

Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer
 , , , ,
, , , , 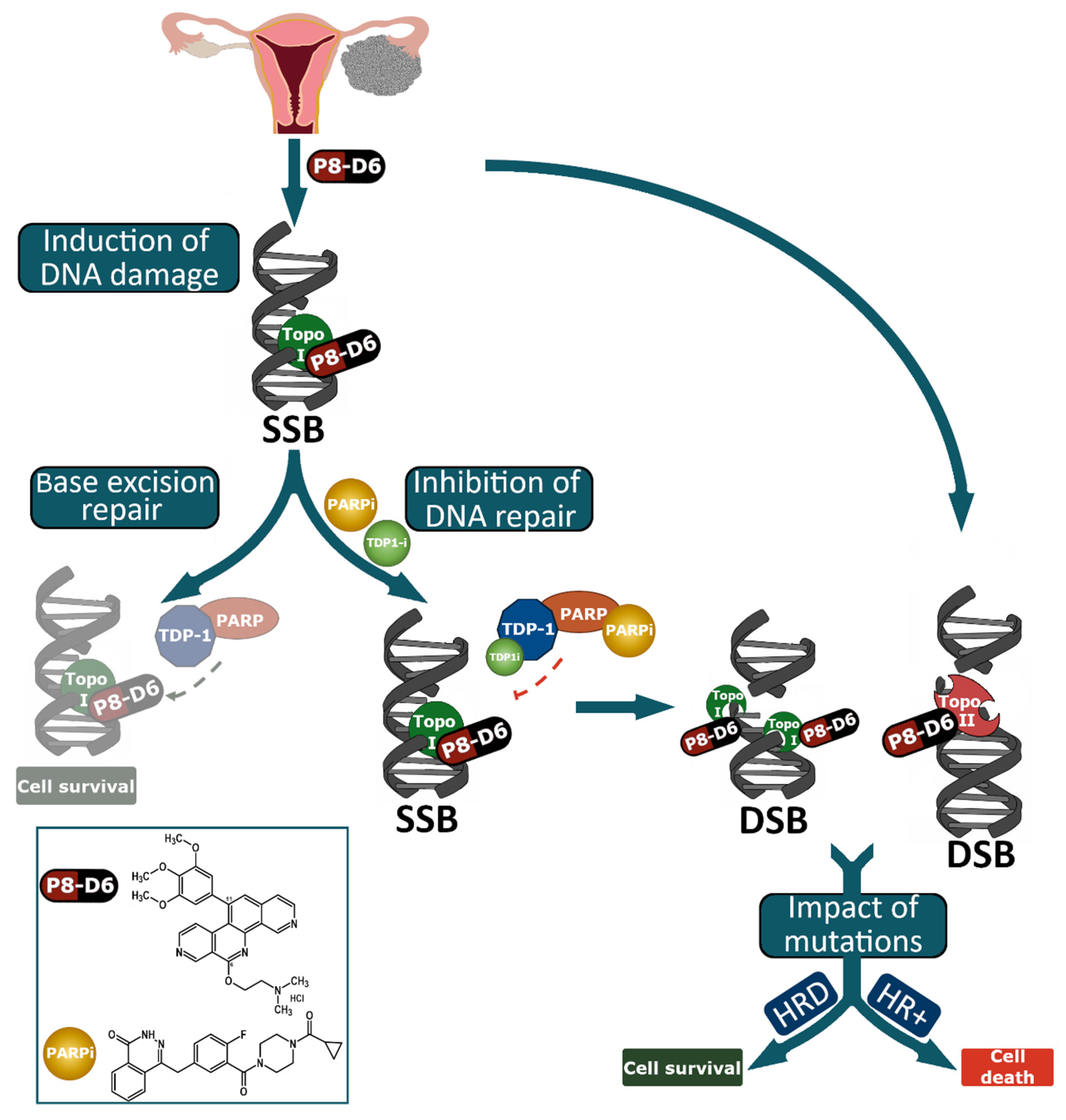{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
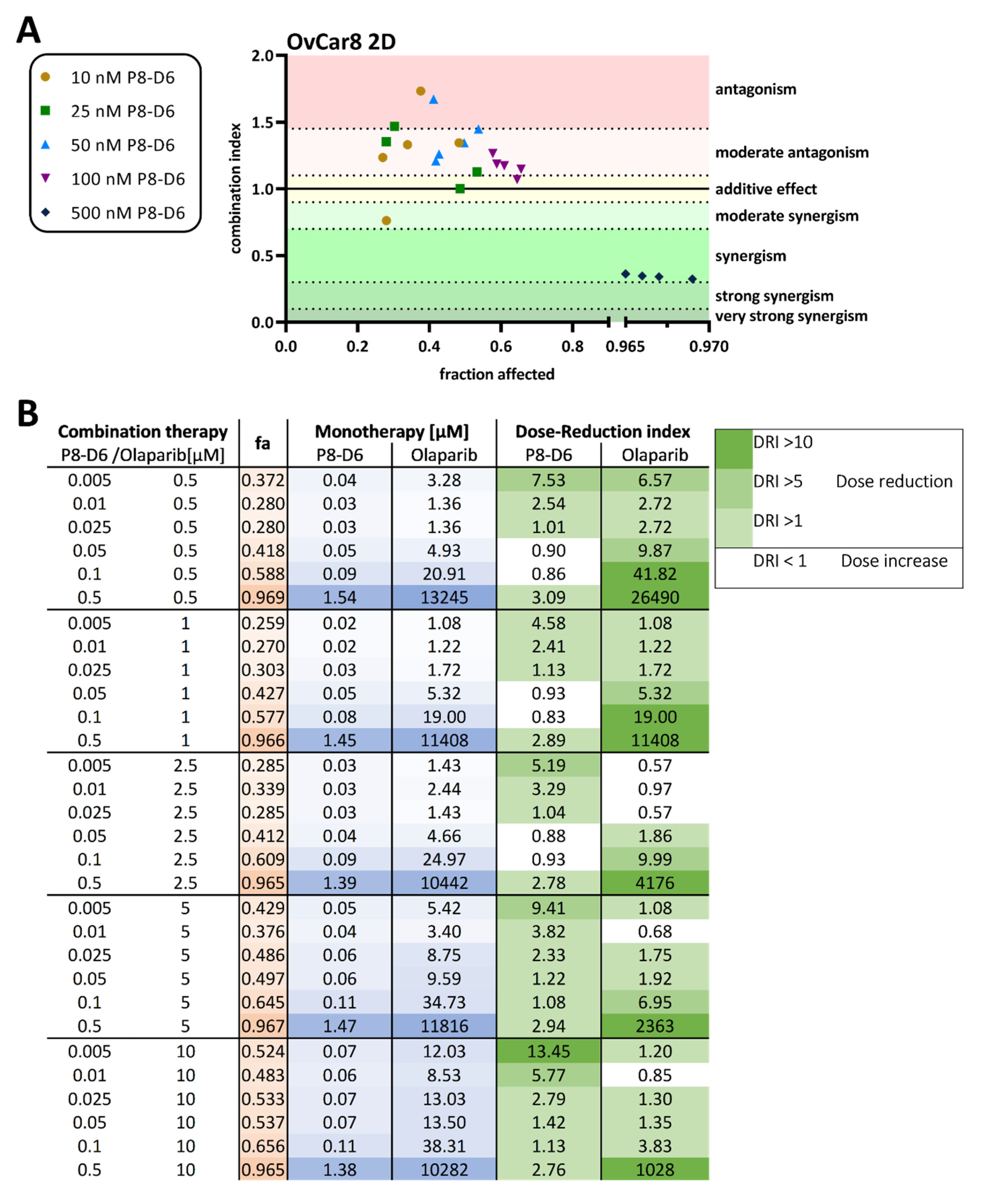
2.1. P8-D6 Sensitizes Ovarian Cancer Cells to Olaparib in 2D Cell Culture
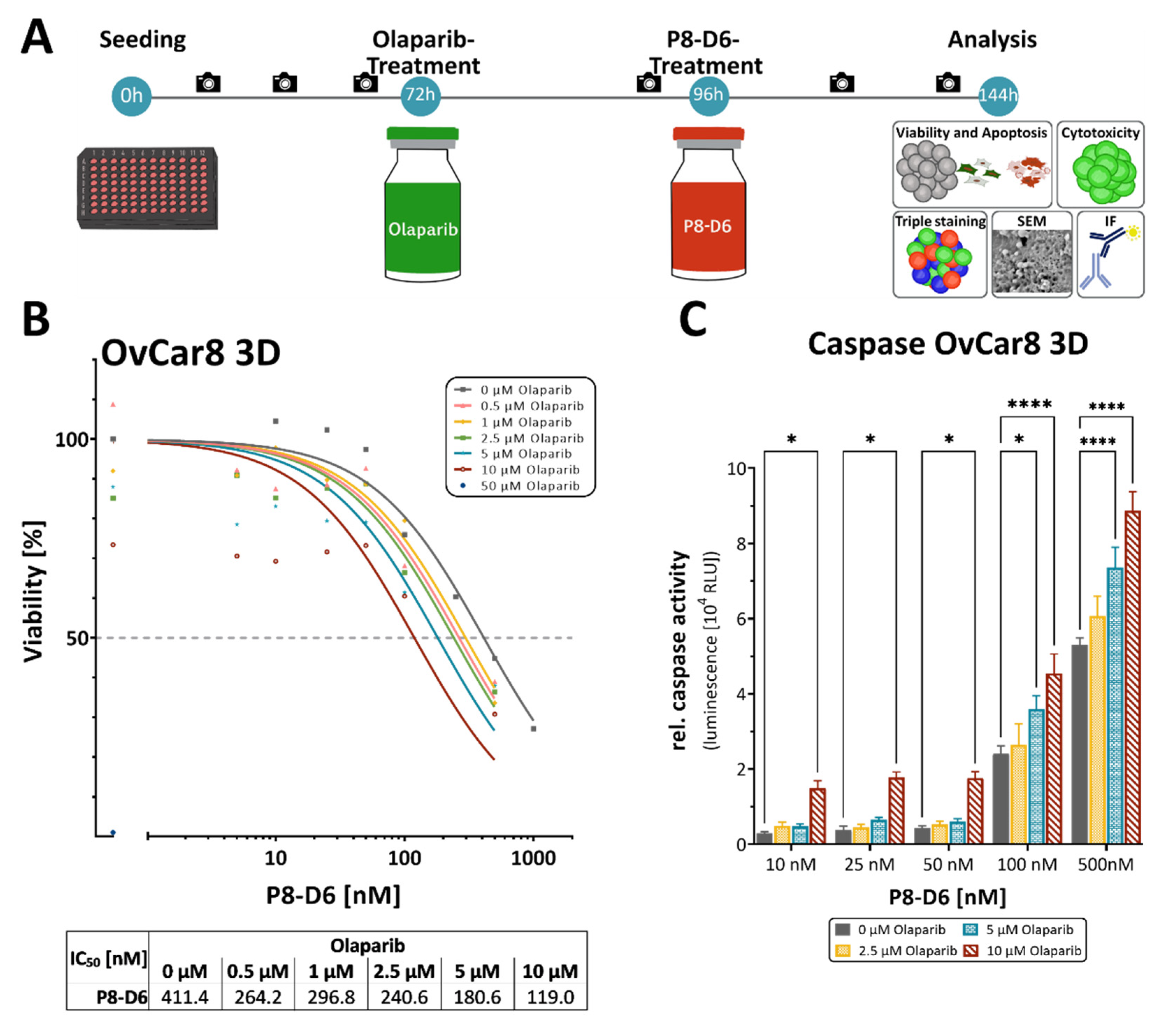
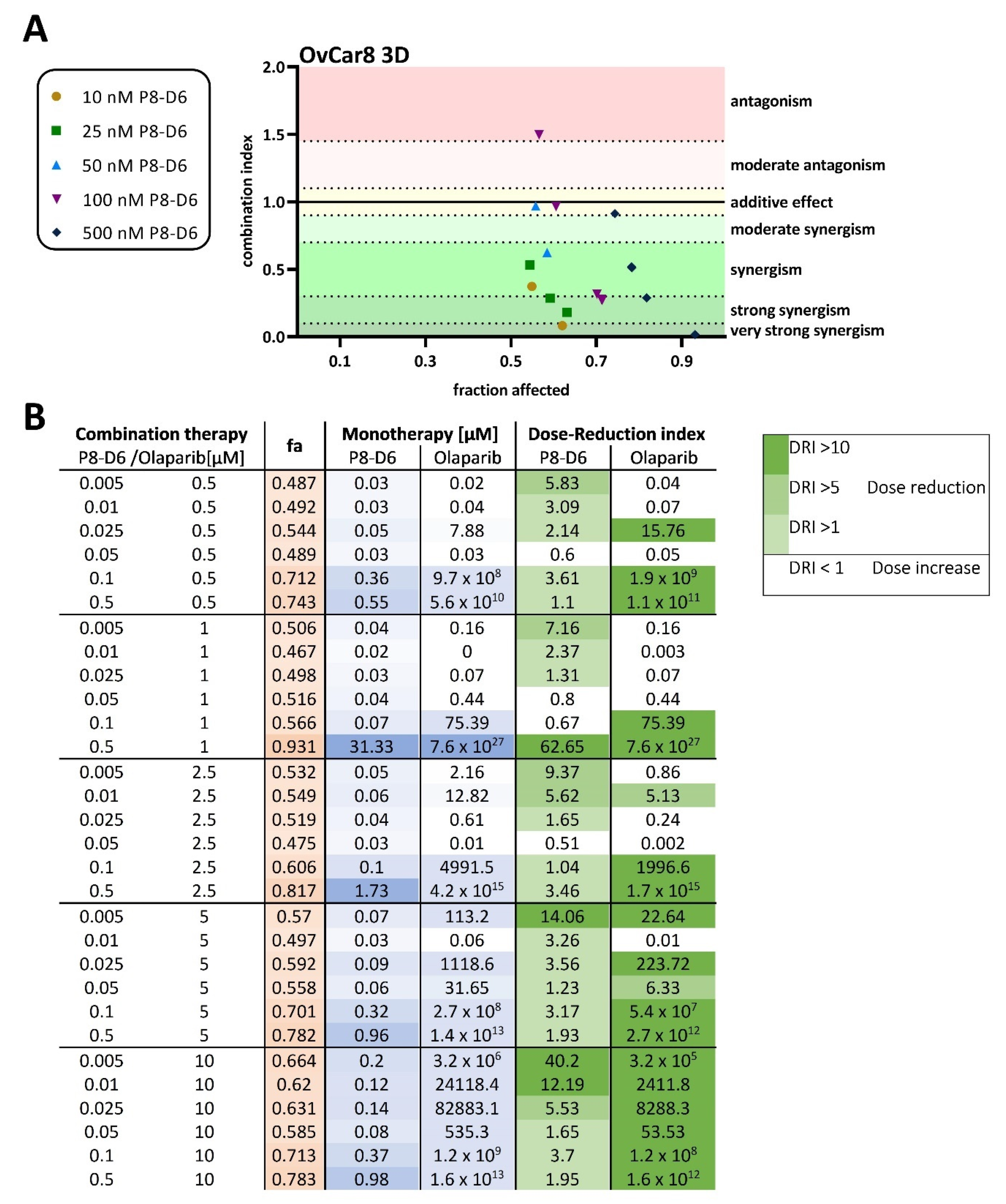
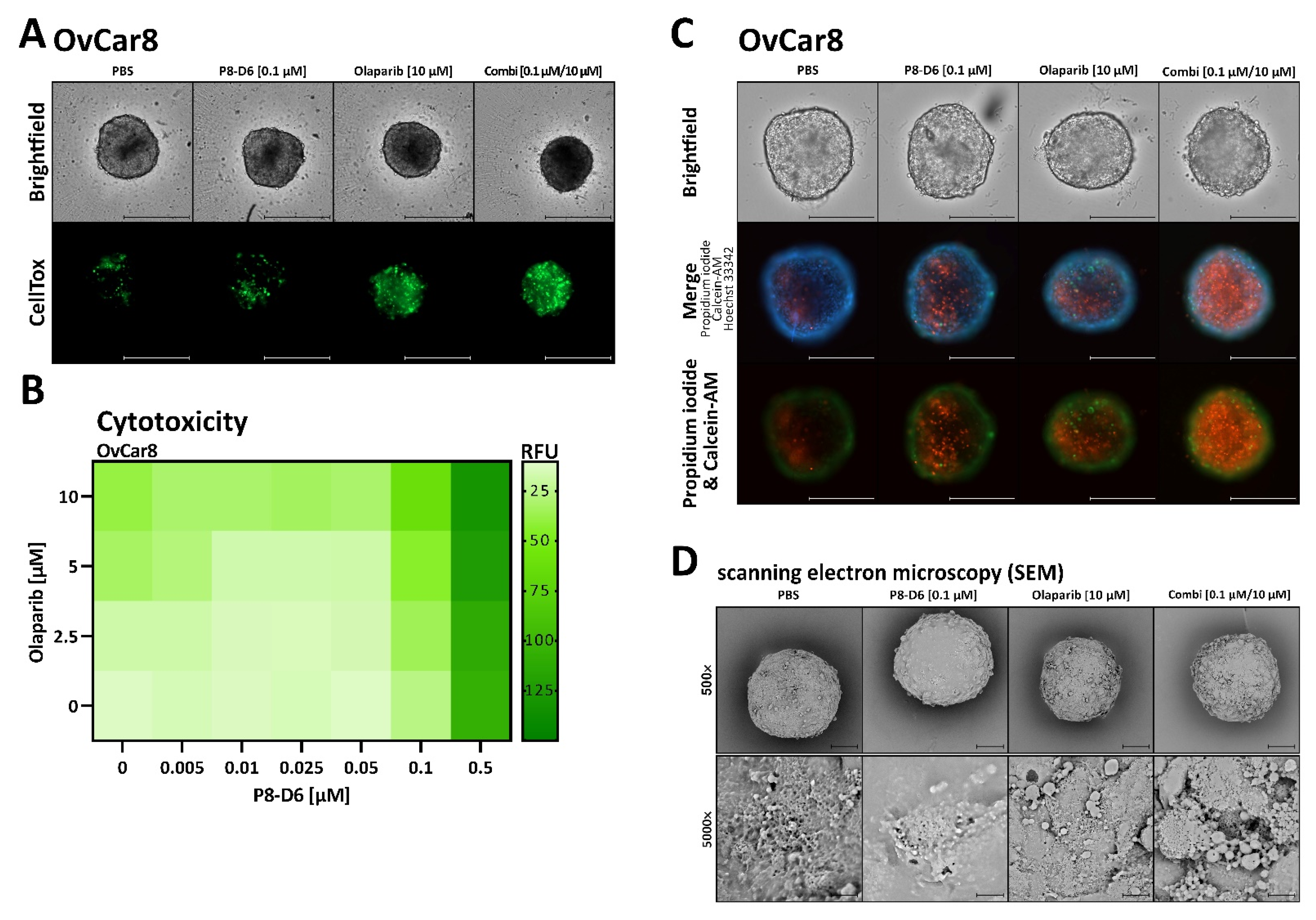
2.2. P8-D6 Sensitizes Ovarian Cancer Cells to Olaparib in 3D Cell Culture
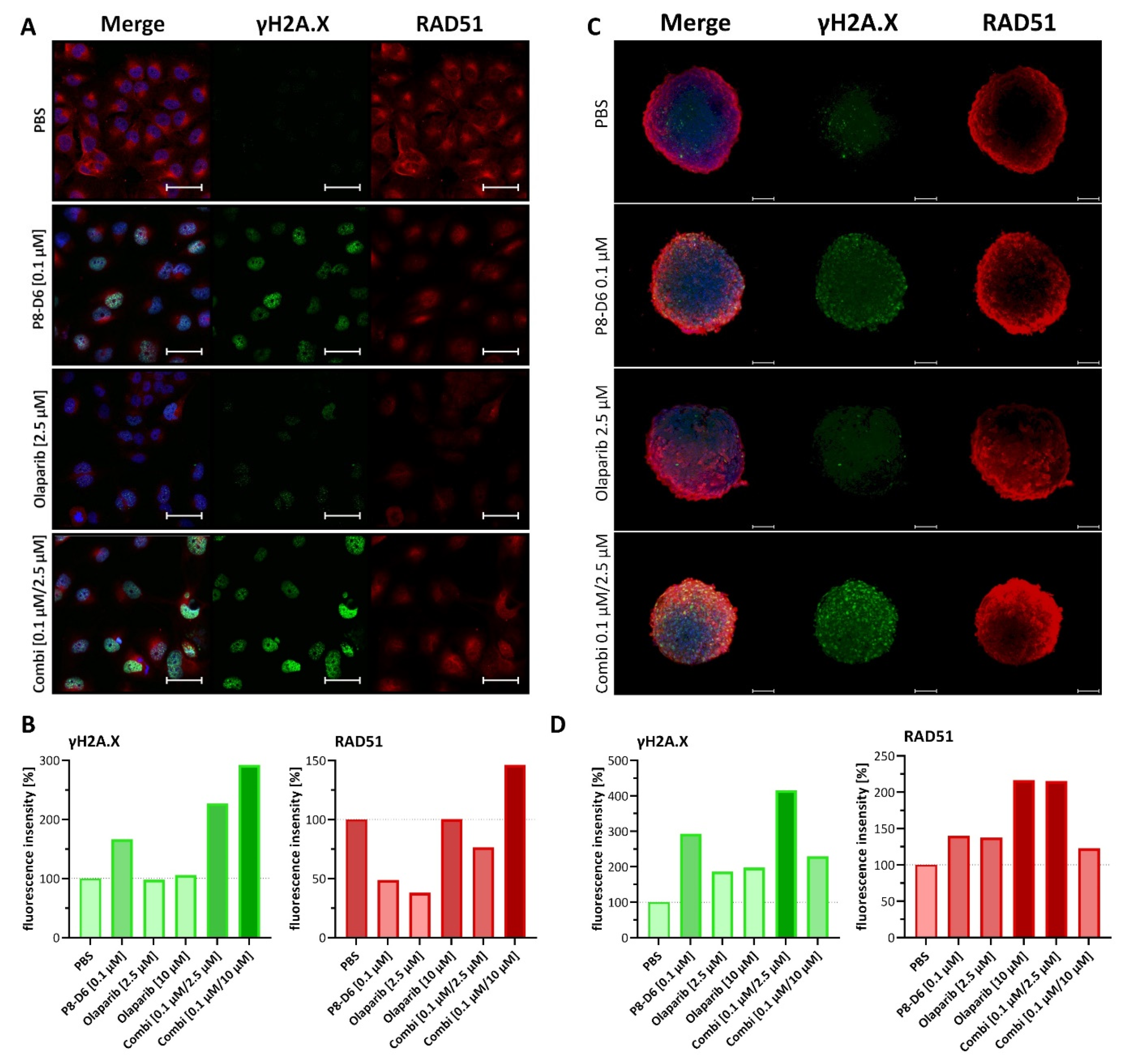
2.3. Combined Treatment of Olaparib and P8-D6 Leads to Genome Instability
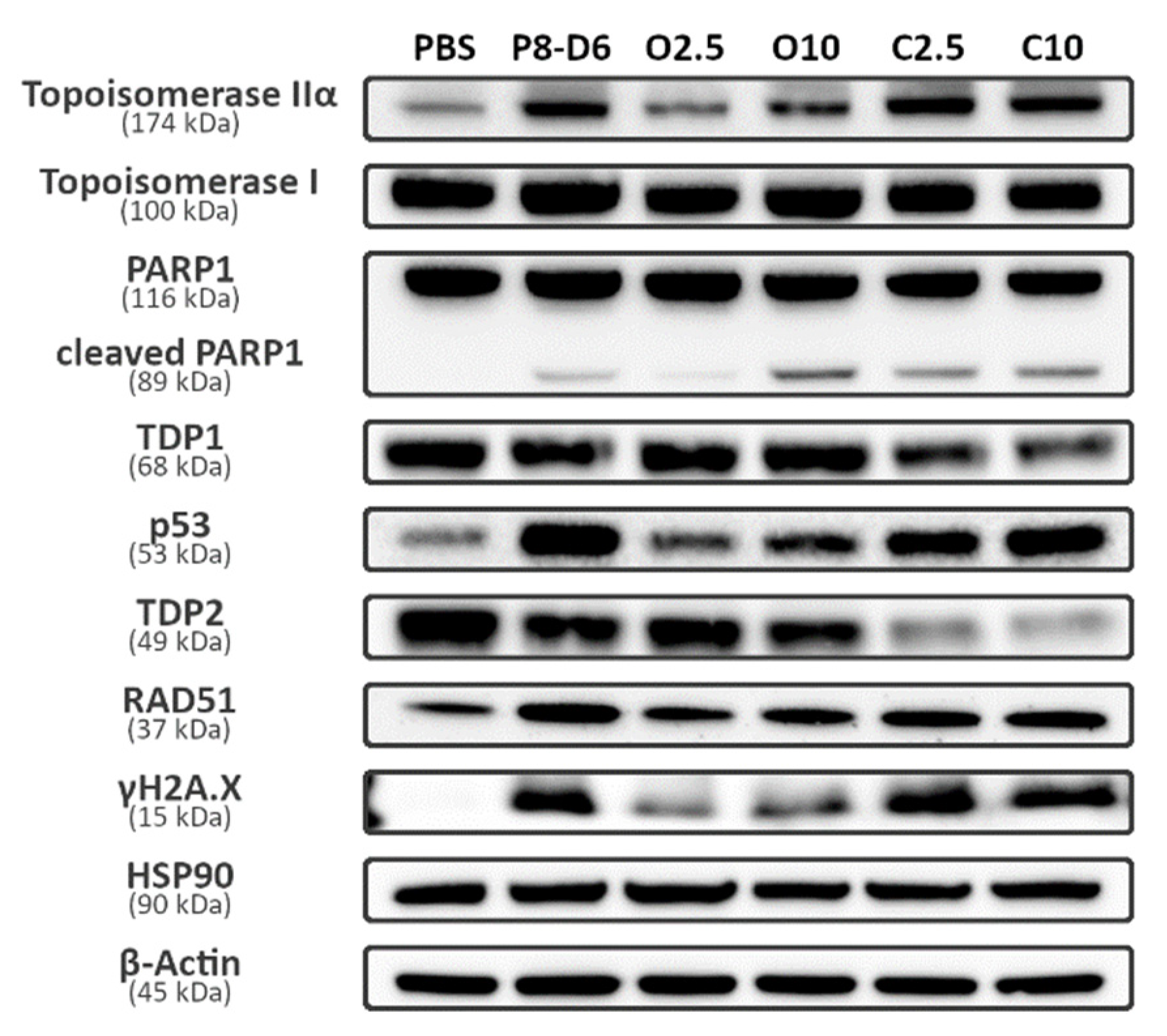
2.4. Combination Therapy Changes Protein Expression of Marker Molecules
3. Discussion
4. Materials and Methods
4.1. Cell Preparation and Culture
4.2. 2D viability and Apoptosis
4.3. 3D Viability, Apoptosis and Cytotoxicity
4.4. Combination Index and Dose-Reduction Index
4.5. Scanning Electron Microscopy
4.6. Fluorescence Imaging (LSM)
4.7. Western Blot Analysis
4.8. HRD by aCGH
4.9. Gene Sequencing
4.10. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA: Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Jayde, V.; White, K.; Blomfield, P. Symptoms and diagnostic delay in ovarian cancer: A summary of the literature. Contemp. Nurse 2009, 34, 55–65. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Ledermann, J.A. First-line treatment of ovarian cancer: Questions and controversies to address. Ther. Adv. Med. Oncol. 2018, 10, 1758835918768232. [Google Scholar] [CrossRef]
- du Bois, A. Treatment of advanced ovarian cancer. Eur. J. Cancer 2001, 37, 1–7. [Google Scholar] [CrossRef]
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019, 8, 7018–7031. [Google Scholar] [CrossRef]
- Flörkemeier, I.; Steinhauer, T.N.; Hedemann, N.; Ölander, M.; Artursson, P.; Clement, B.; Bauerschlag, D.O. Newly developed dual topoisomerase inhibitor P8-D6 is highly active in ovarian cancer. Ther. Adv. Med. Oncol. 2021, 13, 175883592110598. [Google Scholar] [CrossRef]
- Flörkemeier, I.; Steinhauer, T.N.; Hedemann, N.; Weimer, J.P.; Rogmans, C.; van Mackelenbergh, M.T.; Maass, N.; Clement, B.; Bauerschlag, D.O. High Antitumor Activity of the Dual Topoisomerase Inhibitor P8-D6 in Breast Cancer. Cancers 2022, 14, 2. [Google Scholar] [CrossRef]
- Aichinger, G.; Lichtenberger, F.B.; Steinhauer, T.N.; Flörkemeier, I.; Del Favero, G.; Clement, B.; Marko, D. The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties. Molecules 2020, 25, 1524. [Google Scholar] [CrossRef]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.-H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar] [CrossRef]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef]
- Hande, K.R. Topoisomerase II inhibitors. Updat. Cancer Ther. 2008, 3, 13–26. [Google Scholar] [CrossRef]
- Koster, D.A.; Palle, K.; Bot, E.S.M.; Bjornsti, M.-A.; Dekker, N.H. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 2007, 448, 213–217. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Lee, C.-H.; Tsai, H.-P.; An, H.-W.; Lee, C.-M.; Wu, J.-C.; Chen, C.-S.; Huang, S.-H.; Hwang, J.; Cheng, K.-T.; et al. Targeting of Topoisomerase I for Prognoses and Therapeutics of Camptothecin-Resistant Ovarian Cancer. PLoS ONE 2015, 10, e0132579. [Google Scholar] [CrossRef]
- Bai, Y.; Li, L.-D.; Li, J.; Lu, X. Targeting of topoisomerases for prognosis and drug resistance in ovarian cancer. J. Ovarian Res. 2016, 9, 35. [Google Scholar] [CrossRef]
- Meier, C.; Steinhauer, T.N.; Koczian, F.; Plitzko, B.; Jarolim, K.; Girreser, U.; Braig, S.; Marko, D.; Vollmar, A.M.; Clement, B. A Dual Topoisomerase Inhibitor of Intense Pro-Apoptotic and Antileukemic Nature for Cancer Treatment. ChemMedChem 2017, 12, 347–352. [Google Scholar] [CrossRef]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Técher, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks—A new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 591, 1083–1100. [Google Scholar] [CrossRef]
- Znojek, P.; Willmore, E.; Curtin, N.J. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. Br. J. Cancer 2014, 111, 1319–1326. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef]
- Boerner, J.L.; Nechiporchik, N.; Mueller, K.L.; Polin, L.; Heilbrun, L.; Boerner, S.A.; Zoratti, G.L.; Stark, K.; LoRusso, P.M.; Burger, A. Protein expression of DNA damage repair proteins dictates response to topoisomerase and PARP inhibitors in triple-negative breast cancer. PLoS ONE 2015, 10, e0119614. [Google Scholar] [CrossRef]
- Bowman, K.J.; Newell, D.R.; Calvert, A.H.; Curtin, N.J. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br. J. Cancer 2001, 84, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Węsierska-Gądek, J.; Zulehner, N.; Ferk, F.; Składanowski, A.; Komina, O.; Maurer, M. PARP inhibition potentiates the cytotoxic activity of C-1305, a selective inhibitor of topoisomerase II, in human BRCA1-positive breast cancer cells. Biochem. Pharmacol. 2012, 84, 1318–1331. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hedemann, N.; Herz, A.; Schiepanski, J.H.; Dittrich, J.; Sebens, S.; Dempfle, A.; Feuerborn, J.; Rogmans, C.; Tribian, N.; Flörkemeier, I.; et al. ADAM17 Inhibition Increases the Impact of Cisplatin Treatment in Ovarian Cancer Spheroids. Cancers 2021, 13, 2039. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Lagies, S.; Schlimpert, M.; Neumann, S.; Wäldin, A.; Kammerer, B.; Borner, C.; Peintner, L. Cells grown in three-dimensional spheroids mirror in vivo metabolic response of epithelial cells. Commun. Biol. 2020, 3, 246. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; El-Shakankery, K.H.; Lee, J.-Y. PARP inhibitors in ovarian cancer: Overcoming resistance with combination strategies. J. Gynecol. Oncol. 2022, 33, e44. [Google Scholar] [CrossRef]
- Zhou, J.X.; Feng, L.J.; Zhang, X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: A meta-analysis of randomized controlled trials. Drug Des. Devel. Ther. 2017, 11, 3009–3017. [Google Scholar] [CrossRef]
- Guo, X.X.; Wu, H.L.; Shi, H.Y.; Su, L.; Zhang, X. The efficacy and safety of olaparib in the treatment of cancers: A meta-analysis of randomized controlled trials. Cancer Manag. Res. 2018, 10, 2553–2562. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA -mutated (gBRCA m) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef]
- van der Noll, R.; Marchetti, S.; Steeghs, N.; Beijnen, J.H.; Mergui-Roelvink, M.W.J.; Harms, E.; Rehorst, H.; Sonke, G.S.; Schellens, J.H.M. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br. J. Cancer 2015, 113, 396–402. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Z.; Fu, J.; A., J.; Ohno, Y.; Xu, C. Combination treatment with cisplatin, paclitaxel and olaparib has synergistic and dose reduction potential in ovarian cancer cells. Exp. Ther. Med. 2021, 22, 935. [Google Scholar] [CrossRef]
- Perez-Fidalgo, J.A.; Cortés, A.; Guerra, E.; García, Y.; Iglesias, M.; Bohn Sarmiento, U.; Calvo García, E.; Manso Sánchez, L.; Santaballa, A.; Oaknin, A.; et al. Olaparib in combination with pegylated liposomal doxorubicin for platinum-resistant ovarian cancer regardless of BRCA status: A GEICO phase II trial (ROLANDO study). ESMO Open 2021, 6, 100212. [Google Scholar] [CrossRef]
- Dréan, A.; Lord, C.J.; Ashworth, A. PARP inhibitor combination therapy. Crit. Rev. Oncol. Hematol. 2016, 108, 73–85. [Google Scholar] [CrossRef]
- Das, B.B.; Huang, S.-Y.N.; Murai, J.; Rehman, I.; Amé, J.-C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Wang, Y.; Aloyz, R.; Panasci, L. The PARP inhibitor ABT-888 synergizes irinotecan treatment of colon cancer cell lines. Investig. New Drugs 2013, 31, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Inoue, T.; Sato, F.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. The Use of Olaparib (AZD2281) Potentiates SN-38 Cytotoxicity in Colon Cancer Cells by Indirect Inhibition of Rad51-Mediated Repair of DNA Double-Strand Breaks. Mol. Cancer Ther. 2014, 13, 1170–1180. [Google Scholar] [CrossRef]
- Eetezadi, S.; Evans, J.C.; Shen, Y.-T.; de Souza, R.; Piquette-Miller, M.; Allen, C. Ratio-Dependent Synergism of a Doxorubicin and Olaparib Combination in 2D and Spheroid Models of Ovarian Cancer. Mol. Pharm. 2018, 15, 472–485. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Weimer, J.; Wurmb-Schwark, N.; von Fredrik, R.; Arnold, N.; Schem, C. Alteration of STR profiles in ovarian carcinoma cells during primary culture. Arch. Gynecol. Obstet. 2016, 294, 369–376. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Nürnberg, E.; Vitacolonna, M.; Klicks, J.; von Molitor, E.; Cesetti, T.; Keller, F.; Bruch, R.; Ertongur-Fauth, T.; Riedel, K.; Scholz, P.; et al. Routine Optical Clearing of 3D-Cell Cultures: Simplicity Forward. Front. Mol. Biosci. 2020, 7, 20. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Deutsches Konsortium Familiärer Brust- und Eierstockkrebs. TruRisk® Genpanel-Analyse. Available online: https://www.konsortium-familiaerer-brustkrebs.de/betreuungskonzept/molekulare-diagnostik/truriskr-genpanel-analyse/ (accessed on 29 June 2022).








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flörkemeier, I.; Hillmann, J.S.; Weimer, J.P.; Hildebrandt, J.; Hedemann, N.; Rogmans, C.; Dempfle, A.; Arnold, N.; Clement, B.; Bauerschlag, D.O. Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 10503. https://doi.org/10.3390/ijms231810503
Flörkemeier I, Hillmann JS, Weimer JP, Hildebrandt J, Hedemann N, Rogmans C, Dempfle A, Arnold N, Clement B, Bauerschlag DO. Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer. International Journal of Molecular Sciences. 2022; 23(18):10503. https://doi.org/10.3390/ijms231810503
Chicago/Turabian StyleFlörkemeier, Inken, Julia S. Hillmann, Jörg P. Weimer, Jonas Hildebrandt, Nina Hedemann, Christoph Rogmans, Astrid Dempfle, Norbert Arnold, Bernd Clement, and Dirk O. Bauerschlag. 2022. "Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer" International Journal of Molecular Sciences 23, no. 18: 10503. https://doi.org/10.3390/ijms231810503
APA StyleFlörkemeier, I., Hillmann, J. S., Weimer, J. P., Hildebrandt, J., Hedemann, N., Rogmans, C., Dempfle, A., Arnold, N., Clement, B., & Bauerschlag, D. O. (2022). Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer. International Journal of Molecular Sciences, 23(18), 10503. https://doi.org/10.3390/ijms231810503