

Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges

 ,
, 
Abstract
1. Introduction
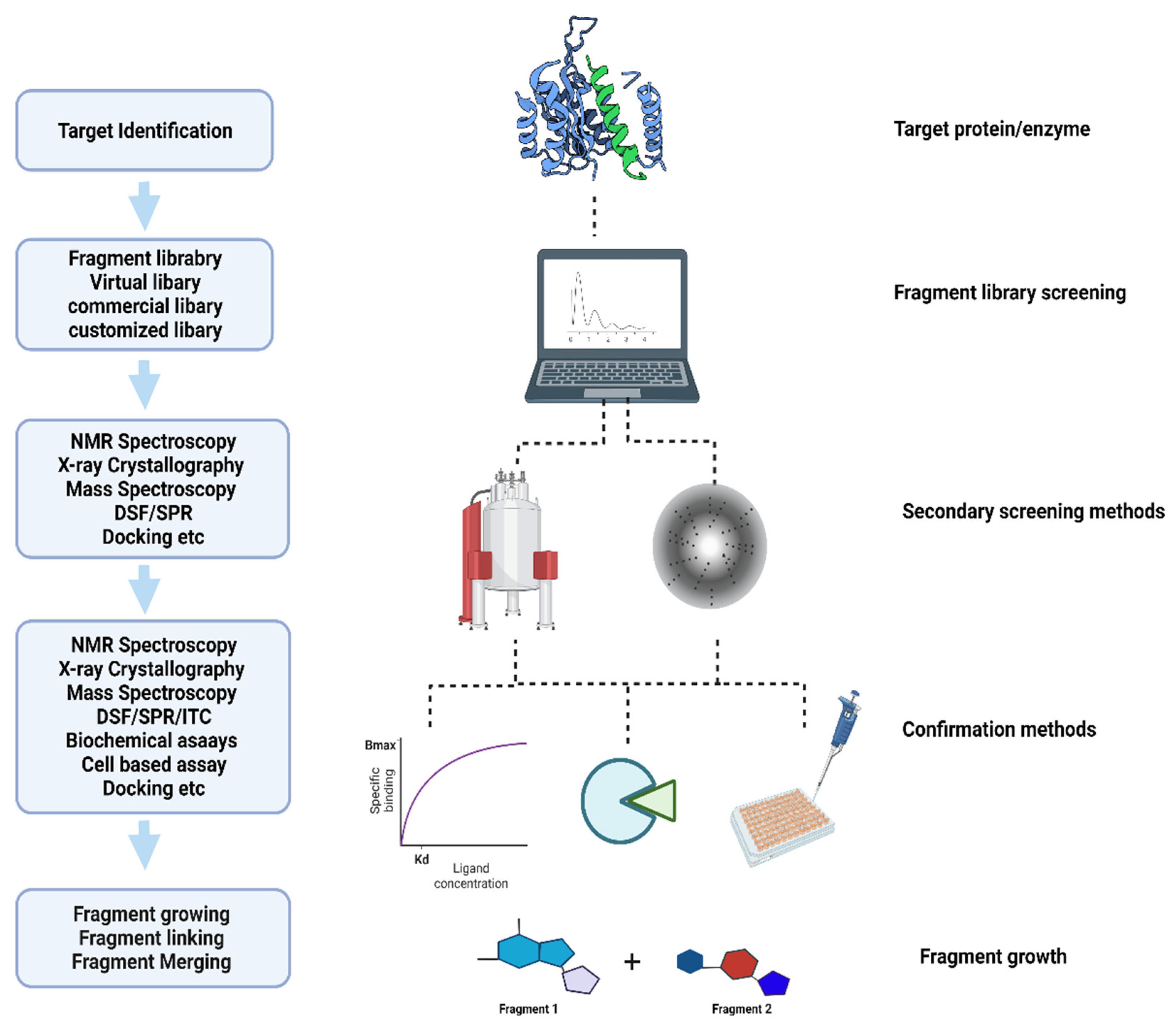
2. Fragment-Based Screening Methods
2.1. Virtual Screening and Computational Methods
2.2. X-ray Crystallography in Fragment-Based Drug Discovery
2.3. Biophysical Detection Methods: Thermal Shift and Mass Spectroscopy
2.4. Cryo-Electron Microscopy
2.5. Surface Plasmon Resonance
2.6. Nuclear Magnetic Resonance
3. Applications
4. Challenges in Fragment-Based Drug Discovery
4.1. Specialized Methods Are Needed to Detect Fragment Binding in Libraries
4.2. Optimization of Fragment Hits Using Computational Tools
4.3. Modeling
4.4. Challenges in Hit Identification and Lead Optimization
4.5. Post-Modeling Expression, Solubilization, Purification, Crystallization, Data Collection, and Structure Solution
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1.
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198. [Google Scholar] [CrossRef]
- Abell, C.; Dagostin, C. Chapter 1. Different Flavours of Fragments. In Fragment-Based Drug Discovery; Howard, S., Abell, C., Eds.; The Royal Society of Chemistry: London, UK, 2015; pp. 1–18. [Google Scholar] [CrossRef]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef]
- Oh, S.; Trifonov, L.; Yadav, V.D.; Barry, C.E.; Boshoff, H.I. Tuberculosis Drug Discovery: A Decade of Hit Assessment for Defined Targets. Front. Cell. Infect. Microbiol. 2021, 11, 611304. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Ayotte, Y.; Bernet, E.; Bilodeau, F.; Cimino, M.; Gagnon, D.; Lebughe, M.; Mistretta, M.; Ogadinma, P.; Ouali, S.L.; Sow, A.A.; et al. Fragment-Based Phenotypic Lead Discovery to Identify New Drug Seeds That Target Infectious Diseases. ACS Chem. Biol. 2021, 16, 2158–2163. [Google Scholar] [CrossRef]
- Charoensutthivarakul, S.; Thomas, S.E.; Curran, A.; Brown, K.P.; Belardinelli, J.M.; Whitehouse, A.J.; Acebrón-García-de-Eulate, M.; Sangan, J.; Gramani, S.G.; Jackson, M.; et al. Development of Inhibitors of SAICAR Synthetase (PurC) from Mycobacterium abscessus Using a Fragment-Based Approach. ACS Infect. Dis. 2022, 8, 296–309. [Google Scholar] [CrossRef]
- Bielska, E.; Lucas, X.; Czerwoniec, A.; Kasprzak, J.M.; Kaminska, K.H.; Bujnicki, J.M. Virtual screening strategies in drug design—Methods and applications. BioTechnologia 2011, 3, 249–264. [Google Scholar] [CrossRef]
- Singh, M.; Tam, B.; Akabayov, B. NMR-Fragment Based Virtual Screening: A Brief Overview. Molecules 2018, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed]
- Keserű, G.M.; Erlanson, D.A.; Ferenczy, G.G.; Hann, M.M.; Murray, C.W.; Pickett, S.D. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem. 2016, 59, 8189–8206. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for “Omics” research on drugs. Nucleic Acids Res. 2011, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef]
- Visini, R.; Awale, M.; Reymond, J.-L. Fragment Database FDB-17. J. Chem. Inf. Model. 2017, 57, 700–709. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Stroud, R.M.; Santi, D.V.; Kuntz, I.D.; Perry, K.M. Structure-Based Discovery of Inhibitors of Thymidylate Synthase. Science 1993, 259, 1445–1450. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for Natural Product Discovery: A Publicly-Accessible Library of Natural Product Fractions for High-Throughput Screening. ACS Chem. Biol. 2018, 13, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Acquah, K.S.; Beukes, D.R.; Seldon, R.; Jordaan, A.; Sunassee, S.N.; Warner, D.F.; Gammon, D.W. Identification of Antimycobacterial Natural Products from a Library of Marine Invertebrate Extracts. Medicines 2022, 9, 9. [Google Scholar] [CrossRef]
- Verlinde, C.L.M.J.; Rudenko, G.; Hol, W.G.J. In search of new lead compounds for trypanosomiasis drug design: A protein structure-based linked-fragment approach. J. Comput. Aided Mol. Des. 1992, 6, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Chau, P.-L.; Dean, P.M. Automated site-directed drug design: Searches of the Cambridge Structural Database for bond lengths in molecular fragments to be used for automated structure assembly. J. Comput. Aided Mol. Des. 1992, 6, 397–406. [Google Scholar] [CrossRef]
- Stultz, C.M.; Karplus, M. Dynamic ligand design and combinatorial optimization: Designing inhibitors to endothiapepsin. Proteins Struct. Funct. Genet. 2000, 40, 258–289. [Google Scholar] [CrossRef]
- Majeux, N.; Carsi, M.; Tenette-Souaille, C.; Caflisch, A. Hydrophobicity Maps and Docking of Molecular Fragments with Solvation. In Virtual Screening: An Alternative or Complement to High Throughput Screening? Perspectivies in Drug Discovery and Design; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 145–169. [Google Scholar] [CrossRef]
- Leach, A.R.; Bryce, R.A.; Robinson, A.J. Synergy between combinatorial chemistry and de novo design22European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom. J. Mol. Graph. Model. 2000, 18, 358–367. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Merz, K., Jr.; Ringe, D.; Reynolds, C.H. (Eds.) Drug Design: Structure- and Ligand-Based Approaches; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef]
- Mountain, V. Astex, Structural Genomix, and Syrrx. Chem. Biol. 2003, 10, 95–98. [Google Scholar] [CrossRef]
- Sharff, A.; Jhoti, H. High-throughput crystallography to enhance drug discovery. Curr. Opin. Chem. Biol. 2003, 7, 340–345. [Google Scholar] [CrossRef]
- Robson-Tull, J. Biophysical screening in fragment-based drug design: A brief overview. Biosci. Horizons Int. J. Stud. Res. 2018, 11, hzy015. [Google Scholar] [CrossRef]
- Thomas, S.E.; Whitehouse, A.J.; Brown, K.; Burbaud, S.; Belardinelli, J.M.; Sangen, J.; Lahiri, R.; Libardo, M.D.J.; Gupta, P.; Malhotra, S.; et al. Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification. Nucleic Acids Res. 2020, 48, 8099–8112. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, M.; Selvam, I.R.; Evangelopoulos, D.; McLean, K.J.; Katariya, M.M.; Tunnicliffe, R.B.; Campbell, B.; Kavanagh, M.E.; Charoensutthivarakul, S.; Blankley, R.T.; et al. A new strategy for hit generation: Novel in cellulo active inhibitors of CYP121A1 from Mycobacterium tuberculosis via a combined X-ray crystallographic and phenotypic screening approach (XP screen). Eur. J. Med. Chem. 2022, 230, 114105. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef]
- Malapati, P.; Krishna, V.S.; Nallangi, R.; Srilakshmi, R.R.; Sriram, D. Identification and development of benzoxazole derivatives as novel bacterial glutamate racemase inhibitors. Eur. J. Med. Chem. 2018, 145, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Flipo, M.; Willand, N.; Lecat-Guillet, N.; Hounsou, C.; Desroses, M.; Leroux, F.; Lens, Z.; Villeret, V.; Wohlkönig, A.; Wintjens, R.; et al. Discovery of Novel N -Phenylphenoxyacetamide Derivatives as EthR Inhibitors and Ethionamide Boosters by Combining High-Throughput Screening and Synthesis. J. Med. Chem. 2012, 55, 6391–6402. [Google Scholar] [CrossRef]
- Dai, R.; Wilson, D.J.; Geders, T.W.; Aldrich, C.C.; Finzel, B.C. Inhibition of Mycobacterium tuberculosis Transaminase BioA by Aryl Hydrazines and Hydrazides. ChemBioChem 2014, 15, 575–586. [Google Scholar] [CrossRef]
- Marchetti, C.; Chan, D.S.H.; Coyne, A.G.; Abell, C. Fragment-based approaches to TB drugs. Parasitology 2018, 145, 184–195. [Google Scholar] [CrossRef]
- Pedro, L.; Quinn, R. Native Mass Spectrometry in Fragment-Based Drug Discovery. Molecules 2016, 21, 984. [Google Scholar] [CrossRef]
- Swayze, E.E.; Jefferson, E.A.; Sannes-Lowery, K.A.; Blyn, L.B.; Risen, L.M.; Arakawa, S.; Osgood, S.A.; Hofstadler, S.A.; Griffey, R.H. SAR by MS: A Ligand Based Technique for Drug Lead Discovery Against Structured RNA Targets. J. Med. Chem. 2002, 45, 3816–3819. [Google Scholar] [CrossRef]
- Kavanagh, M.E.; Coyne, A.G.; McLean, K.J.; James, G.G.; Levy, C.W.; Marino, L.B.; de Carvalho, L.P.S.; Chan, D.S.H.; Hudson, S.A.; Surade, S.; et al. Fragment-Based Approaches to the Development of Mycobacterium tuberculosis CYP121 Inhibitors. J. Med. Chem. 2016, 59, 3272–3302. [Google Scholar] [CrossRef] [PubMed]
- Ockey, D.A.; Dotson, J.L.; Struble, M.E.; Stults, J.T.; Bourell, J.H.; Clark, K.R.; Gadek, T.R. Structure-activity relationships by mass spectrometry: Identification of novel MMP-3 inhibitors. Bioorg. Med. Chem. 2004, 12, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.-A. Direct screening of a dynamic combinatorial library using mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.D.; Wu, H.; Bolaños, B.; Bergqvist, S.; Brooun, A.; Pauly, T.; Nowlin, D. Structural and Biophysical Characterization of XIAP BIR3 G306E Mutant: Insights in Protein Dynamics and Application for Fragment-Based Drug Design. Chem. Biol. Drug Des. 2009, 74, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Vivat Hannah, V.; Atmanene, C.; Zeyer, D.; Van Dorsselaer, A.; Sanglier-Cianférani, S. Native MS: An ’ESI‚ way to support structure- and fragment-based drug discovery. Future Med. Chem. 2010, 2, 35–50. [Google Scholar] [CrossRef]
- Murthy, P.K.; Dixit, S.; Gaur, R.L.; Kumar, R.; Sahoo, M.K.; Shakya, N.; Joseph, S.K.; Palne, S.; Gupta, S. Influence of Brugia malayi life stages and BmAFII fraction on experimental Leishmania donovani infection in hamsters. Acta Trop. 2008, 106, 81–89. [Google Scholar] [CrossRef]
- Vu, H.; Roullier, C.; Campitelli, M.; Trenholme, K.R.; Gardiner, D.L.; Andrews, K.T.; Skinner-Adams, T.; Crowther, G.J.; Van Voorhis, W.C.; Quinn, R.J. Plasmodium Gametocyte Inhibition Identified from a Natural-Product-Based Fragment Library. ACS Chem. Biol. 2013, 8, 2654–2659. [Google Scholar] [CrossRef]
- Woods, L.A.; Dolezal, O.; Ren, B.; Ryan, J.H.; Peat, T.S.; Poulsen, S.-A. Native State Mass Spectrometry, Surface Plasmon Resonance, and X-ray Crystallography Correlate Strongly as a Fragment Screening Combination. J. Med. Chem. 2016, 59, 2192–2204. [Google Scholar] [CrossRef]
- Frank, J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 2017, 12, 209–212. [Google Scholar] [CrossRef]
- Bartesaghi, A.; Merk, A.; Banerjee, S.; Matthies, D.; Wu, X.; Milne, J.L.S.; Subramaniam, S. 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science 2015, 348, 1147–1151. [Google Scholar] [CrossRef]
- Saur, M.; Hartshorn, M.J.; Dong, J.; Reeks, J.; Bunkoczi, G.; Jhoti, H.; Williams, P.A. Fragment-based drug discovery using cryo-EM. Drug Discov. Today 2020, 25, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, W.; Zhou, X.; Zhang, Y.; Lai, Y.; Tang, Y.; Xu, J.; Li, D.; Lin, J.; Yang, X.; et al. Structure of Mycobacterium tuberculosis cytochrome bcc in complex with Q203 and TB47, two anti-TB drug candidates. Elife 2021, 10, e69418. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, S.J.; Chimenti, M.S.; Kelly, M.J.S.; Arkin, M.R. Biophysical Methods for Identifying Fragment-Based Inhibitors of Protein-Protein Interactions. In Protein-Protein Interactions; Meyerkord, C., Fu, H., Eds.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1278, pp. 587–613. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Using Fragment-Based Approaches to Discover New Antibiotics. SLAS Discov. 2018, 23, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.S.; Bhargav, A.G.; Perez, J.G.; Wadajkar, A.S.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Surface plasmon resonance as a high throughput method to evaluate specific and non-specific binding of nanotherapeutics. J. Control. Release 2015, 219, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Chavanieu, A.; Pugnière, M. Developments in SPR Fragment Screening. Expert Opin. Drug Discov. 2016, 11, 489–499. [Google Scholar] [CrossRef]
- Du, H.; Brender, J.R.; Zhang, J.; Zhang, Y. Protein structure prediction provides comparable performance to crystallographic structures in docking-based virtual screening. Methods 2015, 71, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, P.O.; Blaszczyk, M.; Surade, S.; Boshoff, H.I.; Sajid, A.; Delorme, V.; Deboosere, N.; Brodin, P.; Baulard, A.R.; Barry, C.E.; et al. Fragment-Sized EthR Inhibitors Exhibit Exceptionally Strong Ethionamide Boosting Effect in Whole-Cell Mycobacterium tuberculosis Assays. ACS Chem. Biol. 2017, 12, 1390–1396. [Google Scholar] [CrossRef]
- Gupta, P.; Thomas, S.E.; Zaidan, S.A.; Pasillas, M.A.; Cory-Wright, J.; Sebastián-Pérez, V.; Burgess, A.; Cattermole, E.; Meghir, C.; Abell, C.; et al. A fragment-based approach to assess the ligandability of ArgB, ArgC, ArgD and ArgF in the L-arginine biosynthetic pathway of Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3491–3506. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Naik, M.; Raichurkar, A.; Bandodkar, B.S.; Varun, B.V.; Bhat, S.; Kalkhambkar, R.; Murugan, K.; Menon, R.; Bhat, J.; Paul, B.; et al. Structure Guided Lead Generation for M. tuberculosis Thymidylate Kinase (Mtb TMK): Discovery of 3-Cyanopyridone and 1,6-Naphthyridin-2-one as Potent Inhibitors. J. Med. Chem. 2015, 58, 753–766. [Google Scholar] [CrossRef]
- Hung, A.W.; Silvestre, H.L.; Wen, S.; Ciulli, A.; Blundell, T.L.; Abell, C. Application of Fragment Growing and Fragment Linking to the Discovery of Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase. Angew. Chem. Int. Ed. 2009, 48, 8452–8456. [Google Scholar] [CrossRef] [PubMed]
- Sledz, P.; Silvestre, H.L.; Hung, A.W.; Ciulli, A.; Blundell, T.L.; Abell, C. Optimization of the Interligand Overhauser Effect for Fragment Linking: Application to Inhibitor Discovery against Mycobacterium tuberculosis Pantothenate Synthetase. J. Am. Chem. Soc. 2010, 132, 4544–4545. [Google Scholar] [CrossRef] [PubMed]
- Vilchèze, C.; Jacobs, W.R. The Isoniazid Paradigm of Killing, Resistance, and Persistence in Mycobacterium tuberculosis. J. Mol. Biol. 2019, 431, 3450–3461. [Google Scholar] [CrossRef] [PubMed]
- Vilchèze, C.; Jacobs, W.R., Jr. Resistance to Isoniazid and Ethionamide in Mycobacterium tuberculosis: Genes, Mutations, and Causalities. Microbiol. Spectr. 2014, 2, 2–4. [Google Scholar] [CrossRef]
- Thomas, S.E.; Collins, P.; James, R.H.; Mendes, V.; Charoensutthivarakul, S.; Radoux, C.; Abell, C.; Coyne, A.G.; Floto, R.A.; von Delft, F.; et al. Structure-guided fragment-based drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2019, 377, 20180422. [Google Scholar] [CrossRef]
- Thomas, S.E.; Mendes, V.; Kim, S.Y.; Malhotra, S.; Ochoa-Montaño, B.; Blaszczyk, M.; Blundell, T.L. Structural Biology and the Design of New Therapeutics: From HIV and Cancer to Mycobacterial Infections. J. Mol. Biol. 2017, 429, 2677–2693. [Google Scholar] [CrossRef]
- Wu, M.-L.; Aziz, D.B.; Dartois, V.; Dick, T. NTM drug discovery: Status, gaps and the way forward. Drug Discov. Today 2018, 23, 1502–1519. [Google Scholar] [CrossRef]
- Lagutkin, D.; Panova, A.; Vinokurov, A.; Gracheva, A.; Samoilova, A.; Vasilyeva, I. Genome-Wide Study of Drug Resistant Mycobacterium tuberculosis and Its Intra-Host Evolution during Treatment. Microorganisms 2022, 10, 1440. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Thomas, S.E.; Brown, K.P.; Fanourakis, A.; Chan, D.S.-H.; Libardo, M.D.J.; Mendes, V.; Boshoff, H.I.M.; Floto, R.A.; Abell, C.; et al. Development of Inhibitors against Mycobacterium abscessus tRNA (m 1 G37) Methyltransferase (TrmD) Using Fragment-Based Approaches. J. Med. Chem. 2019, 62, 7210–7232. [Google Scholar] [CrossRef]
- Saw, W.-G.; Leow, C.Y.; Harikishore, A.; Shin, J.; Cole, M.S.; Aragaw, W.W.; Ragunathan, P.; Hegde, P.; Aldrich, C.C.; Dick, T.; et al. Structural and Mechanistic Insights into Mycobacterium abscessus Aspartate Decarboxylase PanD and a Pyrazinoic Acid-Derived Inhibitor. ACS Infect. Dis. 2022, 8, 1324–1335. [Google Scholar] [CrossRef]
- Kim, S.Y.; Eun, H.-J.; Lee, J.; Lee, W.; Thomas, S.E.; Brear, P.; Vedithi, S.C.; Abell, C.; Lee, B.J.; Blundell, T.L. Structural-guided fragment-based drug discovery applied to antitoxin, MAB3862 opens a new possibility of exploring the Toxin and Antitoxin for antibiotics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Thomas, S.; Roquet-Banères, F.; Herrmann, J.L.; Pethe, K.; Kremer, L. Bedaquiline inhibits the ATP synthase in mycobacterium abscessus and is effective in infected zebrafish. Antimicrob. Agents Chemother. 2017, 61, e01225-17. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by blocking Arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Roberto Chiarelli, L.; Mori, G.; Esposito, M.; Silvia Orena, B.; Rosalia Pasca, M. New and Old Hot Drug Targets in Tuberculosis. Curr. Med. Chem. 2016, 23, 3813–3846. [Google Scholar] [CrossRef]
- Lee, B.S.; Pethe, K. Therapeutic potential of promiscuous targets in Mycobacterium tuberculosis. Curr. Opin. Pharmacol. 2018, 42, 22–26. [Google Scholar] [CrossRef]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-Thiazoles: A Novel Mode of Inhibition with the Potential to Develop Novel Inhibitors Targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Chung, C.; Ghidelli-Disse, S.; Rullas, J.; Rebollo-López, M.J.; Gurcha, S.S.; Cox, J.A.G.; Mendoza, A.; Jiménez-Navarro, E.; Martínez-Martínez, M.S.; et al. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 12581. [Google Scholar] [CrossRef]
- Schiebel, J.; Kapilashrami, K.; Fekete, A.; Bommineni, G.R.; Schaefer, C.M.; Mueller, M.J.; Tonge, P.J.; Kisker, C. Structural Basis for the Recognition of Mycolic Acid Precursors by KasA, a Condensing Enzyme and Drug Target from Mycobacterium Tuberculosis. J. Biol. Chem. 2013, 288, 34190–34204. [Google Scholar] [CrossRef]
- Harbut, M.B.; Yang, B.; Liu, R.; Yano, T.; Vilchèze, C.; Cheng, B.; Lockner, J.; Guo, H.; Yu, C.; Franzblau, S.G.; et al. Small Molecules Targeting Mycobacterium tuberculosis Type II NADH Dehydrogenase Exhibit Antimycobacterial Activity. Angew. Chem.-Int. Ed. 2018, 57, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, D.; Ray, P.C.; Bayliss, T.; Prosser, G.A.; Harrison, J.R.; Green, K.; Soares de Melo, C.; Feng, T.-S.; Street, L.J.; Chibale, K.; et al. 2-Mercapto-Quinazolinones as Inhibitors of Type II NADH Dehydrogenase and Mycobacterium tuberculosis: Structure-Activity Relationships, Mechanism of Action and Absorption, Distribution, Metabolism, and Excretion Characterization. ACS Infect. Dis. 2018, 4, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, J.; Lee, C.H.; Han, S.K.; Yim, J.J. Progression and treatment outcomes of lung disease caused by mycobacterium abscessus and mycobacterium massiliense. Clin. Infect. Dis. 2017, 64, 301–308. [Google Scholar] [CrossRef]
- Kebbel, F.; Kurz, M.; Arheit, M.; Grütter, M.G.; Stahlberg, H. Structure and Substrate-Induced Conformational Changes of the Secondary Citrate/Sodium Symporter CitS Revealed by Electron Crystallography. Structure 2013, 21, 1243–1250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hudson, S.A.; Surade, S.; Coyne, A.G.; McLean, K.J.; Leys, D.; Munro, A.W.; Abell, C. Overcoming the Limitations of Fragment Merging: Rescuing a Strained Merged Fragment Series Targeting Mycobacterium tuberculosis CYP121. ChemMedChem 2013, 8, 1451–1456. [Google Scholar] [CrossRef]
- Ragunathan, P.; Cole, M.; Latka, C.; Aragaw, W.W.; Hegde, P.; Shin, J.; Subramanian Manimekalai, M.S.; Rishikesan, S.; Aldrich, C.C.; Dick, T.; et al. Mycobacterium tuberculosis PanD Structure–Function Analysis and Identification of a Potent Pyrazinoic Acid-Derived Enzyme Inhibitor. ACS Chem. Biol. 2021, 16, 1030–1039. [Google Scholar] [CrossRef]
- Moffatt, B.A.; Ashihara, H. Purine and Pyrimidine Nucleotide Synthesis and Metabolism. Arab. B. 2002, 1, e0018. [Google Scholar] [CrossRef]
- Manjunath, K.; Jeyakanthan, J.; Sekar, K. Catalytic pathway, substrate binding and stability in SAICAR synthetase: A structure and molecular dynamics study. J. Struct. Biol. 2015, 191, 22–31. [Google Scholar] [CrossRef]
- Tuntland, M.L.; Fung, L.W.-M. Substrate independent ATPase activity may complicate high throughput screening. Anal. Biochem. 2016, 510, 18–20. [Google Scholar] [CrossRef][Green Version]
- Berney, M.; Berney-Meyer, L.; Wong, K.-W.; Chen, B.; Chen, M.; Kim, J.; Wang, J.; Harris, D.; Parkhill, J.; Chan, J.; et al. Essential roles of methionine and S -adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10008–10013. [Google Scholar] [CrossRef]
- Tiwari, S.; van Tonder, A.J.; Vilchèze, C.; Mendes, V.; Thomas, S.E.; Malek, A.; Chen, B.; Chen, M.; Kim, J.; Blundell, T.L.; et al. Arginine-deprivation–induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Viljoen, A.; Dubar, F.; Blaise, M.; Bernut, A.; Pawlik, A.; Bouchier, C.; Brosch, R.; Guérardel, Y.; Lelièvre, J.; et al. A new piperidinol derivative targeting mycolic acid transport in Mycobacterium abscessus. Mol. Microbiol. 2016, 101, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Moreira, W.; Lim, J.J.; Yeo, S.Y.; Ramanujulu, P.M.; Dymock, B.W.; Dick, T. Fragment-Based Whole Cell Screen Delivers Hits against M. tuberculosis and Non-tuberculous Mycobacteria. Front. Microbiol. 2016, 7, 1392. [Google Scholar] [CrossRef]
- Grover, S.; Gangwar, R.; Jamal, S.; Ali, S.; Nisaa, K.; Ehtesham, N.Z.; Hasnain, S.E. Mycobacterial Methyltransferases: Significance in Pathogenesis and Virulence. In Mycobacterium Tuberculosis: Molecular Infection Biology, Pathogenesis, Diagnostics and New Interventions; Springer: Singapore, 2019; pp. 103–122. [Google Scholar]
- Hou, Y.-M.; Matsubara, R.; Takase, R.; Masuda, I.; Sulkowska, J.I. TrmD: A Methyl Transferase for tRNA Methylation with m1G37. Enzymes 2017, 41, 89–115. [Google Scholar] [CrossRef]
- Zhong, W.; Koay, A.; Ngo, A.; Li, Y.; Nah, Q.; Wong, Y.H.; Chionh, Y.H.; Ng, H.Q.; Koh-Stenta, X.; Poulsen, A.; et al. Targeting the Bacterial Epitranscriptome for Antibiotic Development: Discovery of Novel tRNA-(N 1 G37) Methyltransferase (TrmD) Inhibitors. ACS Infect. Dis. 2019, 5, 326–335. [Google Scholar] [CrossRef]
- Chionh, Y.H.; McBee, M.; Babu, I.R.; Hia, F.; Lin, W.; Zhao, W.; Cao, J.; Dziergowska, A.; Malkiewicz, A.; Begley, T.J.; et al. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun. 2016, 7, 13302. [Google Scholar] [CrossRef]
- Gu, C.; Begley, T.J.; Dedon, P.C. tRNA modifications regulate translation during cellular stress. FEBS Lett. 2014, 588, 4287–4296. [Google Scholar] [CrossRef]
- Ahn, H.J. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J. 2003, 22, 2593–2603. [Google Scholar] [CrossRef]
- Ali, S.; Ehtram, A.; Arora, N.; Manjunath, P.; Roy, D.; Ehtesham, N.Z.; Hasnain, S.E. The M. tuberculosis Rv1523 Methyltransferase Promotes Drug Resistance Through Methylation-Mediated Cell Wall Remodeling and Modulates Macrophages Immune Responses. Front. Cell. Infect. Microbiol. 2021, 11, 622487. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Ngeow, Y.F.; Choo, S.W. Support from Phylogenomic Networks and Subspecies Signatures for Separation of Mycobacterium massiliense from Mycobacterium bolletii. J. Clin. Microbiol. 2015, 53, 3042–3046. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Ng, K.P.; Ong, C.S.; Ngeow, Y.F. Genomic Comparisons Reveal Microevolutionary Differences in Mycobacterium abscessus Subspecies. Front. Microbiol. 2017, 8, 2042. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Eisenberg, D. Crystal Structure of the Pantothenate Synthetase from Mycobacterium tuberculosis, Snapshots of the Enzyme in Action. Biochemistry 2006, 45, 1554–1561. [Google Scholar] [CrossRef]
- Zheng, R.; Blanchard, J.S. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry 2001, 40, 12904–12912. [Google Scholar] [CrossRef] [PubMed]
- Sambandamurthy, V.K.; Wang, X.; Chen, B.; Russell, R.G.; Derrick, S.; Collins, F.M.; Morris, S.L.; Jacobs, W.R. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002, 8, 1171–1174. [Google Scholar] [CrossRef]
- Abrahams, G.L.; Kumar, A.; Savvi, S.; Hung, A.W.; Wen, S.; Abell, C.; Barry, C.E.; Sherman, D.R.; Boshoff, H.I.M.; Mizrahi, V. Pathway-Selective Sensitization of Mycobacterium tuberculosis for Target-Based Whole-Cell Screening. Chem. Biol. 2012, 19, 844–854. [Google Scholar] [CrossRef]
- Gupta, A.; Sharma, P.; Singh, T.P.; Sharma, S. Phosphopantetheine Adenylyltransferase: A promising drug target to combat antibiotic resistance. Biochim. Biophys. Acta-Proteins Proteom. 2021, 1869, 140566. [Google Scholar] [CrossRef]
- Thomas, S.E.; McCarthy, W.J.; El Bakali, J.; Brown, K.P.; Kim, S.Y.; Blaszczyk, M.; Mendes, V.; Abell, C.; Floto, R.A.; Coyne, A.G.; et al. Structural Characterization of Mycobacterium abscessus Phosphopantetheine Adenylyl Transferase Ligand Interactions: Implications for Fragment-Based Drug Design. Front. Mol. Biosci. 2022, 9, 880432. [Google Scholar] [CrossRef]
- El Bakali, J.; Blaszczyk, M.; Evans, J.C.; Boland, J.A.; McCarthy, W.J.; Dias, M.V.B.; Coyne, A.G.; Mizrahi, V.; Blundell, T.L.; Abell, C.; et al. Chemical validation of Mycobacterium tuberculosis phosphopantetheine adenylyltransferase using fragment linking and CRISPR interference. bioRxiv 2020. [Google Scholar] [CrossRef]
- Primi, M.C.; Tavares, M.T.; Klein, L.L.; Izard, T.; Sant’Anna, C.M.R.; Franzblau, S.G.; Ferreira, E.I. Design of Novel Phosphopantetheine Adenylyltransferase Inhibitors: A Potential New Approach to Tackle Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2021, 21, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yazidi, A.; Pandya, A.N.; Hegde, P.; Tong, W.; Calado Nogueira de Moura, V.; North, E.J.; Sygusch, J.; Jackson, M. MmpL3 as a Target for the Treatment of Drug-Resistant Nontuberculous Mycobacterial Infections. Front. Microbiol. 2018, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ruyck, J.; Dupont, C.; Lamy, E.; Le Moigne, V.; Biot, C.; Guérardel, Y.; Herrmann, J.; Blaise, M.; Grassin-Delyle, S.; Kremer, L.; et al. Structure-Based Design and Synthesis of Piperidinol-Containing Molecules as New Mycobacterium abscessus Inhibitors. ChemistryOpen 2020, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive Functional Analysis of Mycobacterium tuberculosis Toxin-Antitoxin Systems: Implications for Pathogenesis, Stress Responses, and Evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Hudson, S.A.; McLean, K.J.; Surade, S.; Yang, Y.-Q.; Leys, D.; Ciulli, A.; Munro, A.W.; Abell, C. Application of Fragment Screening and Merging to the Discovery of Inhibitors of the Mycobacterium tuberculosis Cytochrome P450 CYP121. Angew. Chem. Int. Ed. 2012, 51, 9311–9316. [Google Scholar] [CrossRef]
- Ellenbarger, J.F.; Krieger, I.V.; Huang, H.; Gómez-Coca, S.; Ioerger, T.R.; Sacchettini, J.C.; Wheeler, S.E.; Dunbar, K.R. Anion-π Interactions in Computer-Aided Drug Design: Modeling the Inhibition of Malate Synthase by Phenyl-Diketo Acids. J. Chem. Inf. Model. 2018, 58, 2085–2091. [Google Scholar] [CrossRef]
- Krieger, I.V.; Freundlich, J.S.; Gawandi, V.B.; Roberts, J.P.; Gawandi, V.B.; Sun, Q.; Owen, J.L.; Fraile, M.T.; Huss, S.I.; Lavandera, J.-L.; et al. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 2012, 19, 1556–1567. [Google Scholar] [CrossRef]
- Huang, H.-L.; Krieger, I.V.; Parai, M.K.; Gawandi, V.B.; Sacchettini, J.C. Mycobacterium tuberculosis Malate Synthase Structures with Fragments Reveal a Portal for Substrate/Product Exchange. J. Biol. Chem. 2016, 291, 27421–27432. [Google Scholar] [CrossRef]
- Sriram, D.; Aubry, A.; Yogeeswari, P.; Fisher, L.M. Gatifloxacin derivatives: Synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 2006, 16, 2982–2985. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P. Tools for ligand validation in Coot. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.G.; Blower, T.R.; Cacho, M.; Bax, B.; Berger, J.M.; Osheroff, N. Mechanism of Action of Mycobacterium tuberculosis Gyrase Inhibitors: A Novel Class of Gyrase Poisons. ACS Infect. Dis. 2018, 4, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Usha, V.; Gurcha, S.S.; Lovering, A.L.; Lloyd, A.J.; Papaemmanouil, A.; Reynolds, R.C.; Besra, G.S. Identification of novel diphenyl urea inhibitors of Mt-GuaB2 active against Mycobacterium tuberculosis. Microbiology 2011, 157, 290–299. [Google Scholar] [CrossRef]
- Usha, V.; Hobrath, J.V.; Gurcha, S.S.; Reynolds, R.C.; Besra, G.S. Identification of Novel Mt-Guab2 Inhibitor Series Active against M. tuberculosis. PLoS ONE 2012, 7, e33886. [Google Scholar] [CrossRef] [PubMed]
- Makowska-Grzyska, M.; Kim, Y.; Gorla, S.K.; Wei, Y.; Mandapati, K.; Zhang, M.; Maltseva, N.; Modi, G.; Boshoff, H.I.; Gu, M.; et al. Mycobacterium tuberculosis IMPDH in Complexes with Substrates, Products and Antitubercular Compounds. PLoS ONE 2015, 10, e0138976. [Google Scholar] [CrossRef]
- Singh, V.; Donini, S.; Pacitto, A.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Keri, G.; Ascher, D.B.; Mondésert, G.; Vocat, A.; et al. The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 2017, 3, 5–17. [Google Scholar] [CrossRef]
- Cox, J.A.G.; Mugumbate, G.; Del Peral, L.V.-G.; Jankute, M.; Abrahams, K.A.; Jervis, P.; Jackenkroll, S.; Perez, A.; Alemparte, C.; Esquivias, J.; et al. Novel inhibitors of Mycobacterium tuberculosis GuaB2 identified by a target based high-throughput phenotypic screen. Sci. Rep. 2016, 6, 38986. [Google Scholar] [CrossRef]
- Ripoll, F.; Pasek, S.; Schenowitz, C.; Dossat, C.; Barbe, V.; Rottman, M.; Macheras, E.; Heym, B.; Herrmann, J.-L.; Daffé, M.; et al. Non Mycobacterial Virulence Genes in the Genome of the Emerging Pathogen Mycobacterium abscessus. PLoS ONE 2009, 4, e5660. [Google Scholar] [CrossRef]
- Choo, S.W.; Wee, W.Y.; Ngeow, Y.F.; Mitchell, W.; Tan, J.L.; Wong, G.J.; Zhao, Y.; Xiao, J. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci. Rep. 2015, 4, 4061. [Google Scholar] [CrossRef]
- Philley, J.V.; Wallace, R.J.; Benwill, J.L.; Taskar, V.; Brown-Elliott, B.A.; Thakkar, F.; Aksamit, T.R.; Griffith, D.E. Preliminary Results of Bedaquiline as Salvage Therapy for Patients With Nontuberculous Mycobacterial Lung Disease. Chest 2015, 148, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The “rule of three” for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.; Schiebel, J.; Wulsdorf, T.; Grohe, K.; Najbauer, E.E.; Ehrmann, F.R.; Radeva, N.; Zitzer, N.; Linne, U.; Linser, R.; et al. A False-Positive Screening Hit in Fragment-Based Lead Discovery: Watch out for the Red Herring. Angew. Chem. Int. Ed. 2017, 56, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Joseph-McCarthy, D. Challenges of fragment screening. J. Comput.-Aided Mol. Des. 2009, 23, 449–451. [Google Scholar] [CrossRef][Green Version]
- Vangrevelinghe, E.; Rudisser, S. Computational Approaches for Fragment Optimization. Curr. Comput.-Aided Drug Des. 2007, 3, 69–83. [Google Scholar] [CrossRef]
- Mureddu, L.G.; Vuister, G.W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where Can It Be Improved? Front. Mol. Biosci. 2022, 9, 834453. [Google Scholar] [CrossRef]
- Bian, Y. The Research and Development of an Artificial Intelligence Integrated Fragment-Based Drug Design Platform for Small Molecule Drug Discovery. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2021. [Google Scholar]
- Lundquist, K.P.; Panchal, V.; Gotfredsen, C.H.; Brenk, R.; Clausen, M.H. Fragment-Based Drug Discovery for RNA Targets. ChemMedChem 2021, 16, 2588–2603. [Google Scholar] [CrossRef]
- Bhoj, P.S.; Bahekar, S.; Khatri, V.; Singh, N.; Togre, N.S.; Goswami, K.; Chandak, H.S.; Dash, D. Role of Glutathione in Chalcone Derivative Induced Apoptosis of Brugia malayi and its Possible Therapeutic Implication. Acta Parasitol. 2021, 66, 406–415. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [Google Scholar] [CrossRef]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-Based Drug Discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef] [PubMed]
- Konteatis, Z.D. In silico fragment-based drug design. Expert Opin. Drug Discov. 2010, 5, 1047–1065. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P. In Silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef]
- Chiliza, T.E.; Pillay, M.; Pillay, B. Identification of unique essential proteins from a M. tuberculosis F15/LAM4/KZN phage secretome library. Pathog. Dis. 2017, 75, ftx001. [Google Scholar] [CrossRef][Green Version]
- Lamichhane, G. Novel targets in M. tuberculosis: Search for new drugs. Trends Mol. Med. 2011, 17, 25–33. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Zuccotto, F.; Hsinpin, H.; Sandberg, L.; Via, L.E.; Marriner, G.A.; Masquelin, T.; Wyatt, P.; Ray, P.; Dartois, V. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect. Dis. 2016, 2, 552–563. [Google Scholar] [CrossRef]
- Zheng, X.; Av-Gay, Y. New Era of TB Drug Discovery and Its Impact on Disease Management. Curr. Treat. Options Infect. Dis. 2016, 8, 299–310. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002; ISBN 0-8153-3218-1. [Google Scholar]
- Hassell, A.M.; An, G.; Bledsoe, R.K.; Bynum, J.M.; Carter, H.L.; Deng, S.-J.J.; Gampe, R.T.; Grisard, T.E.; Madauss, K.P.; Nolte, R.T.; et al. Crystallization of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 72–79. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls. Int. J. Mol. Sci. 2012, 13, 12857–12879. [Google Scholar] [CrossRef]


{kind=link}
| Target | Analogue | Status | Reference |
|---|---|---|---|
| SAICAR (PurC) | 4-Amino-6-(pyrazol-4-yl)pyrimidine Derivatives | In vitro activity | [11] |
| ArgB | NMR711, NMR446, and L-canavanine | In vitro activity | [63] |
| tRNA (m1G37) methyltransferase | Series of compounds | In vitro activity | [74] |
| Thymidylate Kinase | 4-[3-cyano-2-oxo-7-(1H-pyrazol-4-yl)-5,6-dihydro-1H-benzo[h]quinolin-4-yl] ben zoic acid | In vivo activity | [65] |
| Aspartate decarboxylase (PanD) | (3-(1-naphthamido)pyrazine-2-carboxylic acid) (analogue 2) | In vivo activity | [75] |
| type 2 TA class | Mab3862 | In vitro activity | [76] |
| MmpL3 | PIPD1 | In vivo activity | [77] |
| Decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase | Benzothiazinone, Macozinone | Phase 1 clinical trial | [78,79] |
| Mycobacterial membrane protein Large 3 | SQ109 | In vivo limited activity | [80,81,82] |
| Enoyl Reductase (InhA) | Tetrahydrobenzothieno pyrimidines | In vitro activity | [7] |
| Enoyl Reductase (InhA) | Thiadiazolyl methylthiazoles | In vivo limited activity | [7,83] |
| β-ketoacyl ACP synthase I (KasA) | Indazole sulfonamides (GSK3011724A) | In vitro activity | [84] |
| Cytochrome P450 (CYP121) | 4-(3-amino-1H-pyrazol-4-yl)phenol | In vitro activity | [45] |
| type-2 NADH dehydrogenases | quinolinyl pyrimidine series | In vitro activity | [85] |
| type-2 NADH dehydrogenases | 2−Mercapto quinazolinones | In vitro activity | [86,87] |
| EthR transcriptional repressor | N-phenylphenoxy acetamides | In vitro activity | [7] |
| Inosine-5′-monophosphate dehydrogenase | Indazole sulfonamides | In vitro activity | [88] |
| Structure/X-ray Crystal Structure | Fragment Hit | References |
|---|---|---|
 |  | [11] |
| SAICAR Synthetase (PurC) | 4-Amino-6-(pyrazol-4-yl)pyrimidine Derivatives | |
 |  | [36,74] |
| tRNA (m1G37) methyltransferase | 3-Amino-5-(1-(4-(piperidin-1-ylmethyl)benzyl)-1H-indol-6-yl)-1H-pyrazole-4-carbonitrile derivatives | |
 |  | [65] |
| Thymidylate Kinase | 4-[3-cyano-2-oxo-7-(1H-pyrazol-4-yl)-5,6-dihydro-1H-benzo[h]quinolin-4-yl] benzoic acid | |
 |  | [79] |
| DprE1-PBTZ169 complex | Benzothiazinone, Macozinone | |
 |  | [83,89] |
| Enoyl Reductase (InhA) | Methylthiazoles | |
 |  | [84] |
| β-ketoacyl ACP synthase I (KasA) | Indazole sulfonamides (GSK3011724A) | |
 |  | [45,90] |
| CYP121 in complex with retrofragments 4 | 4-(3-amino-1H-pyrazol-4-yl)phenol | |
 |  | [63] |
| Acetyl Glutamate Kinase (ArgB) | NMR711 and NMR446 | |
 |  | [75,91] |
| Coral model of Mab Aspartate decarboxylase (PanD) | (3-(1-naphthamido)pyrazine-2-carboxylic acid) (analogue 2) |
| Method/Molecules | Challenges |
|---|---|
| X-ray crystallography |
|
| X-ray crystal structures |
|
| Crystal soaking |
|
| Molecular docking with smaller fragments |
|
| Homology modeling |
|
| Stoichiometric approach |
|
| Fragment screening |
|
| Fragments |
|
| Low efficiency fragments |
|
| Fragment potential hits |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Togre, N.S.; Vargas, A.M.; Bhargavi, G.; Mallakuntla, M.K.; Tiwari, S. Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges. Int. J. Mol. Sci. 2022, 23, 10669. https://doi.org/10.3390/ijms231810669
Togre NS, Vargas AM, Bhargavi G, Mallakuntla MK, Tiwari S. Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges. International Journal of Molecular Sciences. 2022; 23(18):10669. https://doi.org/10.3390/ijms231810669
Chicago/Turabian StyleTogre, Namdev S., Ana M. Vargas, Gunapati Bhargavi, Mohan Krishna Mallakuntla, and Sangeeta Tiwari. 2022. "Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges" International Journal of Molecular Sciences 23, no. 18: 10669. https://doi.org/10.3390/ijms231810669
APA StyleTogre, N. S., Vargas, A. M., Bhargavi, G., Mallakuntla, M. K., & Tiwari, S. (2022). Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges. International Journal of Molecular Sciences, 23(18), 10669. https://doi.org/10.3390/ijms231810669