
Therapeutic Potential and Mechanisms of Novel Simple O-Substituted Isoflavones against Cerebral Ischemia Reperfusion



Abstract
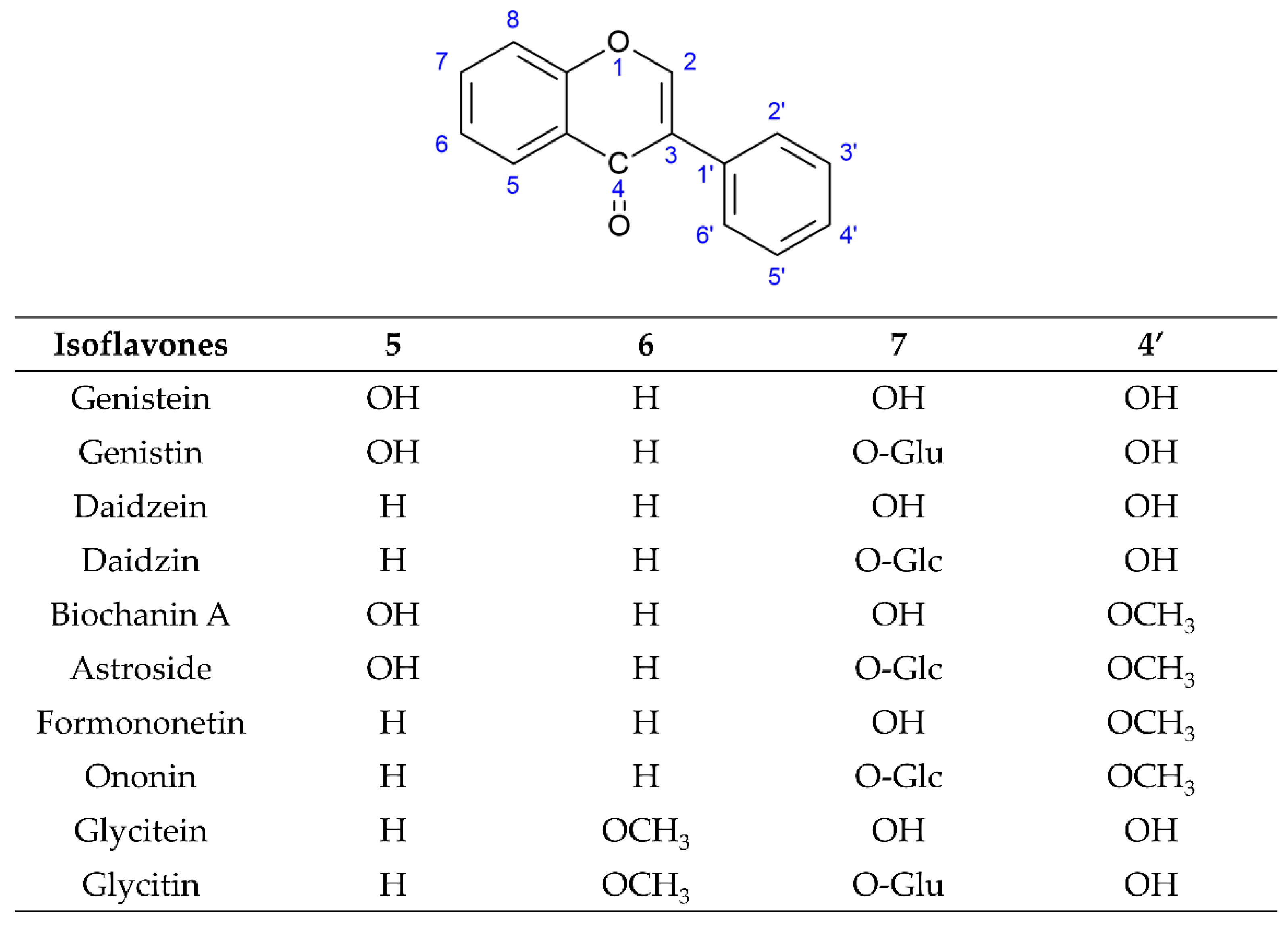
:1. Introduction
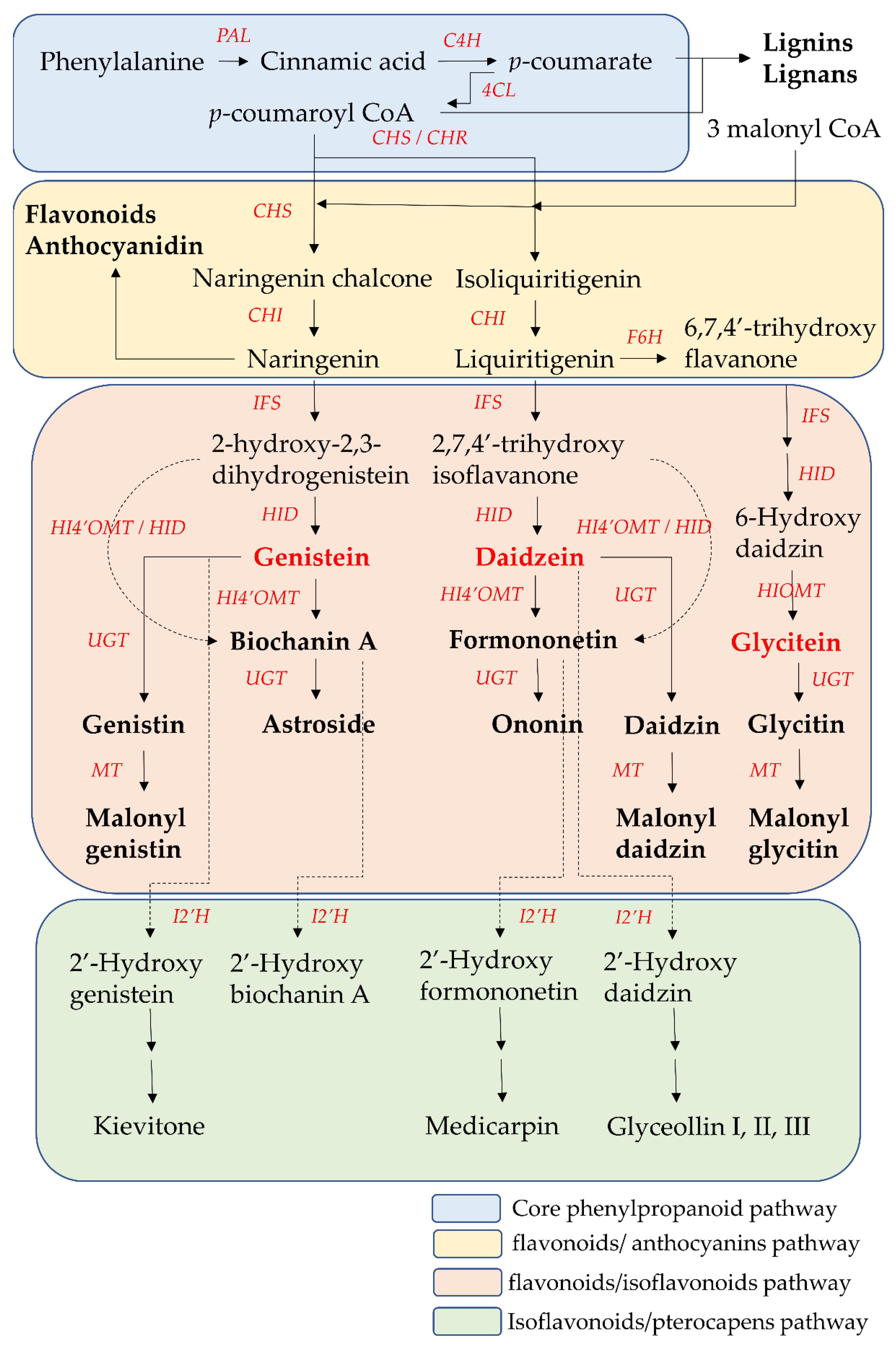
2. The Biosynthesis Pathway of Isoflavones
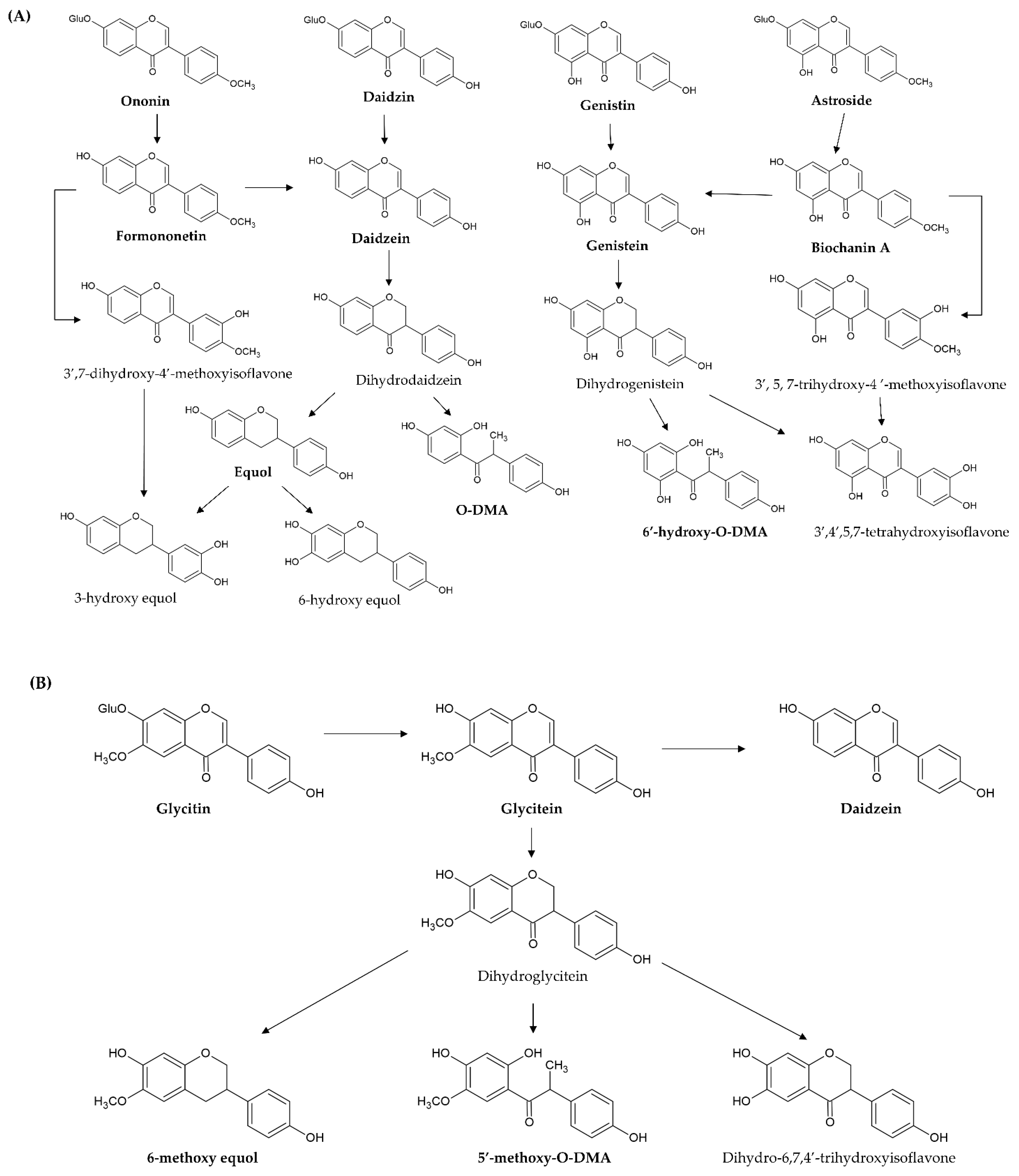
3. The Metabolism of Isoflavones
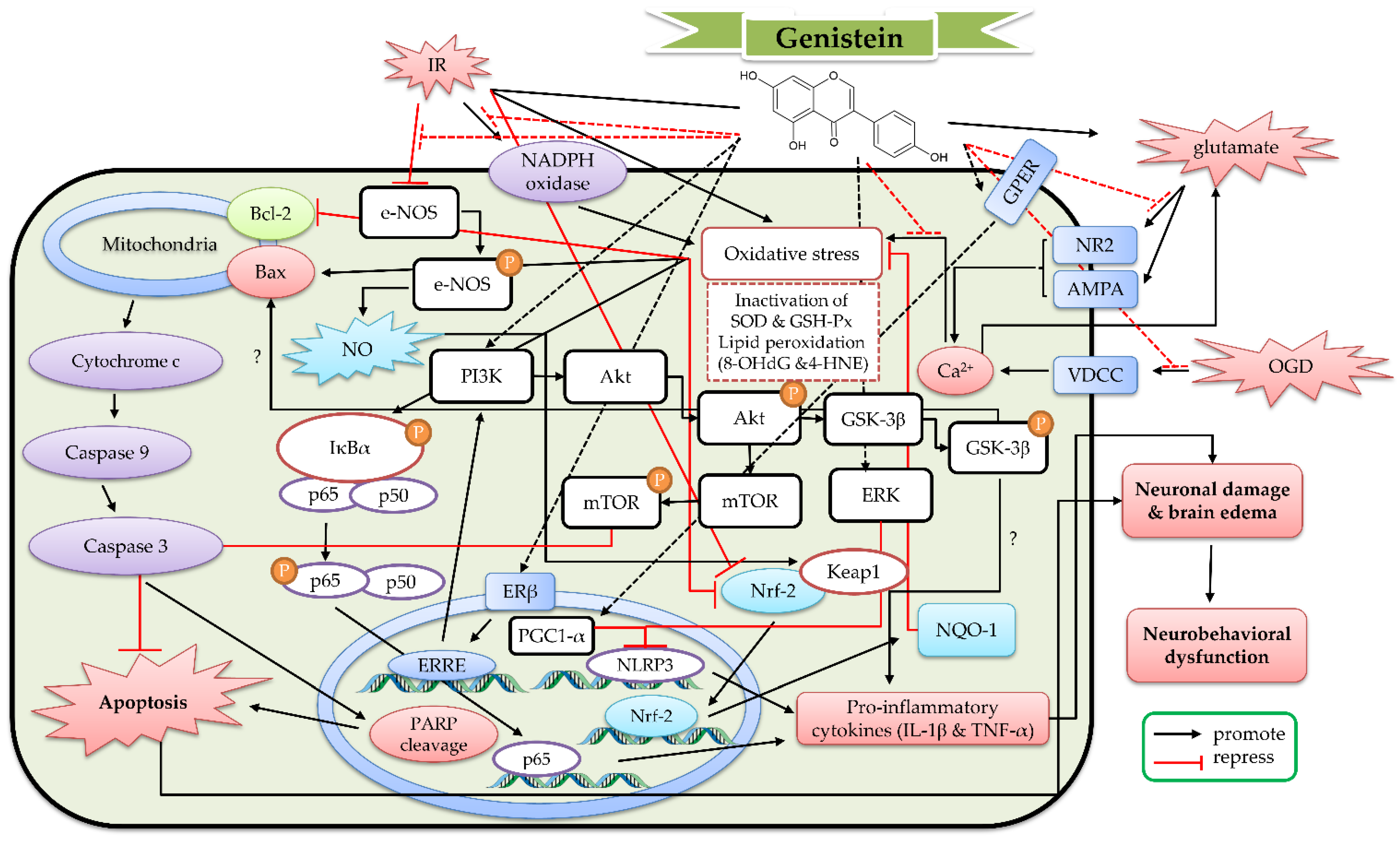
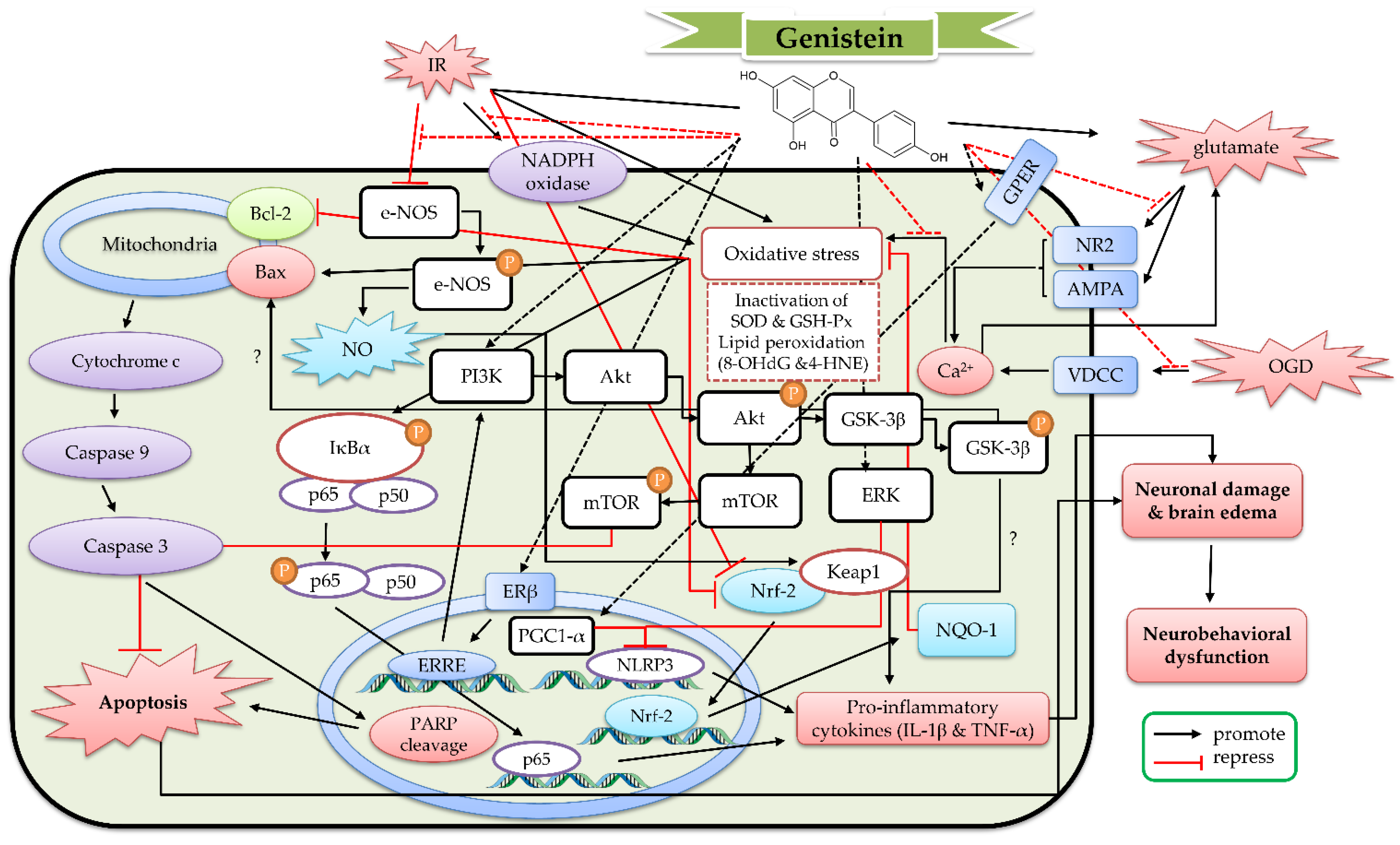
4. The Protective Mechanism of Genistein and Genistin against Cerebral IR
4.1. The Protective Effects of Genistein In Vitro Models
4.2. The Protective Effects of Geinistein In Vivo Cerebral IR Models
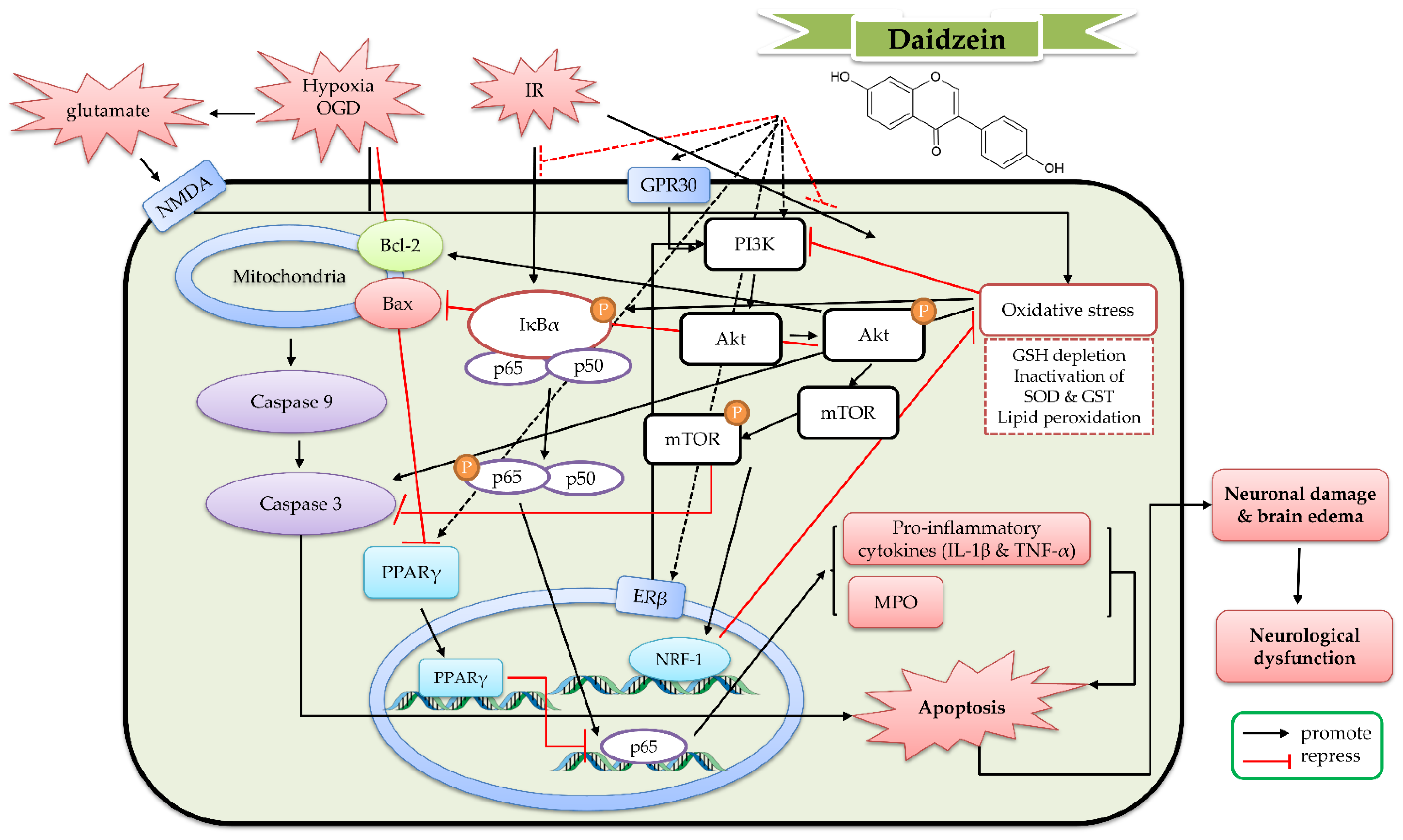
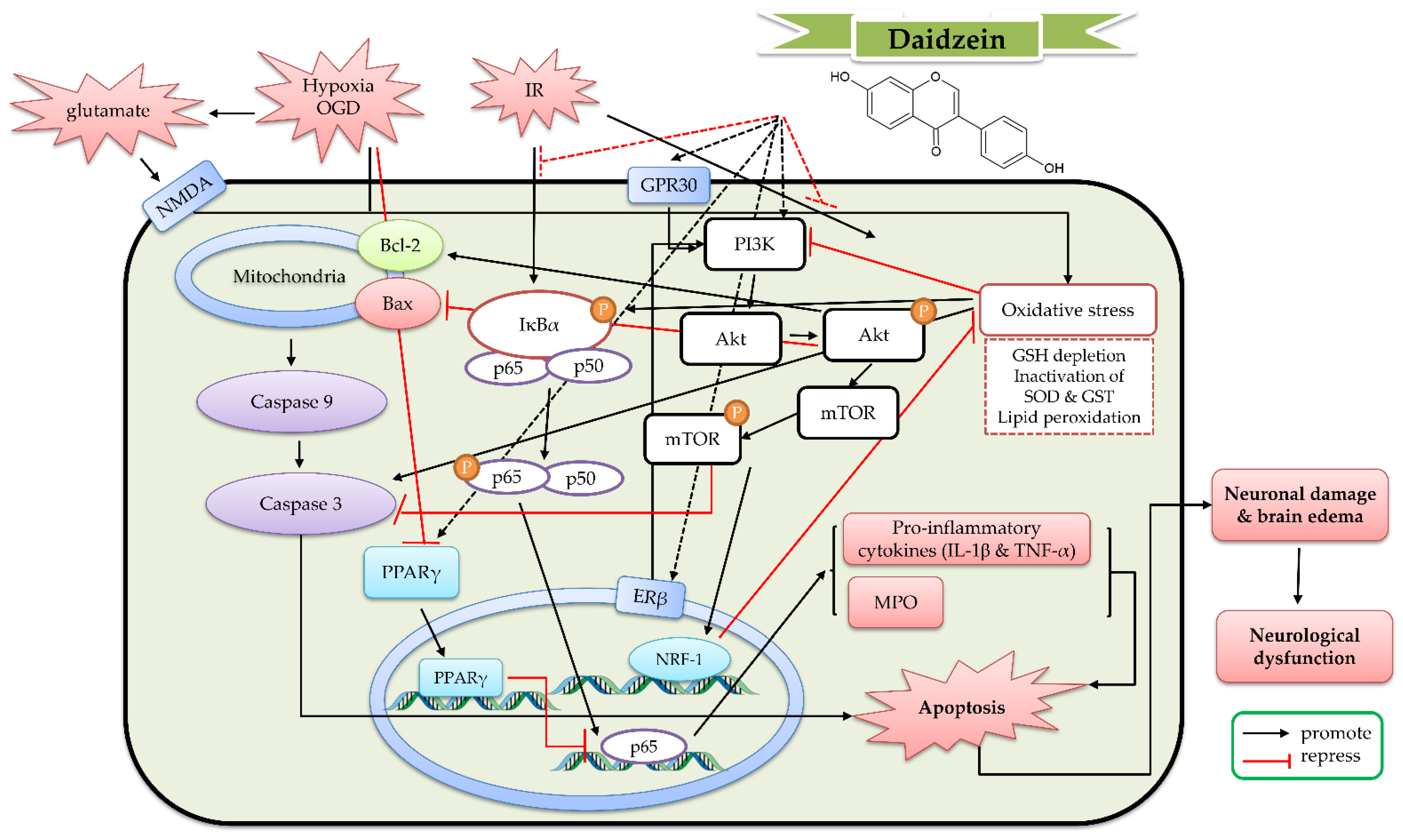
5. The Protective Mechanism of Daidzein and Daidzin against Cerebral IR
5.1. The Protective Effects of Daidzein In Vitro Models
5.2. The Protective Effects of Daidzein In Vivo Cerebral IR Models
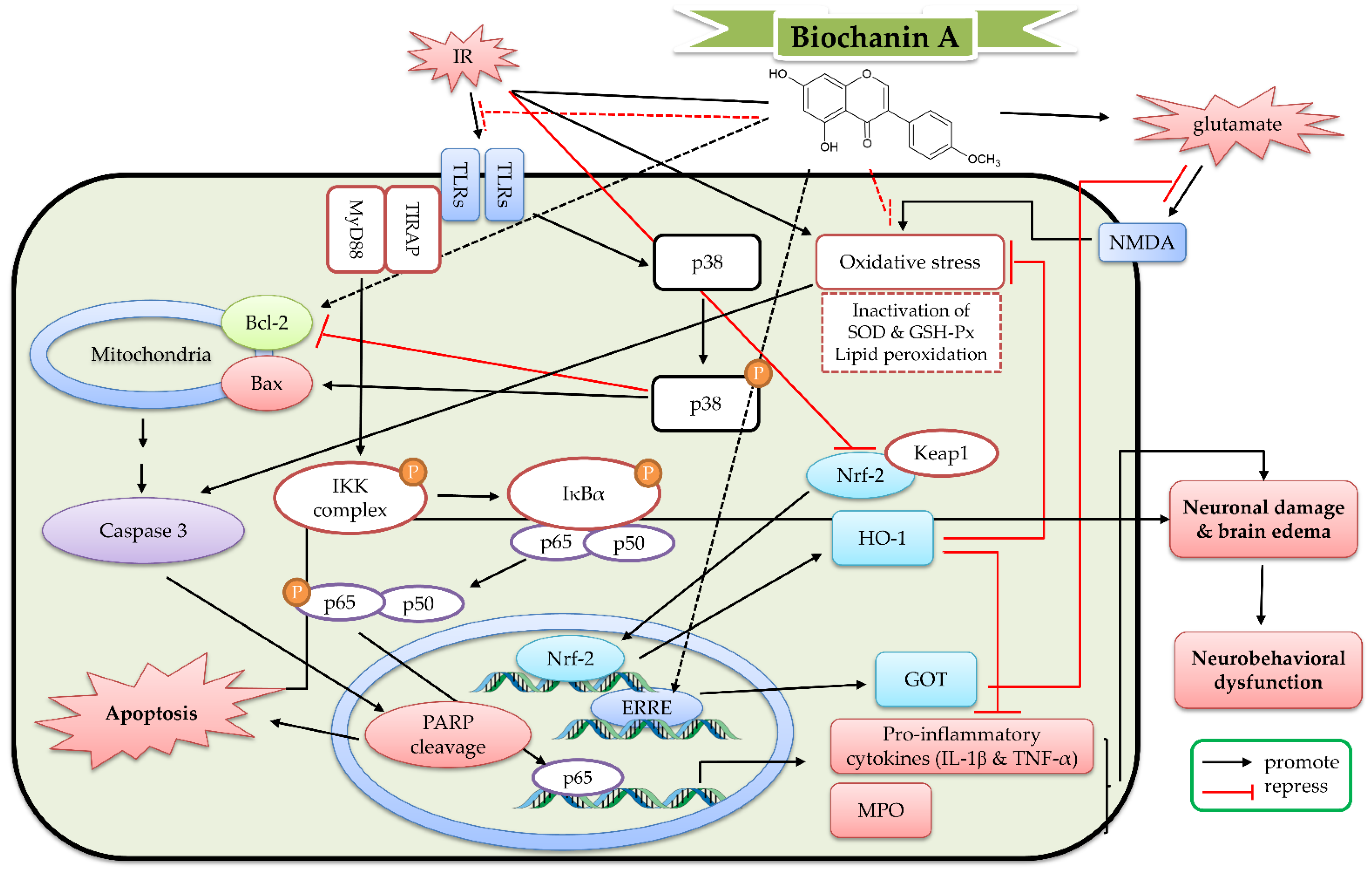
6. The Protective Mechanism of Biochanin A and Astroside against Cerebral IR
6.1. The Protective Effects of Biochanin A In Vitro Models
6.2. The Protective Effects of Biochanin A In Vivo Cerebral IR Models
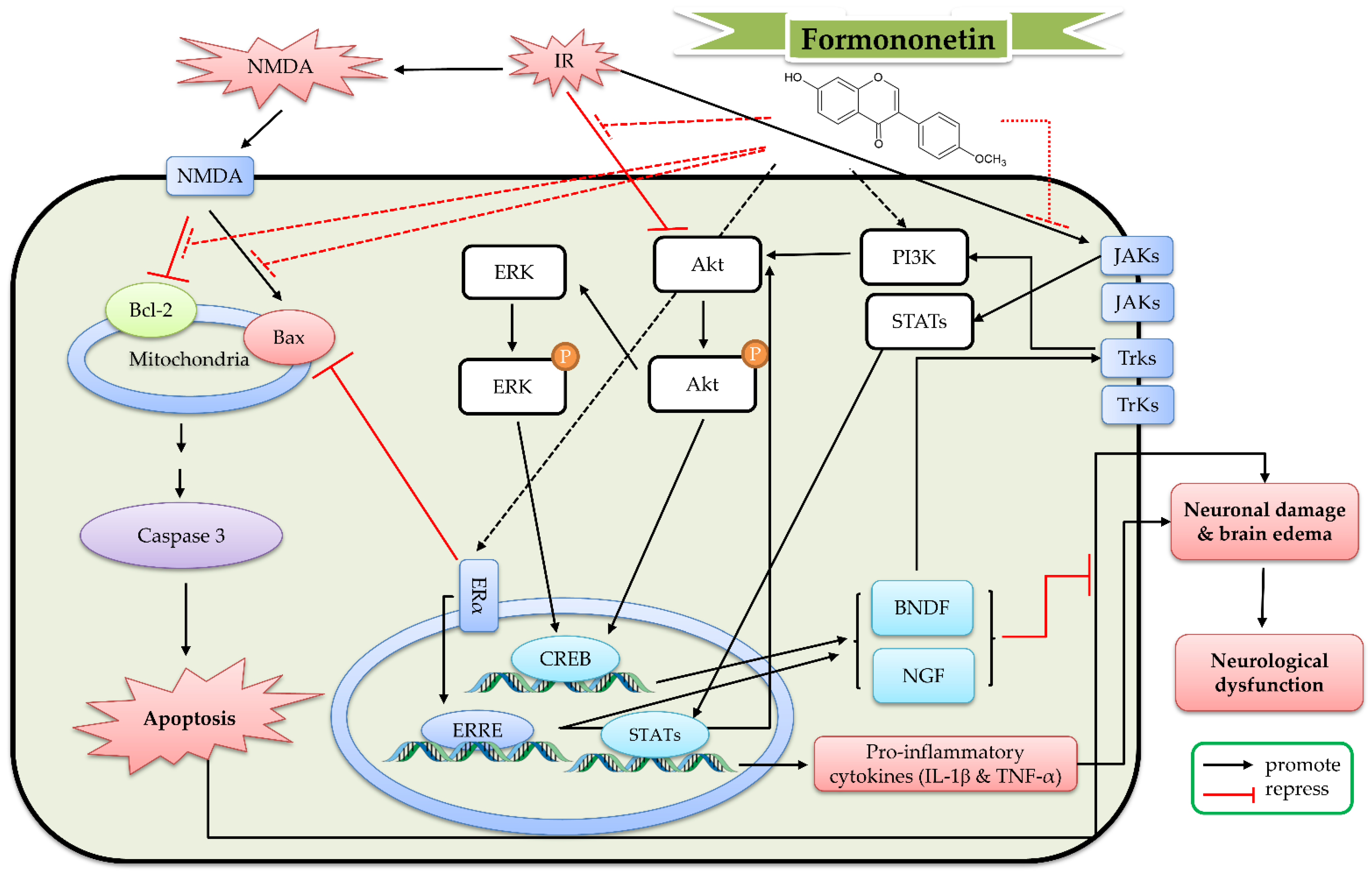
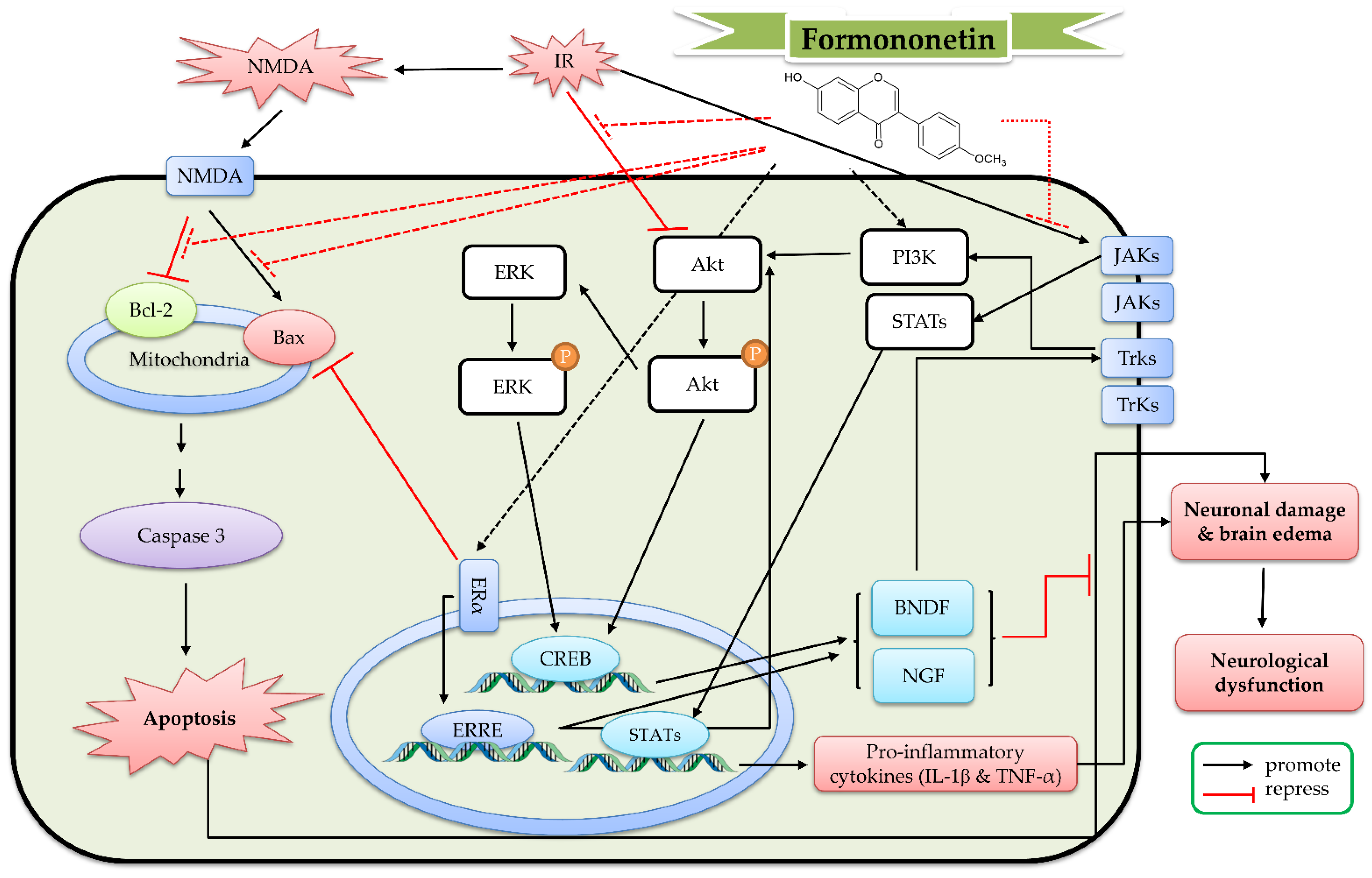
7. The Protective Mechanism of Formononetin and Ononin against Cerebral IR
8. The Protective Mechanism of Equol, O-DMA, and Other Metabolites against Cerebral IR
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- GBD 2016 Stroke Collaborators Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [CrossRef]
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Whitaker, E.E.; Cipolla, M.J. Perinatal stroke. Handb. Clin. Neurol. 2020, 171, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Martinez-Lara, E.; Siles, E.; Peinado, M.A. New Strategies for Stroke Therapy: Nanoencapsulated Neuroglobin. Pharmaceutics 2022, 14, 1737. [Google Scholar] [CrossRef]
- Ahnstedt, H.; Sweet, J.; Cruden, P.; Bishop, N.; Cipolla, M.J. Effects of Early Post-Ischemic Reperfusion and tPA on Cerebrovascular Function and Nitrosative Stress in Female Rats. Transl. Stroke Res. 2016, 7, 228–238. [Google Scholar] [CrossRef]
- Vongsfak, J.; Pratchayasakul, W.; Apaijai, N.; Vaniyapong, T.; Chattipakorn, N.; Chattipakorn, S.C. The Alterations in Mitochondrial Dynamics Following Cerebral Ischemia/Reperfusion Injury. Antioxidants 2021, 10, 1384. [Google Scholar] [CrossRef]
- Przykaza, L. Understanding the Connection Between Common Stroke Comorbidities, Their Associated Inflammation, and the Course of the Cerebral Ischemia/Reperfusion Cascade. Front. Immunol. 2021, 12, 782569. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Zhang, X.; Ye, Y.; Xiong, X.; Zhang, S.; Gu, L.; Jian, Z.; Wang, H. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cerebral Ischemia/Reperfusion Injury. Front. Cell. Neurosci. 2022, 16, 864426. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Ardelean, A.I. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines 2022, 10, 574. [Google Scholar] [CrossRef]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2021, 23, 14. [Google Scholar] [CrossRef]
- Hou, B.; Liu, R.; Wu, Y.; Huang, S. Astragaloside IV Reduces Cerebral Ischemia/Reperfusion-Induced Blood-Brain Barrier Permeability in Rats by Inhibiting ER Stress-Mediated Apoptosis. Evid.-Based Complement. Altern. Med. 2020, 2020, 9087873. [Google Scholar] [CrossRef]
- Venkat, P.; Chopp, M.; Chen, J. Blood-Brain Barrier Disruption, Vascular Impairment, and Ischemia/Reperfusion Damage in Diabetic Stroke. J. Am. Hear. Assoc. 2017, 6, e005819. [Google Scholar] [CrossRef]
- Kamada, H.; Yu, F.; Nito, C.; Chan, P.H. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: Relation to blood-brain barrier dysfunction. Stroke 2007, 38, 1044–1049. [Google Scholar] [CrossRef]
- Aronowski, J.; Strong, R.; Grotta, J.C. Reperfusion injury: Demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J. Cereb. Blood Flow Metab. 1997, 17, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Hou, P.P.; Shao, S.; Manaenko, A.; Xiao, Z.P.; Chen, Y.; Zhao, B.; Jia, F.; Zhang, X.H.; Mei, Q.Y.; et al. microRNA-455-5p alleviates neuroinflammation in cerebral ischemia/reperfusion injury. Neural Regen. Res. 2022, 17, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Zeng, Z.; Liu, Y.; Zeng, L.; Fang, J.; Ma, Y. Activation of Wnt/Beta-Catenin Signaling Pathway as a Promising Therapeutic Candidate for Cerebral Ischemia/Reperfusion Injury. Front. Pharmacol. 2022, 13, 914537. [Google Scholar] [CrossRef]
- Trotman-Lucas, M.; Gibson, C.L. A review of experimental models of focal cerebral ischemia focusing on the middle cerebral artery occlusion model. F1000Research 2021, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.L. Experimental models of focal and multifocal cerebral ischemia: A review. Rev. Neurosci. 2018, 29, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Xie, Q.; Li, Y.; Chen, Z.; Ren, M.; Chen, H.; Li, H.; Li, J.; Wang, J. Animal models of cerebral ischemia: A review. Biomed. Pharmacother. 2020, 131, 110686. [Google Scholar] [CrossRef]
- Yang, L.; Wen, K.S.; Ruan, X.; Zhao, Y.X.; Wei, F.; Wang, Q. Response of Plant Secondary Metabolites to Environmental Factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef]
- Wink, M. Modes of Action of Herbal Medicines and Plant Secondary Metabolites. Medicines 2015, 2, 251–286. [Google Scholar] [CrossRef]
- Nakayama, T.; Takahashi, S.; Waki, T. Formation of Flavonoid Metabolons: Functional Significance of Protein-Protein Interactions and Impact on Flavonoid Chemodiversity. Front. Plant Sci. 2019, 10, 821. [Google Scholar] [CrossRef]
- Veitch, N.C. Isoflavonoids of the leguminosae. Nat. Prod. Rep. 2013, 30, 988–1027. [Google Scholar] [CrossRef]
- Veitch, N.C. Isoflavonoids of the leguminosae. Nat. Prod. Rep. 2007, 24, 417–464. [Google Scholar] [CrossRef] [PubMed]
- Lapcik, O. Isoflavonoids in non-leguminous taxa: A rarity or a rule? Phytochemistry 2007, 68, 2909–2916. [Google Scholar] [CrossRef]
- Mackova, Z.; Koblovska, R.; Lapcik, O. Distribution of isoflavonoids in non-leguminous taxa—An update. Phytochemistry 2006, 67, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, J.; Guilet, D.; Terreux, R.; Lussignol, M.; Walchshofer, N. Isoflavonoids in non-leguminous families: An update. Nat. Prod. Rep. 2005, 22, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Mayo, B.; Vazquez, L.; Florez, A.B. Equol: A Bacterial Metabolite from The Daidzein Isoflavone and Its Presumed Beneficial Health Effects. Nutrients 2019, 11, 2231. [Google Scholar] [CrossRef] [PubMed]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Pilsakova, L.; Riecansky, I.; Jagla, F. The physiological actions of isoflavone phytoestrogens. Physiol. Res. 2010, 59, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Frankenfeld, C.L. O-desmethylangolensin: The importance of equol’s lesser known cousin to human health. Adv. Nutr. 2011, 2, 317–324. [Google Scholar] [CrossRef]
- Luo, M.; Yang, Z.Q.; Huang, J.C.; Wang, Y.S.; Guo, B.; Yue, Z.P. Genistein protects ovarian granulosa cells from oxidative stress via cAMP-PKA signaling. Cell Biol. Int. 2020, 44, 433–445. [Google Scholar] [CrossRef]
- Weng, L.; Zhang, F.; Wang, R.; Ma, W.; Song, Y. A review on protective role of genistein against oxidative stress in diabetes and related complications. Chem. Biol. Interact. 2019, 310, 108665. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T. Emerging Signal Regulating Potential of Genistein Against Alzheimer’s Disease: A Promising Molecule of Interest. Front. Cell Dev. Biol. 2019, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Sharma, A.K.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, P.; Puga-Olguin, A.; Rodriguez-Landa, J.F.; Zepeda, R.C. Genistein as Potential Therapeutic Candidate for Menopausal Symptoms and Other Related Diseases. Molecules 2019, 24, 3892. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, D.A.; Oppong-Gyebi, A. Genistein: Mechanisms of action for a pleiotropic neuroprotective agent in stroke. Nutr. Neurosci. 2019, 22, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Sanches Silva, A.; Sanchez-Machado, D.I.; Lopez-Cervantes, J.; Daglia, M.; Nabavi, S.F.; Nabavi, S.M. Hypotensive effects of genistein: From chemistry to medicine. Chem. Biol. Interact. 2017, 268, 37–46. [Google Scholar] [CrossRef]
- Mukund, V.; Mukund, D.; Sharma, V.; Mannarapu, M.; Alam, A. Genistein: Its role in metabolic diseases and cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 13–22. [Google Scholar] [CrossRef]
- Devi, K.P.; Shanmuganathan, B.; Manayi, A.; Nabavi, S.F.; Nabavi, S.M. Molecular and Therapeutic Targets of Genistein in Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 7028–7041. [Google Scholar] [CrossRef]
- de Oliveira, M.R. Evidence for genistein as a mitochondriotropic molecule. Mitochondrion 2016, 29, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Daglia, M.; Tundis, R.; Loizzo, M.R.; Sobarzo-Sanchez, E.; Orhan, I.E.; Nabavi, S.M. Genistein: A Boon for Mitigating Ischemic Stroke. Curr. Top. Med. Chem. 2015, 15, 1714–1721. [Google Scholar] [CrossRef]
- Islam, A.; Islam, M.S.; Uddin, M.N.; Hasan, M.M.I.; Akanda, M.R. The potential health benefits of the isoflavone glycoside genistin. Arch. Pharmacal Res. 2020, 43, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Javed, S.; Tariq, A.; Budzynska, B.; D’Onofrio, G.; Daglia, M.; Nabavi, S.F.; Nabavi, S.M. Daidzein and its Effects on Brain. Curr. Med. Chem. 2017, 24, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Ganai, A.A.; Farooqi, H. Bioactivity of genistein: A review of in vitro and in vivo studies. Biomed. Pharmacother. 2015, 76, 30–38. [Google Scholar] [CrossRef]
- Sarfraz, A.; Javeed, M.; Shah, M.A.; Hussain, G.; Shafiq, N.; Sarfraz, I.; Riaz, A.; Sadiqa, A.; Zara, R.; Zafar, S.; et al. Biochanin A: A novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020, 722, 137907. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, P.; Lou, L.; Wang, Y. Perspectives Regarding the Role of Biochanin A in Humans. Front. Pharmacol. 2019, 10, 793. [Google Scholar] [CrossRef]
- Sekikawa, A.; Ihara, M.; Lopez, O.; Kakuta, C.; Lopresti, B.; Higashiyama, A.; Aizenstein, H.; Chang, Y.F.; Mathis, C.; Miyamoto, Y.; et al. Effect of S-equol and Soy Isoflavones on Heart and Brain. Curr. Cardiol. Rev. 2019, 15, 114–135. [Google Scholar] [CrossRef]
- Ong, S.K.L.; Shanmugam, M.K.; Fan, L.; Fraser, S.E.; Arfuso, F.; Ahn, K.S.; Sethi, G.; Bishayee, A. Focus on Formononetin: Anticancer Potential and Molecular Targets. Cancers 2019, 11, 611. [Google Scholar] [CrossRef]
- Jiang, D.; Rasul, A.; Batool, R.; Sarfraz, I.; Hussain, G.; Mateen Tahir, M.; Qin, T.; Selamoglu, Z.; Ali, M.; Li, J.; et al. Potential Anticancer Properties and Mechanisms of Action of Formononetin. BioMed Res. Int. 2019, 2019, 5854315. [Google Scholar] [CrossRef]
- Wang, X. Structure, function, and engineering of enzymes in isoflavonoid biosynthesis. Funct. Integr. Genom. 2011, 11, 13–22. [Google Scholar] [CrossRef]
- Du, H.; Huang, Y.; Tang, Y. Genetic and metabolic engineering of isoflavonoid biosynthesis. Appl. Microbiol. Biotechnol. 2010, 86, 1293–1312. [Google Scholar] [CrossRef]
- Biala, W.; Jasinski, M. The Phenylpropanoid Case—It Is Transport That Matters. Front. Plant Sci. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Zabala, G.; Zou, J.; Tuteja, J.; Gonzalez, D.O.; Clough, S.J.; Vodkin, L.O. Transcriptome changes in the phenylpropanoid pathway of Glycine max in response to Pseudomonas syringae infection. BMC Plant Biol. 2006, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Anguraj Vadivel, A.K.; Renaud, J.; Kagale, S.; Dhaubhadel, S. GmMYB176 Regulates Multiple Steps in Isoflavonoid Biosynthesis in Soybean. Front. Plant Sci. 2019, 10, 562. [Google Scholar] [CrossRef]
- Goel, A.; Kumar, A.; Raghuvanshi, A. Synthesis, stereochemistry, structural classification, and chemical reactivity of natural pterocarpans. Chem. Rev. 2013, 113, 1614–1640. [Google Scholar] [CrossRef]
- Lundh, T. Metabolism of estrogenic isoflavones in domestic animals. Proc. Soc. Exp. Biol. Med. 1995, 208, 33–39. [Google Scholar] [CrossRef]
- Batterham, T.J.; Hart, N.K.; Lamberton, J.A. Metabolism of oestrogenic isoflavones in sheep. Nature 1965, 206, 509. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.R.; Haak, S.J.; Bohn, T.; Tian, Q.; Schwartz, S.J.; Failla, M.L. Isoflavonoid glucosides are deconjugated and absorbed in the small intestine of human subjects with ileostomies. Am. J. Clin. Nutr. 2007, 85, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Zubik, L.; Meydani, M. Bioavailability of soybean isoflavones from aglycone and glucoside forms in American women. Am. J. Clin. Nutr. 2003, 77, 1459–1465. [Google Scholar] [CrossRef]
- Gu, L.; House, S.E.; Prior, R.L.; Fang, N.; Ronis, M.J.; Clarkson, T.B.; Wilson, M.E.; Badger, T.M. Metabolic phenotype of isoflavones differ among female rats, pigs, monkeys, and women. J. Nutr. 2006, 136, 1215–1221. [Google Scholar] [CrossRef]
- Setchell, K.D.; Brown, N.M.; Desai, P.; Zimmer-Nechemias, L.; Wolfe, B.E.; Brashear, W.T.; Kirschner, A.S.; Cassidy, A.; Heubi, J.E. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J. Nutr. 2001, 131, 1362S–1375S. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Kulkarni, K.; Zhu, W.; Hu, M. Bioavailability and pharmacokinetics of genistein: Mechanistic studies on its ADME. Anti-Cancer Agents Med. Chem. 2012, 12, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Sfakianos, J.; Coward, L.; Kirk, M.; Barnes, S. Intestinal uptake and biliary excretion of the isoflavone genistein in rats. J. Nutr. 1997, 127, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.B.; de Avila, A.R.A.; de Queiros, L.D.; Macedo, J.A.; Macedo, G.A. Bioconversion of Isoflavones into Bioactive Equol: State of the Art. Recent Pat. Food Nutr. Agric. 2016, 8, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Gaya, P.; Medina, M.; Sanchez-Jimenez, A.; Landete, J.M. Phytoestrogen Metabolism by Adult Human Gut Microbiota. Molecules 2016, 21, 1034. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, X.; Zhang, Y.; Ma, Y.; Li, L.; Li, D.; Zhang, L.; Zhang, Z. Comprehensive Study of the in Vivo and in Vitro Metabolism of Dietary Isoflavone Biochanin A Based on UHPLC-Q-TOF-MS/MS. J. Agric. Food Chem. 2019, 67, 12481–12495. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.Y.; Fan, M.X.; Zhao, H.Y.; Li, M.X.; Wu, X.; Gao, W.Y. Pharmacokinetics and Bioavailability of the Isoflavones Formononetin and Ononin and Their in Vitro Absorption in Ussing Chamber and Caco-2 Cell Models. J. Agric. Food Chem. 2018, 66, 2917–2924. [Google Scholar] [CrossRef]
- Tolleson, W.H.; Doerge, D.R.; Churchwell, M.I.; Marques, M.M.; Roberts, D.W. Metabolism of biochanin A and formononetin by human liver microsomes in vitro. J. Agric. Food Chem. 2002, 50, 4783–4790. [Google Scholar] [CrossRef]
- Simons, A.L.; Renouf, M.; Hendrich, S.; Murphy, P.A. Metabolism of glycitein (7,4′-dihydroxy-6-methoxy-isoflavone) by human gut microflora. J. Agric. Food Chem. 2005, 53, 8519–8525. [Google Scholar] [CrossRef]
- Pereira, D.B.; Carvalho, A.P.; Duarte, C.B. Genistein inhibits Ca2+ influx and glutamate release from hippocampal synaptosomes: Putative non-specific effects. Neurochem. Int. 2003, 42, 179–188. [Google Scholar] [CrossRef]
- Kajta, M.; Domin, H.; Grynkiewicz, G.; Lason, W. Genistein inhibits glutamate-induced apoptotic processes in primary neuronal cell cultures: An involvement of aryl hydrocarbon receptor and estrogen receptor/glycogen synthase kinase-3beta intracellular signaling pathway. Neuroscience 2007, 145, 592–604. [Google Scholar] [CrossRef]
- Qian, Y.; Cao, L.; Guan, T.; Chen, L.; Xin, H.; Li, Y.; Zheng, R.; Yu, D. Protection by genistein on cortical neurons against oxidative stress injury via inhibition of NF-kappaB, JNK and ERK signaling pathway. Pharm. Biol. 2015, 53, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Linford, N.J.; Dorsa, D.M. 17beta-Estradiol and the phytoestrogen genistein attenuate neuronal apoptosis induced by the endoplasmic reticulum calcium-ATPase inhibitor thapsigargin. Steroids 2002, 67, 1029–1040. [Google Scholar] [CrossRef]
- Ma, X.L.; Zhang, F.; Wang, Y.X.; He, C.C.; Tian, K.; Wang, H.G.; An, D.; Heng, B.; Liu, Y.Q. Genistein inhibition of OGD-induced brain neuron death correlates with its modulation of apoptosis, voltage-gated potassium and sodium currents and glutamate signal pathway. Chem. Biol. Interact. 2016, 254, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Tian, K.; He, C.C.; Ma, X.L.; Zhang, F.; Wang, H.G.; An, D.; Heng, B.; Jiang, Y.G.; Liu, Y.Q. Genistein inhibits hypoxia, ischemic-induced death, and apoptosis in PC12 cells. Environ. Toxicol. Pharmacol. 2017, 50, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.; Perez-Basterrechea, M.; Garrido, P.; Diaz, E.; Alonso, A.; Otero, J.; Colado, E.; Gonzalez, C. Effects of Estrogen and Phytoestrogen Treatment on an In Vitro Model of Recurrent Stroke on HT22 Neuronal Cell Line. Cell. Mol. Neurobiol. 2017, 37, 405–416. [Google Scholar] [CrossRef]
- Huang, R.; Singh, M.; Dillon, G.H. Genistein directly inhibits native and recombinant NMDA receptors. Neuropharmacology 2010, 58, 1246–1251. [Google Scholar] [CrossRef]
- Trieu, V.N.; Dong, Y.; Zheng, Y.; Uckun, F.M. In vivo antioxidant activity of genistein in a murine model of singlet oxygen-induced cerebral stroke. Radiat. Res. 1999, 152, 508–516. [Google Scholar] [CrossRef]
- Rumman, M.; Pandey, S.; Singh, B.; Gupta, M.; Ubaid, S.; Mahdi, A.A. Genistein Prevents Hypoxia-Induced Cognitive Dysfunctions by Ameliorating Oxidative Stress and Inflammation in the Hippocampus. Neurotox. Res. 2021, 39, 1123–1133. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.J.; Chen, R.J.; Chen, L.; Chen, S.; Yang, X.F.; Min, J.W. Genistein mitigates oxidative stress and inflammation by regulating Nrf2/HO-1 and NF-kappaB signaling pathways in hypoxic-ischemic brain damage in neonatal mice. Ann. Transl. Med. 2022, 10, 32. [Google Scholar] [CrossRef]
- Li, H.C.; Zhang, G.Y. Inhibitory effect of genistein on activation of STAT3 induced by brain ischemia/reperfusion in rat hippocampus. Acta Pharmacol. Sin. 2003, 24, 1131–1136. [Google Scholar]
- Liang, H.W.; Qiu, S.F.; Shen, J.; Sun, L.N.; Wang, J.Y.; Bruce, I.C.; Xia, Q. Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci. Lett. 2008, 438, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tu, J.; Zhang, Q.; Zhang, X.; Zhu, Y.; Ma, W.; Cheng, C.; Brann, D.W.; Yang, F. Genistein attenuates ischemic oxidative damage and behavioral deficits via eNOS/Nrf2/HO-1 signaling. Hippocampus 2013, 23, 634–647. [Google Scholar] [CrossRef]
- Donzelli, A.; Braida, D.; Finardi, A.; Capurro, V.; Valsecchi, A.E.; Colleoni, M.; Sala, M. Neuroprotective effects of genistein in Mongolian gerbils: Estrogen receptor-beta involvement. J. Pharmacol. Sci. 2010, 114, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Rajput, M.S.; Sarkar, P.D.; Nirmal, N.P. Inhibition of DPP-4 Activity and Neuronal Atrophy with Genistein Attenuates Neurological Deficits Induced by Transient Global Cerebral Ischemia and Reperfusion in Streptozotocin-Induced Diabetic Mice. Inflammation 2017, 40, 623–635. [Google Scholar] [CrossRef]
- Qian, Y.; Guan, T.; Huang, M.; Cao, L.; Li, Y.; Cheng, H.; Jin, H.; Yu, D. Neuroprotection by the soy isoflavone, genistein, via inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen induced-NF-kappaB activation in a cerebral ischemia mouse model. Neurochem. Int. 2012, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sullivan, J.C.; Schreihofer, D.A. Dietary genistein and equol (4′, 7 isoflavandiol) reduce oxidative stress and protect rats against focal cerebral ischemia. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R871–R877. [Google Scholar] [CrossRef]
- Aras, A.B.; Guven, M.; Akman, T.; Alacam, H.; Kalkan, Y.; Silan, C.; Cosar, M. Genistein exerts neuroprotective effect on focal cerebral ischemia injury in rats. Inflammation 2015, 38, 1311–1321. [Google Scholar] [CrossRef]
- Cortina, B.; Torregrosa, G.; Castello-Ruiz, M.; Burguete, M.C.; Moscardo, A.; Latorre, A.; Salom, J.B.; Valles, J.; Santos, M.T.; Alborch, E. Improvement of the circulatory function partially accounts for the neuroprotective action of the phytoestrogen genistein in experimental ischemic stroke. Eur. J. Pharmacol. 2013, 708, 88–94. [Google Scholar] [CrossRef]
- Wang, S.; Wei, H.; Cai, M.; Lu, Y.; Hou, W.; Yang, Q.; Dong, H.; Xiong, L. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of ERK activity in ovariectomized mice. Int. J. Biol. Sci. 2014, 10, 457–465. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Wei, H.; Gu, T.; Wang, J.; Wu, Z.; Yang, Q. Genistein Attenuates Acute Cerebral Ischemic Damage by Inhibiting the NLRP3 Inflammasome in Reproductively Senescent Mice. Front. Aging Neurosci. 2020, 12, 153. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Wang, J.; Ma, L.; Zhao, J.; Wang, J.; Fang, Z.; Hou, W.; Guo, H. Neuronal GPER Participates in Genistein-Mediated Neuroprotection in Ischemic Stroke by Inhibiting NLRP3 Inflammasome Activation in Ovariectomized Female Mice. Mol. Neurobiol. 2022, 59, 5024–5040. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.Y.; Xia, X.; Che, L.; Song, Y.T. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 2018, 40, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Y.; Liu, Y.; Gong, Y.F.; Zheng, X.Y. A preliminary report: Genistein attenuates cerebral ischemia injury in ovariectomized rats via regulation of the PI3K-Akt-mTOR pathway. Gen. Physiol. Biophys. 2019, 38, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, S.; Qi, X.; Chen, S.; Chen, P.; Zhang, Q. Lose dose genistein inhibits glucocorticoid receptor and ischemic brain injury in female rats. Neurochem. Int. 2014, 65, 14–22. [Google Scholar] [CrossRef]
- Schreihofer, D.A.; Redmond, L. Soy phytoestrogens are neuroprotective against stroke-like injury in vitro. Neuroscience 2009, 158, 602–609. [Google Scholar] [CrossRef]
- Hurtado, O.; Ballesteros, I.; Cuartero, M.I.; Moraga, A.; Pradillo, J.M.; Ramirez-Franco, J.; Bartolome-Martin, D.; Pascual, D.; Torres, M.; Sanchez-Prieto, J.; et al. Daidzein has neuroprotective effects through ligand-binding-independent PPARgamma activation. Neurochem. Int. 2012, 61, 119–127. [Google Scholar] [CrossRef]
- Kajta, M.; Rzemieniec, J.; Litwa, E.; Lason, W.; Lenartowicz, M.; Krzeptowski, W.; Wojtowicz, A.K. The key involvement of estrogen receptor beta and G-protein-coupled receptor 30 in the neuroprotective action of daidzein. Neuroscience 2013, 238, 345–360. [Google Scholar] [CrossRef]
- Subedi, L.; Ji, E.; Shin, D.; Jin, J.; Yeo, J.H.; Kim, S.Y. Equol, a Dietary Daidzein Gut Metabolite Attenuates Microglial Activation and Potentiates Neuroprotection In Vitro. Nutrients 2017, 9, 207. [Google Scholar] [CrossRef]
- Torregrosa, G.; Burguete, M.C.; Perez-Asensio, F.J.; Salom, J.B.; Gil, J.V.; Alborch, E. Pharmacological profile of phytoestrogens in cerebral vessels: In vitro study with rabbit basilar artery. Eur. J. Pharmacol. 2003, 482, 227–234. [Google Scholar] [CrossRef]
- Wang, P.; Jeng, C.J.; Chien, C.L.; Wang, S.M. Signaling mechanisms of daidzein-induced axonal outgrowth in hippocampal neurons. Biochem. Biophys. Res. Commun. 2008, 366, 393–400. [Google Scholar] [CrossRef]
- Pan, M.; Han, H.; Zhong, C.; Geng, Q. Effects of genistein and daidzein on hippocampus neuronal cell proliferation and BDNF expression in H19-7 neural cell line. J. Nutr. Health Aging 2012, 16, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Aras, A.B.; Guven, M.; Akman, T.; Ozkan, A.; Sen, H.M.; Duz, U.; Kalkan, Y.; Silan, C.; Cosar, M. Neuroprotective effects of daidzein on focal cerebral ischemia injury in rats. Neural Regen. Res. 2015, 10, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zhou, M.; Chen, M.; Lu, Y.; Shi, D.; Wang, J.; Liu, C. Neuroprotective Effect of Daidzein Extracted From Pueraria lobate Radix in a Stroke Model Via the Akt/mTOR/BDNF Channel. Front. Pharmacol. 2021, 12, 772485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ru, N.; Shang, Z.H.; Chen, J.F.; Yan, C.; Li, Y.; Liang, J. Daidzein ameliorates spinal cord ischemia/reperfusion injury-induced neurological function deficits in Sprague-Dawley rats through PI3K/Akt signaling pathway. Exp. Ther. Med. 2017, 14, 4878–4886. [Google Scholar] [CrossRef]
- Sobey, C.G.; Weiler, J.M.; Boujaoude, M.; Woodman, O.L. Effect of short-term phytoestrogen treatment in male rats on nitric oxide-mediated responses of carotid and cerebral arteries: Comparison with 17beta-estradiol. J. Pharmacol. Exp. Ther. 2004, 310, 135–140. [Google Scholar] [CrossRef]
- Rodrigues, F.; Almeida, I.; Sarmento, B.; Amaral, M.H.; Oliveira, M.B.P.P. Study of the isoflavone content of different extracts of Medicago spp. as potential active ingredient. Ind. Crop. Prod. 2014, 57, 110–115. [Google Scholar] [CrossRef]
- Tan, J.W.; Tham, C.L.; Israf, D.A.; Lee, S.H.; Kim, M.K. Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem. Res. 2013, 38, 512–518. [Google Scholar] [CrossRef]
- Khanna, S.; Stewart, R.; Gnyawali, S.; Harris, H.; Balch, M.; Spieldenner, J.; Sen, C.K.; Rink, C. Phytoestrogen isoflavone intervention to engage the neuroprotective effect of glutamate oxaloacetate transaminase against stroke. FASEB J. 2017, 31, 4533–4544. [Google Scholar] [CrossRef]
- Berkoz, M.; Krosniak, M.; Ozkan-Yilmaz, F.; Ozluer-Hunt, A. Prophylactic effect of Biochanin A in lipopolysaccharide-stimulated BV2 microglial cells. Immunopharmacol. Immunotoxicol. 2020, 42, 330–339. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.A. Biochanin A inhibits lipopolysaccharide-induced inflammatory cytokines and mediators production in BV2 microglia. Neurochem. Res. 2015, 40, 165–171. [Google Scholar] [CrossRef]
- Wu, W.Y.; Wu, Y.Y.; Huang, H.; He, C.; Li, W.Z.; Wang, H.L.; Chen, H.Q.; Yin, Y.Y. Biochanin A attenuates LPS-induced pro-inflammatory responses and inhibits the activation of the MAPK pathway in BV2 microglial cells. Int. J. Mol. Med. 2015, 35, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, X.; Ding, M.; Zhai, B.; Xiao, L.; Piao, T.; Liu, M. Biochanin A inhibits lipopolysaccharide-induced inflammation in human umbilical vein endothelial cells. Life Sci. 2015, 136, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Lu, H.; Qin, J.; Qu, S.; Wang, W.; Guo, Y.; Liao, W.; Song, M.; Chen, J.; Wang, Y. Biochanin A Provides Neuroprotection Against Cerebral Ischemia/Reperfusion Injury by Nrf2-Mediated Inhibition of Oxidative Stress and Inflammation Signaling Pathway in Rats. Med Sci. Monit. 2019, 25, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, L.; Li, Y.; Wang, Y. Biochanin A protects against focal cerebral ischemia/reperfusion in rats via inhibition of p38-mediated inflammatory responses. J. Neurol. Sci. 2015, 348, 121–125. [Google Scholar] [CrossRef]
- Guo, M.M.; Qu, S.B.; Lu, H.L.; Wang, W.B.; He, M.L.; Su, J.L.; Chen, J.; Wang, Y. Biochanin A Alleviates Cerebral Ischemia/Reperfusion Injury by Suppressing Endoplasmic Reticulum Stress-Induced Apoptosis and p38MAPK Signaling Pathway In Vivo and In Vitro. Front. Endocrinol. 2021, 12, 646720. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Y.; Ye, Z.N.; Zhuang, Z.; Gao, Y.; Tang, C.; Zhou, C.H.; Wang, C.X.; Zhang, X.S.; Xie, G.B.; Liu, J.P.; et al. Biochanin A Reduces Inflammatory Injury and Neuronal Apoptosis following Subarachnoid Hemorrhage via Suppression of the TLRs/TIRAP/MyD88/NF-kappaB Pathway. Behav. Neurol. 2018, 2018, 1960106. [Google Scholar] [CrossRef]
- Tay, K.C.; Tan, L.T.; Chan, C.K.; Hong, S.L.; Chan, K.G.; Yap, W.H.; Pusparajah, P.; Lee, L.H.; Goh, B.H. Formononetin: A Review of Its Anticancer Potentials and Mechanisms. Front. Pharmacol. 2019, 10, 820. [Google Scholar] [CrossRef]
- Luo, L.; Zhou, J.; Zhao, H.; Fan, M.; Gao, W. The anti-inflammatory effects of formononetin and ononin on lipopolysaccharide-induced zebrafish models based on lipidomics and targeted transcriptomics. Metabolomics 2019, 15, 153. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, S.B.; Wang, Y.C.; Li, X.Q.; Zheng, L.H.; Zhao, M.G. Neuroprotective effects of formononetin against NMDA-induced apoptosis in cortical neurons. Phytotherapy Res. 2013, 27, 1770–1775. [Google Scholar] [CrossRef]
- Wu, Q.L.; Cheng, Y.Q.; Liu, A.J.; Zhang, W.D. Formononetin recovered injured nerve functions by enhancing synaptic plasticity in ischemic stroke rats. Biochem. Biophys. Res. Commun. 2020, 525, 67–72. [Google Scholar] [CrossRef]
- Liang, K.; Ye, Y.; Wang, Y.; Zhang, J.; Li, C. Formononetin mediates neuroprotection against cerebral ischemia/reperfusion in rats via downregulation of the Bax/Bcl-2 ratio and upregulation PI3K/Akt signaling pathway. J. Neurol. Sci. 2014, 344, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, Y.; Chen, Q.; He, Y.; Zhou, H.; Wan, H.; Yang, J. Formononetin protects against inflammation associated with cerebral ischemia-reperfusion injury in rats by targeting the JAK2/STAT3 signaling pathway. Biomed. Pharmacother. 2022, 149, 112836. [Google Scholar] [CrossRef] [PubMed]
- Kostelac, D.; Rechkemmer, G.; Briviba, K. Phytoestrogens modulate binding response of estrogen receptors alpha and beta to the estrogen response element. J. Agric. Food Chem. 2003, 51, 7632–7635. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Deng, X.; Ma, Z.; Wang, Y. Equol protects PC12 neuronal cells against hypoxia/reoxygenation injury in vitro by reducing reactive oxygen species production. J. South. Med. Univ. 2016, 36, 1–7. [Google Scholar]
- Yu, W.; Wang, Y.; Zhou, D.X.; Zhao, L.M.; Li, G.R.; Deng, X.L. Equol is neuroprotective during focal cerebral ischemia and reperfusion that involves p-Src and gp91(phox). Curr. Neurovascular Res. 2014, 11, 367–377. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| |||||||
|---|---|---|---|---|---|---|---|
| O-Substituted Isoflavones | Applied Concentration | Application | Employed Cells | Employed Model | Observed Effects | Possible Mechanism | References |
| Genistein | 0.1–1 μM | Pre-treatment for 24 h | primary cortical cells | Glutamate (300 μM) for 10 min Thapsigargin (50 nM) for 48 h Hypoxia for 16 h OGD for 2 or 5 h | ↓ apoptosis | ↑ ER, PI3K and MAPK | [105] |
| 10–10,000 nM | Co-treatment | Primary hippocampal, neocortical, and cerebellar cell | Glutamate (1 mM) for 3, 6, 24 h | ↓ apoptosis | ↓ aryl hydrocarbon receptor and ER/GSK3β | [80] | |
| 10–500 nM | Co-treatment | primary cortical cells | Thapsigargin (50 nM) for 48 h | ↓ apoptosis | ERβ | [82] | |
| 0.01–1 μM | Pre-treatment for 24 h | primary cortical cells | H2O2 (500 μM) for 1 h | ↓ apoptosis and neurotoxicity | ↓ MAPK/ NF-κB pathway | [81] | |
| 1 mM | Co-treament | Primary neurons | OGD for 3, 6, 24, 48 h | ↓ apoptosis and neurotoxicity | ↑ Kv currents ↓ Nav currents and glutamate pathway | [83] | |
| 30 μM | Co-treament | PC12 cells | OGD for 1, 3, 6, 24 h | ↓ apoptosis and neurotoxicity | ↑ Kv currents ↓ glutamate pathway | [84] | |
| 1 μM | Pretreatment for 1 h | HT22 cells | OGD/R for 18 h | ↓ apoptosis and neurotoxicity | - | [85] | |
| Daidzein | 0.1–1 μM | Pre-treatment for 24 h | primary cortical cells | Glutamate (300 μM) for 10 min Thapsigargin (50 nM) for 48 h Hypoxia for 16 h OGD for 2 or 5 h | ↓ apoptosis | ↑ ER, PI3K | [105] |
| 0.05–5 μM | Pre-treatment for 24 h | primary cortical cells | OGD for 2 or 5 h | ↓ apoptosis and neurotoxicity | ↑ PPARγ | [106] | |
| 1–10 μM | Co-treatment | primary neocortical, cerebellar, and hippocampal cell | Glutamate (1 mM) for 6 or 24 h | ↓ apoptosis and neurotoxicity | ↓ ERβ, GPR30 and caspase-3 | [107] | |
| Biochanin A | 1–100 μM | Pre-treatment for 2 h | PC12 cells | Glutamate (10 mM) for 6 or 24 h | ↓ apoptosis | ↓ caspase-3 | [117] |
| 25–50 μM | Pre-treatment for 24 h | HT4 neural cells | Glutamate (5 mM) for 24 h | ↓ neurotoxicity ↓ inflammation | ↑ GOT ↓ TLR4/MyD88/NF-κB, MAPK, and Akt | [118] | |
| pirmary cortical cells | |||||||
| 2.5–100 μM | Co-treatment | BV2 cells | LPS (100 ng/mL) for 24 h | ↓ inflammation | ↑ PPARγ ↓ NF-κB | [119] | |
| 5–20 μM | Pre-treatment for 1 h | BV2 cells | LPS (0.5 μg/mL) for 24 h | ↓ inflammation | ↓ MAPK | [120] | |
| 1.25–5 μM | Co-treatment | BV2 cells | LPS (1 μg/mL) for 36 h | ↓ inflammation and adhesion | ↑ PPARγ ↓ NF-κB | [121] | |
| 10–40 μM | Pre-treatment for 12 h | HUVEC cells | LPS (1 μg/mL) for 36 h | ↓ neurotoxicity | ↑ GRP78 ↓ CHOP and p38 | [122] | |
| 2–4 μM | Pre-treatment for 24 h | primary cortical cells | OGD for 2 h | ↓ apoptosis | ↓ caspase-3 | [125] | |
| Formononetin | 1–10 μM | Pre-treatment for 12 h | primary cortical cells | NMDA (200 μM) plus glycine (10 μM) for 40 min | ↓ excitotoxic injury | ↑ Bcl-2 | [129] |
| Equol | 0.1–1 μM | Pre-treatment for 24 h | primary cortical cells | Hypoxia for 16 h OGD for 2 or 5 h | ↓ apoptosis | - | [105] |
| 5–20 μM | Pre-treatment for 30 min | BV2, C6, and N2a cells | LPS (100 ng/mL) for 24 h | ↓ neuroinflammation and activated microglia-induced neurotoxicity | ↓ TLR4/JNK/NF-κB pathway | [108] | |
| 0.1–10 μM | Pre-treatment for 1 h | PC12 cells | Hypoxia for 12 h | ↓ cell death and ROS production | ↓ gp91phox and Src phosphorylation | [134,135] | |
| |||||||
| O-Substituted Isoflavones | Applied dose | Application | Species | Employed Model | Observed Effects | Possible Mechanism | References |
| Genistein | 5–20 mg/kg, ip | Pre- and post-treatment | SD rats | 4-VO | - | ↓ STAT3 activation | [90] |
| 15 mg/kg, ip | Pre- and post-treatment | SD rats | 4-VO | ↓ hippocampal neuronal damage | ↑ Bcl-2-related pathway | [91] | |
| 3–10 mg/kg, | Post-treatment | Mongolian gerbils | common carotid occlusion | ↓ neuronal damage and neurological deficits | ERβ | [93] | |
| 500 ppm, po | Pre-treatment for 2 wk | SD rats (OVA) | MCAO for 90 min Followed by 24 h of reperfusion | ↓ infarct size | ↓ gp91phox | [96] | |
| 2.5–10 mg/kg, po | Pre-treatment for 2 wk | C57/ BL6J mice | MCAO for 60 min | ↓ infarct volume, neurological deficit, and apoptosis | ↓ mitochondria-dependent apoptosis pathway and NF-κB | [95] | |
| 1 mg/kg, iv | Post-treatment | SD rats | 4-VO | ↓ hippocampal neuronal damage, and neurological deficits | ↑ eNOS/Nrf2/HO-1 pathway | [92] | |
| 10 mg/kg, sc | Pre-treatment | SD rats | MCAO for 90 min | ↓ infarct volume, neurological deficit, and vascular reactivity | ↑ circulatory function | [98] | |
| 10 mg/kg, ip | Pre-treatment for 2 wk | Kunming mice (OVA) | MCAO for 90 min | ↓ infarct volume, neurological deficit, and apoptosis | ↑ ERK activation | [99] | |
| 0.1 mg/kg/day, sc | Pre-treatment for 2 wk | SD rats (OVA) | MCAO for 10 min | ↓ hippocampal neuronal damage | ↓ GR–Mdm2 interaction | [104] | |
| 10 mg/kg, ip | Post-treatment | SD rats | MCAO for 5 min | ↓ neuronal damage, brain edema, and neurological deficits | ↑ NRF-1 ↓ mitochondria-mediated apoptotic pathway | [97] | |
| 2.5–10 mg/kg, ip | Pre-treatment for 2 wk | STZ-induced Swiss mice | MCAO for 30 min | ↓ infarct size, neurological deficits and glucose levels | ↑ GLP-1 ↓ DPP-4 | [94] | |
| 10 mg/kg, ip | Pre-treatment for 2 wk | SD rats (OVA) | MCAO for 90 min | ↓ infarct volume, neuronal damage, and neurological deficits | ↑ Nrf-2/NQO-1 ↑ PI3K/Akt/mTOR pathway | [102,103] | |
| 10 mg/kg, ip | Pre-treatment for 2 wk | senescent C57BL/6J mice | MCAO for 60 min | ↓ infarct volume, neuronal damage, and neurological deficits | ↓ NLRP3 Inflammasome | [100] | |
| 10–30 mg/kg, po | Co-treatment for 2 wk | Swiss mice | Hypoxia for 2 wk | ↓ cognition deficits | ↑ IGF-1/CREB/BDNF | [88] | |
| 10 mg/kg, ip | Pre-treatment for 3 days | neonatal C57BL/6 mice | MCAO for 60 min | ↓ infarct volume, neuronal damage, and neurological deficits | ↑ Nrf2/HO-1 pathway ↓ NF-κB pathway | [89] | |
| 10 mg/kg, ip | Post-treatment | C57BL/6 mice (OVA) | MCAO for 10 min | ↓ infarct volume, apoptosis, and neurological deficits | ↑ GPER/PGC-1α pathway ↓ NLRP3 Inflammasome | [101] | |
| Daidzein | 10 mg/kg, ip | Post-treatment | SD rats | MCAO | ↓ apoptosis, neuronal damage, and neurological deficits | ↓ caspase-3 | [112] |
| 20 mg/kg, ip | Pre-treatment for 1 wk | SD rats | Spinal cord IR for 25 min | ↓ apoptosis, neuronal damage, and neurological deficits | ↓ caspase-3 and NF-κB ↑ PI3K/Akt pathway | [114] | |
| 10–30 mg/kg, ip | Post-treatment | ICR mice | MCAO for 2 h | ↓ infarct volume, brain edema, and neurological deficits | ↑ PI3K/Akt/mTOR pathway ↑ Akt/mTOR/BDNF pathway | [113] | |
| Biochanin A | 5–10 mg/kg, ip | Pre-treatment for 4 wk | C57BL/6 mice | MCAO for 60 min | ↓ infarcted volume and sensorimotor deficit | ↑ ERRE and GOT | [118] |
| 10–40 mg/kg, ip | Pre-treatment for 2 wk | SD rats | MCAO for 2 h Followed by 24 h of reperfusion | ↓ infarct volume, brain edema, and neurological deficits | ↑ Nrf-2/HO-1 pathway ↓ NF-κB | [123] | |
| 10–40 mg/kg, ip | Pre-treatment for 2 wk | SD rats | MCAO for 2 h Followed by 24 h of reperfusion | ↓ infarct volume, brain edema, and neurological deficits | ↑ GRP78 ↓ CHOP and p38 | [125] | |
| 10–40 mg/kg, ip | Pre-treatment for 2 wk | SD rats | MCAO for 2 h | ↓ infarct volume, brain edema, and neurological deficits | ↓ p38 pathway | [124] | |
| 10–40 μg/kg, icv | Post-treatment | SD rats | Subarachnoid Hemorrhage | ↓ mortality, brain edema, and neurological deficits | ↓ TLRs/TIRAP/MyD88/NF-κB pathway | [126] | |
| Formononetin | 12.5–25 mg/kg, ip | Pre-treatment for 2 wk | SD rats | MCAO for 2 h Followed by 24 h of reperfusion | ↓ infarcted volume, brain edema, and neurological deficit | ↑ PI3K/Akt and Bcl-2 | [131] |
| 30 mg/kg | Post-treatment for 2 wk | SD rats | MCAO for 2 h Followed by 14 days of reperfusion | ↑ injured neurological functions, neuronal differentiation, and synaptic plasticity. | ↑ BDNF or NGF/Trk/PI3K/Akt/ERK | [130] | |
| 30 mg/kg | Post-treatment for 3 d | SD rats | MCAO for 1 h Followed by 3 days of reperfusion | ↓ neurological deficit and neuroinflammation | ↓ JAK2/STAT3 pathway | [132] | |
| Equol | 250 ppm, po | Pre-treatment for 2 wk | SD rats | MCAO for 90 min Followed by 24 h of reperfusion | ↓ infarct size | ↓ gp91phox | [96] |
| 0.625–2.5 mg/kg, po | Pre-treatment for 3 d | SD rats | MCAO for 2 h Followed by 22 h of reperfusion | ↓ mortality, neurological deficit, brain damage, and infarct volume | ↓ gp91phox and Src phosphorylation | [135] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-E.; Lien, J.-C.; Tsai, C.-W.; Wu, C.-R. Therapeutic Potential and Mechanisms of Novel Simple O-Substituted Isoflavones against Cerebral Ischemia Reperfusion. Int. J. Mol. Sci. 2022, 23, 10394. https://doi.org/10.3390/ijms231810394
Yang S-E, Lien J-C, Tsai C-W, Wu C-R. Therapeutic Potential and Mechanisms of Novel Simple O-Substituted Isoflavones against Cerebral Ischemia Reperfusion. International Journal of Molecular Sciences. 2022; 23(18):10394. https://doi.org/10.3390/ijms231810394
Chicago/Turabian StyleYang, Shu-Er, Jin-Cherng Lien, Chia-Wen Tsai, and Chi-Rei Wu. 2022. "Therapeutic Potential and Mechanisms of Novel Simple O-Substituted Isoflavones against Cerebral Ischemia Reperfusion" International Journal of Molecular Sciences 23, no. 18: 10394. https://doi.org/10.3390/ijms231810394
APA StyleYang, S.-E., Lien, J.-C., Tsai, C.-W., & Wu, C.-R. (2022). Therapeutic Potential and Mechanisms of Novel Simple O-Substituted Isoflavones against Cerebral Ischemia Reperfusion. International Journal of Molecular Sciences, 23(18), 10394. https://doi.org/10.3390/ijms231810394