
Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols

 ,
,  , and
, and 
Abstract
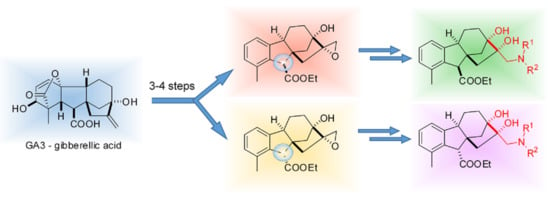
:
1. Introduction
2. Results
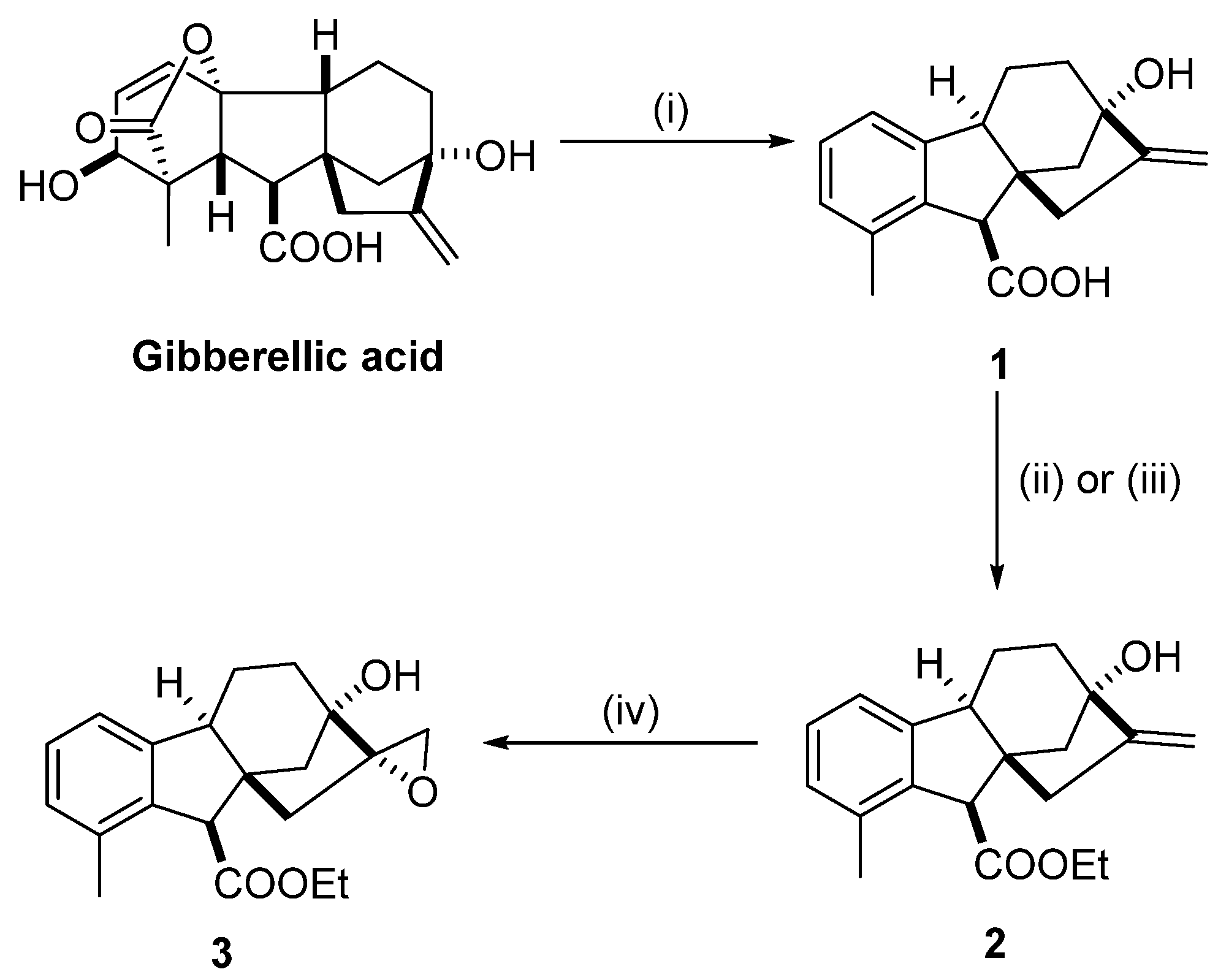
2.1. Preparation of Epoxyalcohol 3 Based on Allo-Gibberic Acid
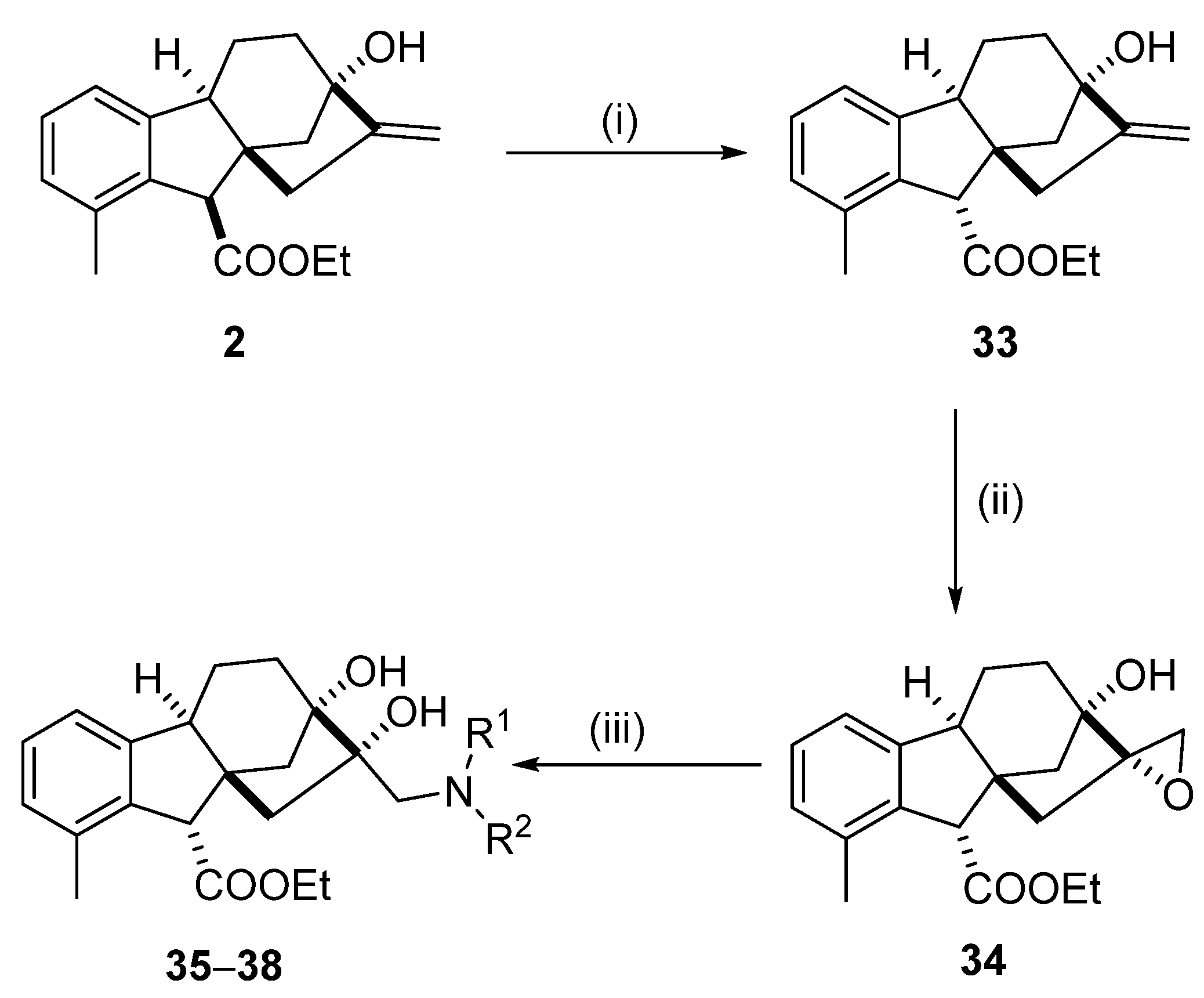
2.2. Synthesis of Allo-Gibberic Acid-Based Aminodiol Derivatives
2.3. Synthesis of Azole Derivatives Based on Allo-Gibberic Acid
2.4. Isomerization of the Ester Group
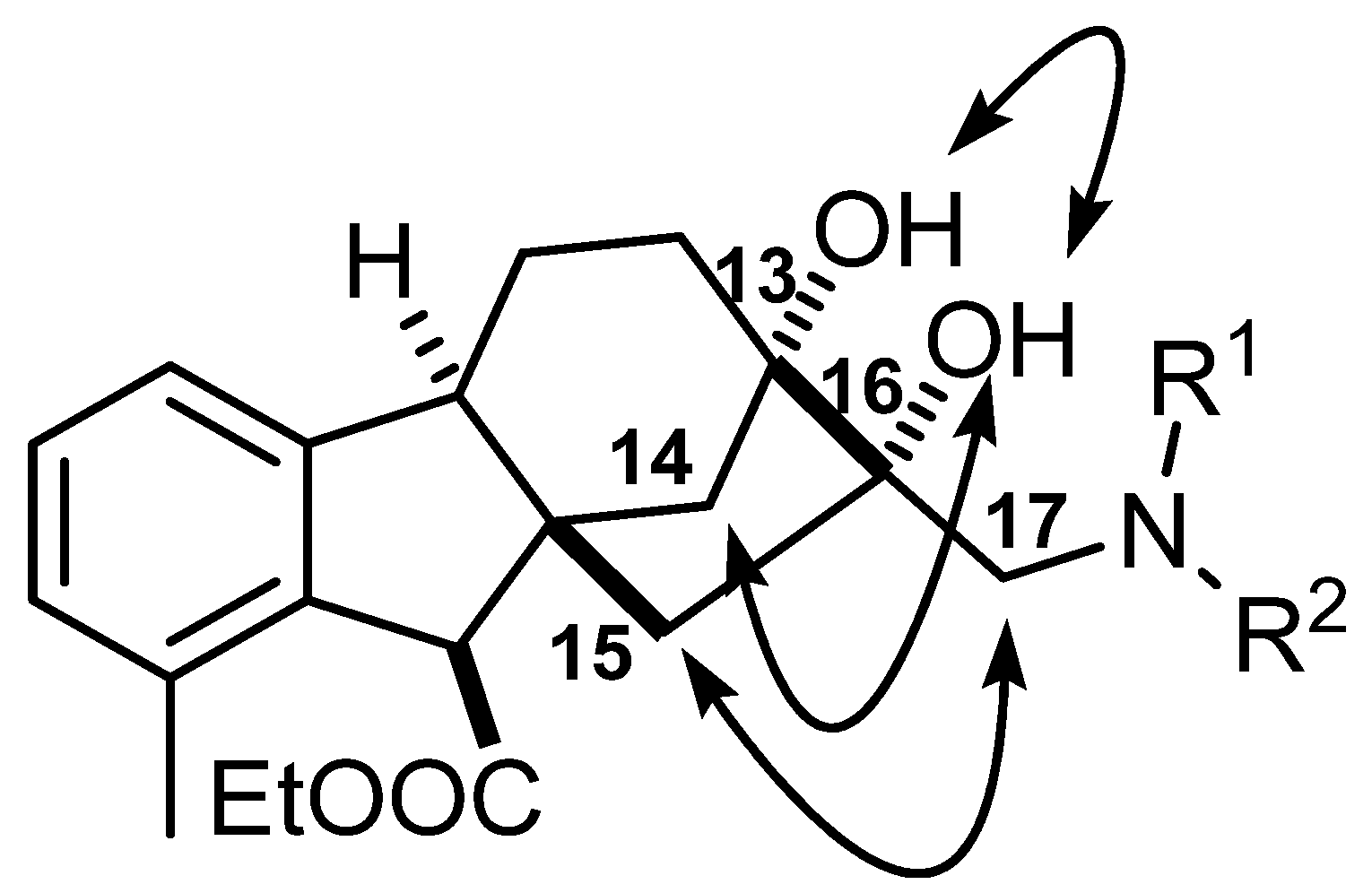
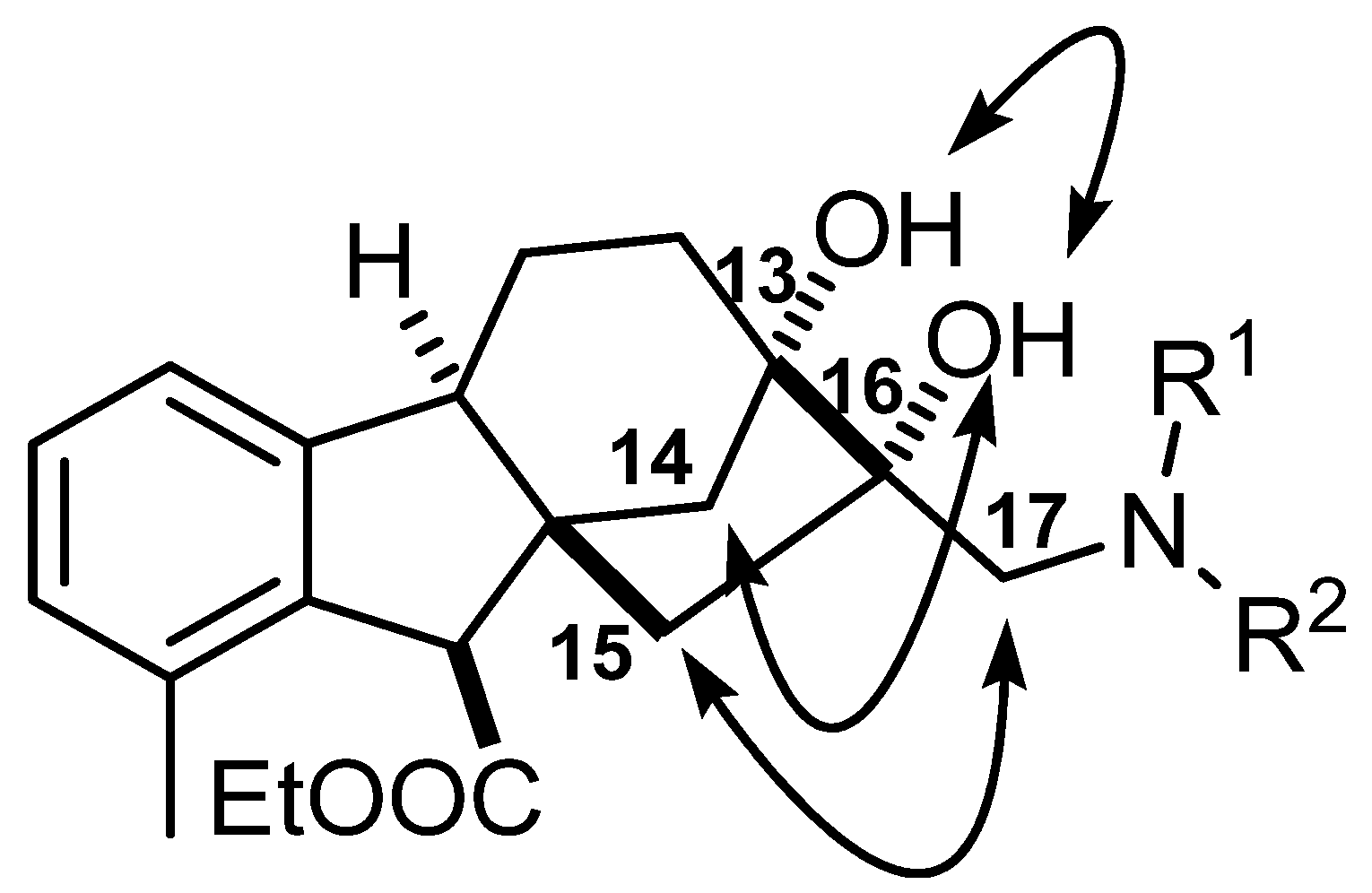
2.5. Determination of Relative Configuration of Allo-Gibberic Acid Derivatives
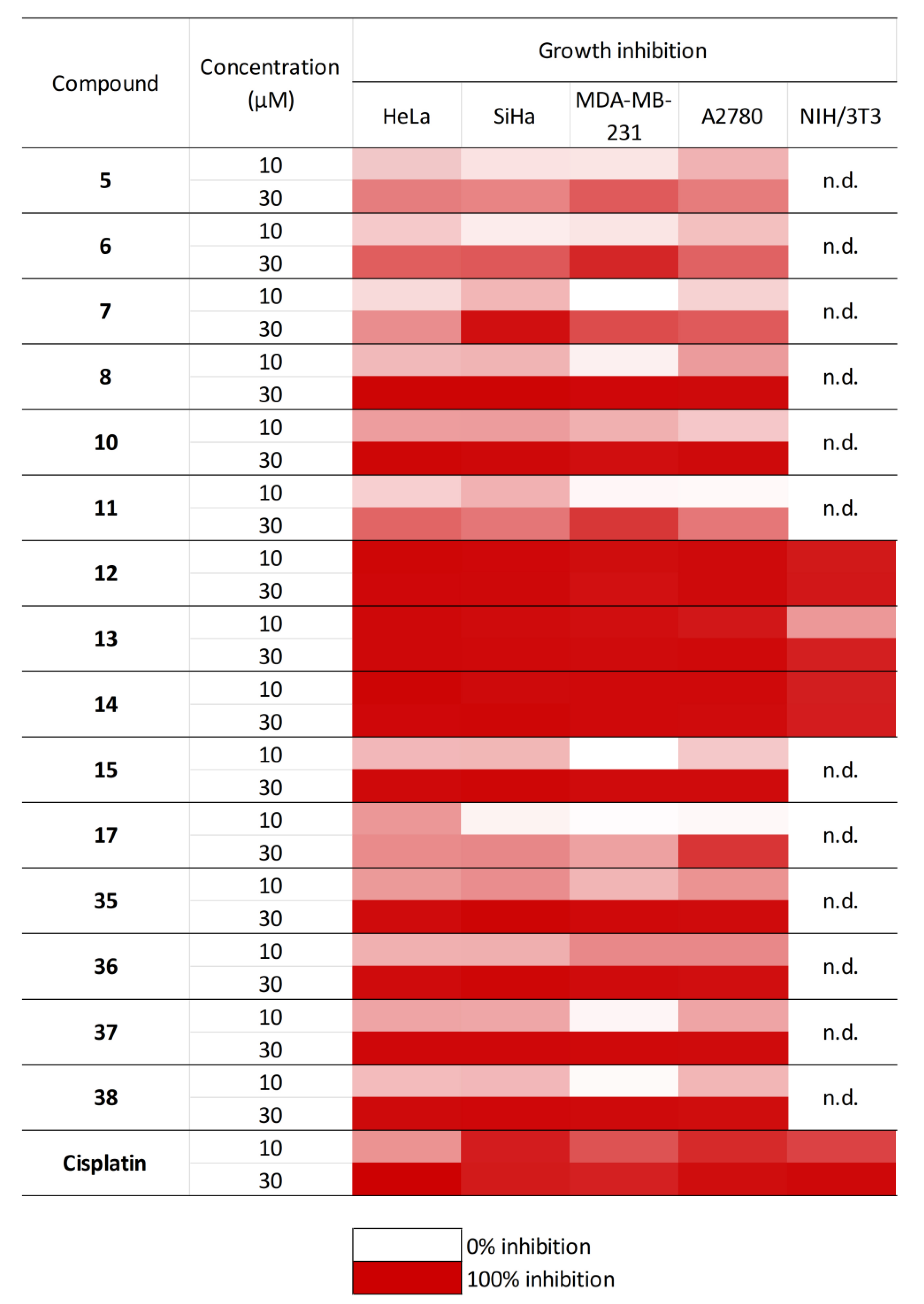
2.6. In Vitro Antiproliferative Studies of Gibberellic Acid-Based Aminodiols
2.7. In Vitro Antioxidant Activity Studies of Gibberellic Acid-Based Aminodiols
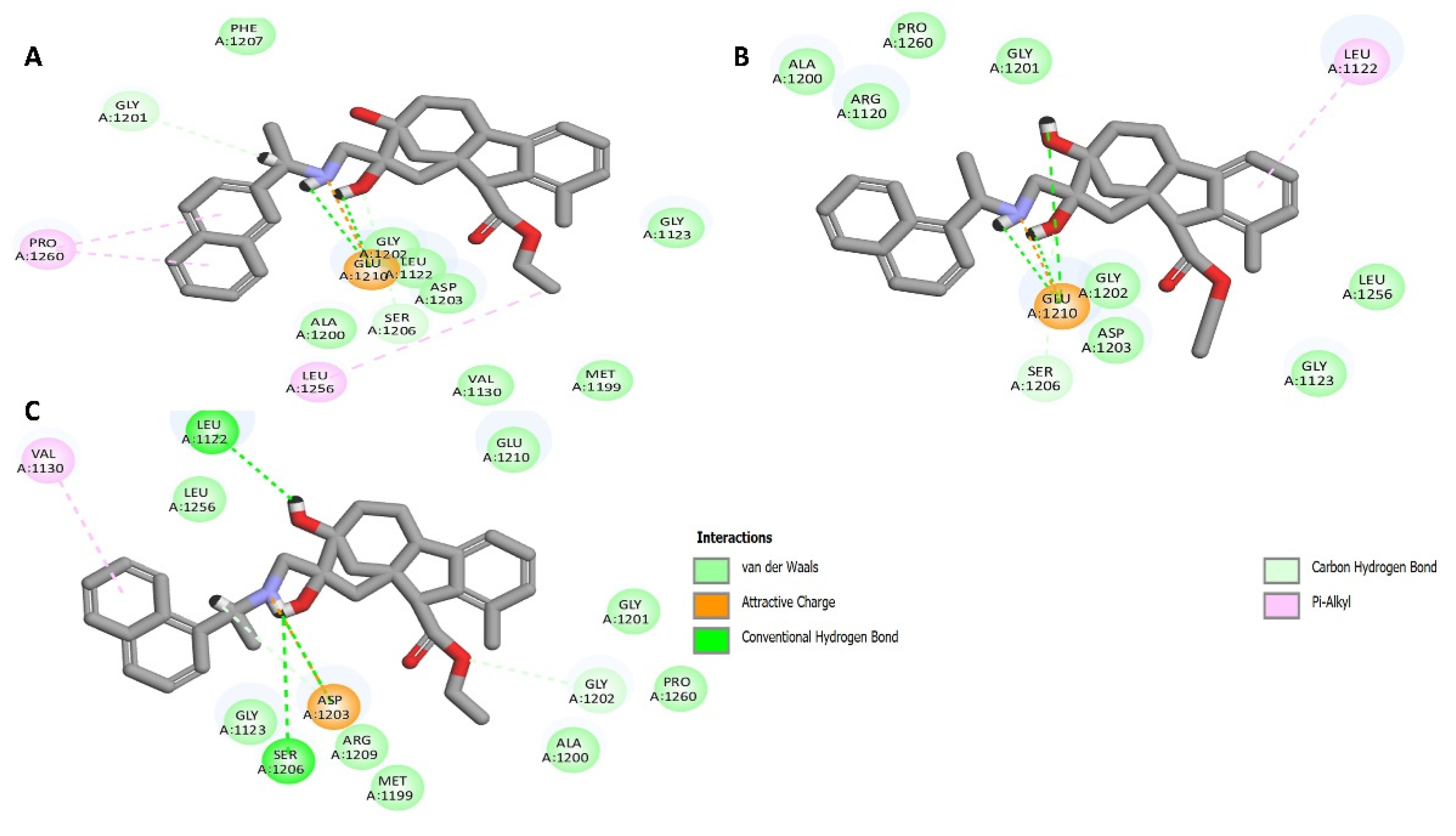
2.8. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. General Procedure for Epoxidation
4.3. General Procedure for Ring-Opening of Epoxide with Amines
4.4. General Procedure for Ring-Opening of Epoxide with Azoles
4.5. General Procedure for Dipolar Cycloaddition
4.6. Determination of Antiproliferative Effects of Aminodiol Derivatives
4.7. Determination of Antioxidant Effects by Free Radical Scavenging (DPPH Assay)
4.8. Docking Study
4.8.1. Preparation of the Crystal Structure of ALK
4.8.2. Docking Study (CDocker)
4.8.3. In-Silico ADMET Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kroll, D.J. Natural Compounds in Cancer Therapy: Promising Nontoxic Antitumor Agents from Plants and Other Natural Sources. J. Nat. Prod. 2001, 64, 1605–1606. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzym. Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Csermely, P. Heat shock proteins in the regulation of apoptosis: New strategies in tumor therapy. Pharmacol. Ther. 2004, 101, 227–257. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.G.; Golfis, G.; Knox, A.J.; Fayne, D.; Meegan, M.J.; Oprea, T.I. Oncology exploration: Charting cancer medicinal chemistry space. Drug Discov. Today 2006, 11, 149–159. [Google Scholar] [CrossRef]
- Raskin, I.; Ribnicky, D.M.; Komarnytsky, S.; Ilic, N.; Poulev, A.; Borisjuk, N.; Brinker, A.; Moreno, D.Á.; Ripoll, C.; Yakoby, N.; et al. Plants and human health in the twenty-first century. Trends Biotechnol. 2002, 20, 522–531. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Zhao, S.; Wang, X.; Liu, B.; Xu, H. Click Chemistry in Natural Product Modification. Front. Chem. 2021, 9, 774977. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Urbanová, T.; Tarkowská, D.; Strnad, M.; Hedden, P. Gibberellins—terpenoid plant hormones: Biological importance and chemical analysis. Collect. Czechoslov. Chem. Commun. 2011, 76, 1669–1686. [Google Scholar] [CrossRef]
- Pérez, F.J.; Vecchiola, A.; Pinto, M.; Agosin, E. Gibberellic acid decomposition and its loss of biological activity in aqueous solutions. Phytochemistry 1996, 41, 675–679. [Google Scholar] [CrossRef]
- Chen, H.; Liu, X.; Yang, D.; Yin, P. Degradation pattern of gibberellic acid during the whole process of tea production. Food Chem. 2013, 138, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; de Souza Vandenberghe, L.P.; De Oliveira, J.; Soccol, C.R. New perspectives of gibberellic acid production: A review. Crit. Rev. Biotechnol. 2011, 32, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Alsudays, I.M.A.; Sayed, H.E.S.A.E. Influence of Ascorbic Acid, Gibberellic Acid and Moringa oleifera Extract for Alleviating Salinity Stress by Enhancing Antioxidant Enzymatic Activity and Some Physiological Studies on Two Tomato Cultivars. J. Appl. Life Sci. Int. 2020, 23, 39–62. [Google Scholar] [CrossRef]
- Bose, S.K.; Yadav, R.K.; Mishra, S.; Sangwan, R.S.; Singh, A.K.; Mishra, B.; Srivastava, A.K.; Sangwan, N.S. Effect of gibberellic acid and calliterpenone on plant growth attributes, trichomes, essential oil biosynthesis and pathway gene expression in differential manner in Mentha arvensis L. Plant Physiol. Biochem. 2013, 66, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Camara, M.C.; Vandenberghe, L.P.S.; Rodrigues, C.; de Oliveira, J.; Faulds, C.; Bertrand, E.; Soccol, C.R. Current advances in gibberellic acid (GA3) production, patented technologies and potential applications. Planta 2018, 248, 1049–1062. [Google Scholar] [CrossRef]
- El-Sayed, M.Y.; Fetooh, H.; Refat, M.S.; Eldaroti, H.H.; Adam, A.M.A.; Saad, H.A. Complexes of the plant hormone gibberellic acid with the Pt(II), Au(III), Ru(III), V(III), and Se(IV) ions: Preparation, characterization, and in vitro evaluation of biological activity. J. Mol. Liq. 2019, 296, 111895. [Google Scholar] [CrossRef]
- Pereira, A.E.S.; Sandoval-Herrera, I.E.; Zavala-Betancourt, S.A.; Oliveira, H.C.; Ledezma-Pérez, A.S.; Romero, J.; Fraceto, L.F. γ-Polyglutamic acid/chitosan nanoparticles for the plant growth regulator gibberellic acid: Characterization and evaluation of biological activity. Carbohydr. Polym. 2017, 157, 1862–1873. [Google Scholar] [CrossRef]
- Seleem, A.A.; Hussein, B.H.M. Synthesis and effect of a new Terbium gibberellic complex on the histopathological alteration induced by Gibberellic acid on liver and kidney of mice Mus musculus. Chem. Biol. Drug Des. 2018, 92, 1288–1300. [Google Scholar] [CrossRef]
- Karihtala, P.; Soini, Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies: Review Article. Acta Path. Micro. Im. 2007, 115, 81–103. [Google Scholar] [CrossRef]
- Troudi, A.; Mahjoubi Samet, A.; Zeghal, N. Hepatotoxicity induced by gibberellic acid in adult rats and their progeny. Exp. Toxicol. Pathol. 2010, 62, 637–642. [Google Scholar] [CrossRef]
- Fath, A.; Bethke, P.C.; Jones, R.L. Enzymes That Scavenge Reactive Oxygen Species Are Down-Regulated Prior to Gibberellic Acid-Induced Programmed Cell Death in Barley Aleurone. Plant Physiol. 2001, 126, 156–166. [Google Scholar] [CrossRef] [PubMed]
- El-Mofty, M.M.; Sakr, S.A.; Rizk, A.M.; Moussa, E.A. Carcinogenic effect of gibberellin A3in Swiss Albino Mice. Nutr. Cancer 1994, 21, 183–190. [Google Scholar] [CrossRef] [PubMed]
- El-Mofty, M.M.; Sakr, S.A. Induction of Neoplasms in the Egyptian Toad Bufo regularis by Gibberellin A3. Oncology 1988, 45, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, S.; Srikumar, K. Metabolic dysregulation and inhibition of spermatogenesis by gibberellic acid in rat testicular cells. J. Environ. Biol. 2005, 26, 567–569. [Google Scholar]
- Soliman, M.M.; Aldhahrani, A.; Gaber, A.; Alsanie, W.F.; Shukry, M.; Mohamed, W.A.; Metwally, M.M.M.; Mohamed, A.A. Impacts of n-acetyl cysteine on gibberellic acid-induced testicular dysfunction through regulation of inflammatory cytokines, steroid and antioxidant activity. Andrologia 2021, 53, e14036. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Z.; Zhang, Y.; Zeng, X.; Qing, C.; Liu, J.; Li, L.; Zhang, H. Synthesis of gibberellin derivatives with anti-tumor bioactivities. Bioorganic Med. Chem. Lett. 2009, 19, 5496–5499. [Google Scholar] [CrossRef]
- Zhu, S.-Y.; Luo, F.-Z.; Sun, P.-H. Synthesis and antitumor activity of novel gibberellin derivatives with tetracyclic diterpenoid skeletons. Med. Chem. Res. 2020, 29, 1341–1354. [Google Scholar] [CrossRef]
- Egbewande, F.A.; Sadowski, M.C.; Levrier, C.; Tousignant, K.D.; White, J.M.; Coster, M.J.; Nelson, C.C.; Davis, R.A. Identification of Gibberellic Acid Derivatives That Deregulate Cholesterol Metabolism in Prostate Cancer Cells. J. Nat. Prod. 2018, 81, 838–845. [Google Scholar] [CrossRef]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [Google Scholar] [CrossRef]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. Int. J. Mol. Sci. 2019, 21, 184. [Google Scholar] [CrossRef]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Synthesis and Biological Application of Isosteviol-Based 1,3-Aminoalcohols. Int. J. Mol. Sci. 2021, 22, 11232. [Google Scholar] [CrossRef] [PubMed]
- Huigens Iii, R.W.; Morrison, K.C.; Hicklin, R.W.; Flood, T.A., Jr.; Richter, M.F.; Hergenrother, P.J. A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 2013, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Allam, B.K.; Raghuvanshi, D.S.; Singh, K.N. An Efficient Tetrabutylammonium Fluoride (TBAF)-Catalyzed Three-Component Synthesis of 3-Substituted Indole Derivatives under Solvent-Free Conditions. Adv. Synth. Catal. 2013, 355, 1840–1848. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shimazaki, H.; Miyamoto, Y.; Shimada, K.; Haga, F.; Yamada, Y.; Miyazawa, H.; Nishiwaki, K.; Kashimura, S. Simple and Convenient Synthesis of Esters from Carboxylic Acids and Alkyl Halides Using Tetrabutylammonium Fluoride. J. Oleo Sci. 2014, 63, 539–544. [Google Scholar] [CrossRef]
- Rabie, R.; Hammouda, M.M.; Elattar, K.M. Cesium carbonate as a mediated inorganic base in some organic transformations. Res. Chem. Intermed. 2017, 43, 1979–2015. [Google Scholar] [CrossRef]
- Lee, J.C.; Oh, Y.S.; Cho, S.H.; Lee, J.D. Efficientin Situesterification of Carboxylic Acids Using Cesium Carbonate. Org. Prep. Proced. Int. 1996, 28, 480–483. [Google Scholar] [CrossRef]
- Parrish, J.P.; Dueno, E.E.; Kim, S.-I.; Jung, K.W. Improved Cs2CO3Promoted O-Alkylation of Acids. Synth. Commun. 2000, 30, 2687–2700. [Google Scholar] [CrossRef]
- Henbest, H.B.; Wilson, R.A.L. 376. Aspects of stereochemistry. Part I. Stereospecificity in formation of epoxides from cyclic allylic alcohols. J. Chem. Soc. 1957, 376, 1958–1965. [Google Scholar] [CrossRef]
- Azizi, N.; Mirmashhori, B.; Saidi, M.R. Lithium perchlorate promoted highly regioselective ring opening of epoxides under solvent-free conditions. Catal. Commun. 2007, 8, 2198–2203. [Google Scholar] [CrossRef]
- Assadieskandar, A.; Yu, C.; Maisonneuve, P.; Kurinov, I.; Sicheri, F.; Zhang, C. Rigidification Dramatically Improves Inhibitor Selectivity for RAF Kinases. ACS Med. Chem. Lett. 2019, 10, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Viana, J.; Monteiro, A.F.M.; Filho, J.M.B.; Scotti, L.; Scotti, M.T. The Azoles in Pharmacochemistry: Perspectives on the Synthesis of New Compounds and Chemoinformatic Contributions. Curr. Pharm. Des. 2020, 25, 4702–4716. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, Z.; Li, M.; Wang, C. K2CO3-Promoted Ring-Opening/Cyclization Reactions of Multi-substituted Donor-Acceptor Cyclopropanes with Thiourea: Access to 2-Amino-4,6-diarylnicotinonitrile Derivatives. ChemistrySelect 2020, 5, 6011–6015. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide–alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Breugst, M.; Reissig, H. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Sillanpää, R.; Fülöp, F. Stereoselective synthesis of perillaldehyde-based chiral β-amino acid derivatives through conjugate addition of lithium amides. Beilstein J. Org. Chem. 2014, 10, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Brian, P.W.; Grove, J.F.; MacMillan, J. The Gibberellins. In Progress in the Chemistry of Organic Natural Products; Zechmeister, L., Ed.; Springer: Vienna, Austria, 1960; pp. 350–433. ISBN 978-3-7091-7161-5. [Google Scholar]
- Grove, J.F.; Mulholland, T.P.C. 605. Gibberellic acid. Part XII. The stereochemistry of allogibberic acid. J. Chem. Soc. 1960, 605, 3007–3022. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Trueba, G.P.; Sánchez, G.M.; Giuliani, A. Oxygen free radical and antioxidant defense mechanism in cancer. Front. Biosci. 2004, 9, 2029–2044. [Google Scholar] [CrossRef]
- Fabbro, D.; Ruetz, S.; Buchdunger, E.; Cowan-Jacob, S.W.; Fendrich, G.; Liebetanz, J.; Mestan, J.; O’Reilly, T.; Traxler, P.; Chaudhuri, B.; et al. Protein kinases as targets for anticancer agents: From inhibitors to useful drugs. Pharmacol. Ther. 2002, 93, 79–98. [Google Scholar] [CrossRef]
- Cicenas, J.; Račienė, A. Anti-Cancer Drugs Targeting Protein Kinases Approved by FDA in 2020. Cancers 2021, 13, 947. [Google Scholar] [CrossRef]
- Liu, M.; Wang, W.-G.; Sun, H.-D.; Pu, J.-X. Diterpenoids from Isodon species: An update. Nat. Prod. Rep. 2017, 34, 1090–1140. [Google Scholar] [CrossRef] [PubMed]
- Meade-Tollin, L.C.; Wijeratne, E.M.K.; Cooper, D.; Guild, M.; Jon, E.; Fritz, A.; Zhou, G.-X.; Whitesell, L.; Liang, J.-Y.; Gunatilaka, A.A.L. Ponicidin and Oridonin Are Responsible for the Antiangiogenic Activity of Rabdosia rubescens, a Constituent of the Herbal Supplement PC SPES. J. Nat. Prod. 2004, 67, 2–4. [Google Scholar] [CrossRef]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a Selective ALK Inhibitor Capable of Blocking the Resistant Gatekeeper Mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Sanchez-Moreno, C. Review: Methods Used to Evaluate the Free Radical Scavenging Activity in Foods and Biological Systems. Food Sci. Technol. Int. 2002, 8, 121–137. [Google Scholar] [CrossRef]
- Rampogu, S.; Baek, A.; Son, M.; Park, C.; Yoon, S.; Parate, S.; Lee, K.W. Discovery of Lonafarnib-Like Compounds: Pharmacophore Modeling and Molecular Dynamics Studies. ACS Omega 2020, 5, 1773–1781. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and Toxicity Prediction of Ceftazidime and Its Impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | R1 | R2 | Yield (%) |
|---|---|---|---|---|
| 1 | 4 | H | Phenylmethyl | 70 |
| 2 | 5 | H | (1S)-1-Phenylethyl | 88 |
| 3 | 6 | H | (1R)-1-Phenylethyl | 72 |
| 4 | 7 | H | (1S)-1-Phenyl-1-propyl | 72 |
| 5 | 8 | H | (1R)-1-Phenyl-1-propyl | 72 |
| 6 | 9 | H | (4-Fluorophenyl)methyl | 85 |
| 7 | 10 | H | (1R)-1-(4-Fluorophenyl)ethyl | 92 |
| 8 | 11 | H | (4-Methoxyphenyl)methyl | 60 |
| 9 | 12 | H | (1S)-1-Naphthalen-2-ylethyl | 85 |
| 10 | 13 | H | (1S)-1-Naphthalen-1-ylethyl | 93 |
| 11 | 14 | H | (1R)-1-Naphthalen-1-ylethyl | 86 |
| 12 | 15 | H | Naphthalen-1-ylmethyl | 75 |
| 13 | 16 | Methyl | Phenylmethyl | 92 |
| 14 | 17 | Benzyl | Phenylmethyl | 95 |
| 15 | 18 | Ethyl | Ethyl | 50 |
| 16 | 19 | H | i-Propyl | 80 |
| 17 | 20 | H | Propargyl | 86 |
| 18 | 21 | Piperidine | 80 | |
| 19 | 22 | N-Methylpiperazine | 55 | |
| 20 | 23 | Morpholine | 80 | |
| Entry | Compound | R | Yield (%) |
|---|---|---|---|
| 1 | 26 | Imidazole | 60 |
| 2 | 27 | Benzimidazole | 66 |
| 3 | 28 | 1,2,4-Triazole | 75 |
| 4 | 29 | 1,2,3-Triazole | 50 |
| 5 | 30 | Benzotriazole | 45 |
| Entry | Compound | R1 | R2 | Yield (%) |
|---|---|---|---|---|
| 1 | 35 | H | (1S)-1-Naphthalen-2-ylethyl | 86 |
| 2 | 36 | H | (1S)-1-Naphthalen-1-ylethyl | 60 |
| 3 | 37 | H | (1R)-1-Naphthalen-1-ylethyl | 87 |
| 4 | 38 | H | Naphthalen-1-ylmethyl | 78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khdar, Z.A.; Le, T.M.; Schelz, Z.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols. Int. J. Mol. Sci. 2022, 23, 10366. https://doi.org/10.3390/ijms231810366
Khdar ZA, Le TM, Schelz Z, Zupkó I, Szakonyi Z. Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols. International Journal of Molecular Sciences. 2022; 23(18):10366. https://doi.org/10.3390/ijms231810366
Chicago/Turabian StyleKhdar, Zein Alabdeen, Tam Minh Le, Zsuzsanna Schelz, István Zupkó, and Zsolt Szakonyi. 2022. "Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols" International Journal of Molecular Sciences 23, no. 18: 10366. https://doi.org/10.3390/ijms231810366
APA StyleKhdar, Z. A., Le, T. M., Schelz, Z., Zupkó, I., & Szakonyi, Z. (2022). Stereoselective Synthesis and Application of Gibberellic Acid-Derived Aminodiols. International Journal of Molecular Sciences, 23(18), 10366. https://doi.org/10.3390/ijms231810366