Other S. spp. | ||||
| Sscpd5 | TCTTCTTTATTCTTCGAATTGATTGC TGCGTATTTGATTCCATTATCGTTCAT | rbcL-psaI | TaqI | 201/A | 216/G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Duan, W.; Zhao, J.; Jing, Y.; Feng, M.; Kuang, B.; Wei, N.; Chen, B.; Yang, X. Comparative Analysis of Chloroplast Genome in Saccharum spp. and Related Members of ‘Saccharum Complex’. Int. J. Mol. Sci. 2022, 23, 7661. https://doi.org/10.3390/ijms23147661
Li S, Duan W, Zhao J, Jing Y, Feng M, Kuang B, Wei N, Chen B, Yang X. Comparative Analysis of Chloroplast Genome in Saccharum spp. and Related Members of ‘Saccharum Complex’. International Journal of Molecular Sciences. 2022; 23(14):7661. https://doi.org/10.3390/ijms23147661
Chicago/Turabian StyleLi, Sicheng, Weixing Duan, Jihan Zhao, Yanfen Jing, Mengfan Feng, Bowen Kuang, Ni Wei, Baoshan Chen, and Xiping Yang. 2022. "Comparative Analysis of Chloroplast Genome in Saccharum spp. and Related Members of ‘Saccharum Complex’" International Journal of Molecular Sciences 23, no. 14: 7661. https://doi.org/10.3390/ijms23147661
APA StyleLi, S., Duan, W., Zhao, J., Jing, Y., Feng, M., Kuang, B., Wei, N., Chen, B., & Yang, X. (2022). Comparative Analysis of Chloroplast Genome in Saccharum spp. and Related Members of ‘Saccharum Complex’. International Journal of Molecular Sciences, 23(14), 7661. https://doi.org/10.3390/ijms23147661