
Deciphering the Regulatory Circuits of RA3 Replication Module - Mechanisms of the Copy Number Control


Abstract
1. Introduction
2. Results
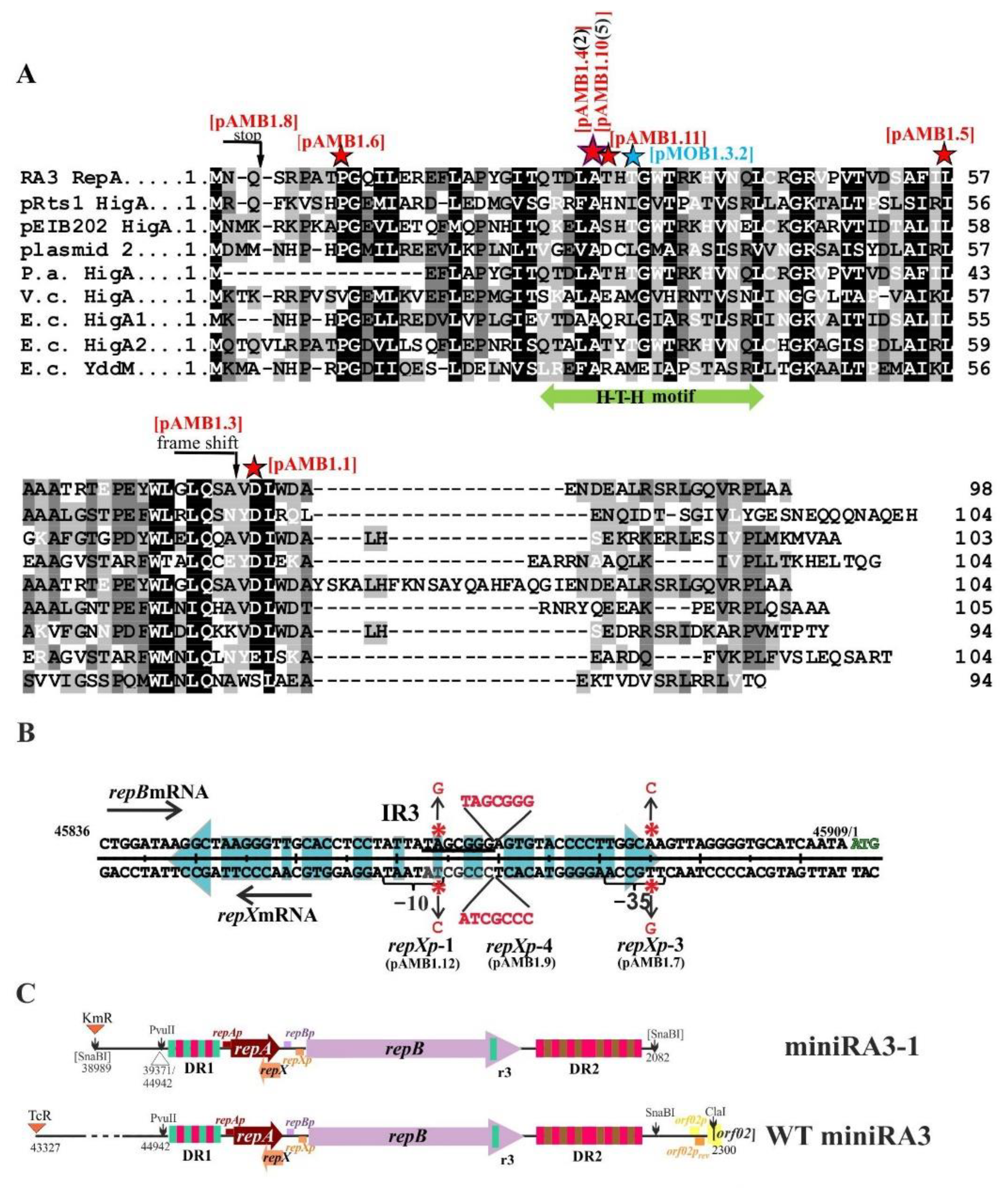
2.1. Wild Type RA3 Minireplicon Encompasses DR1-repA-repB-DR2 and the Junction Region between the Replication and Stability Modules
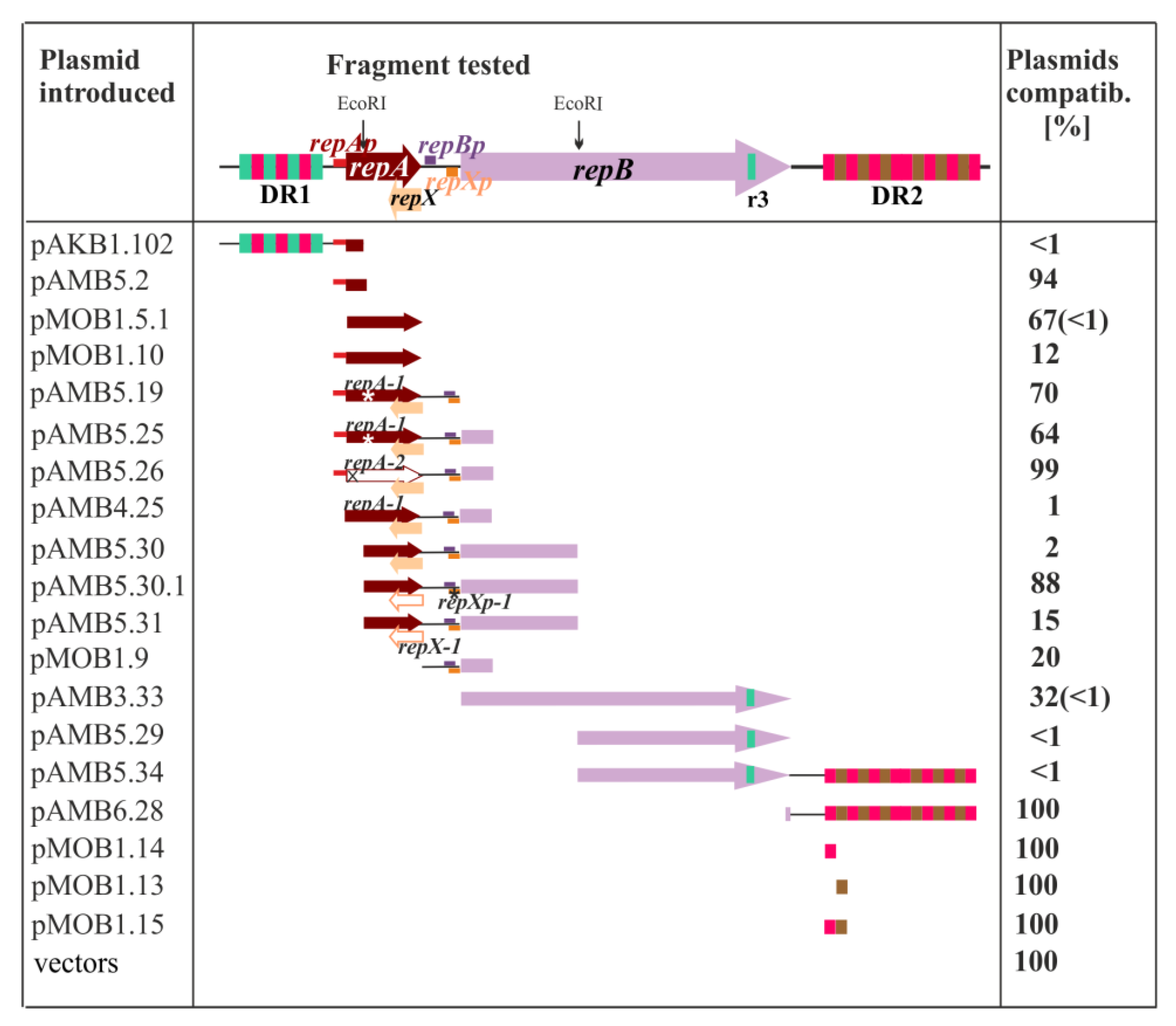
2.2. Search for the Plasmid Components Vital for the Minireplicon Function by Use of the Incompatibility Test
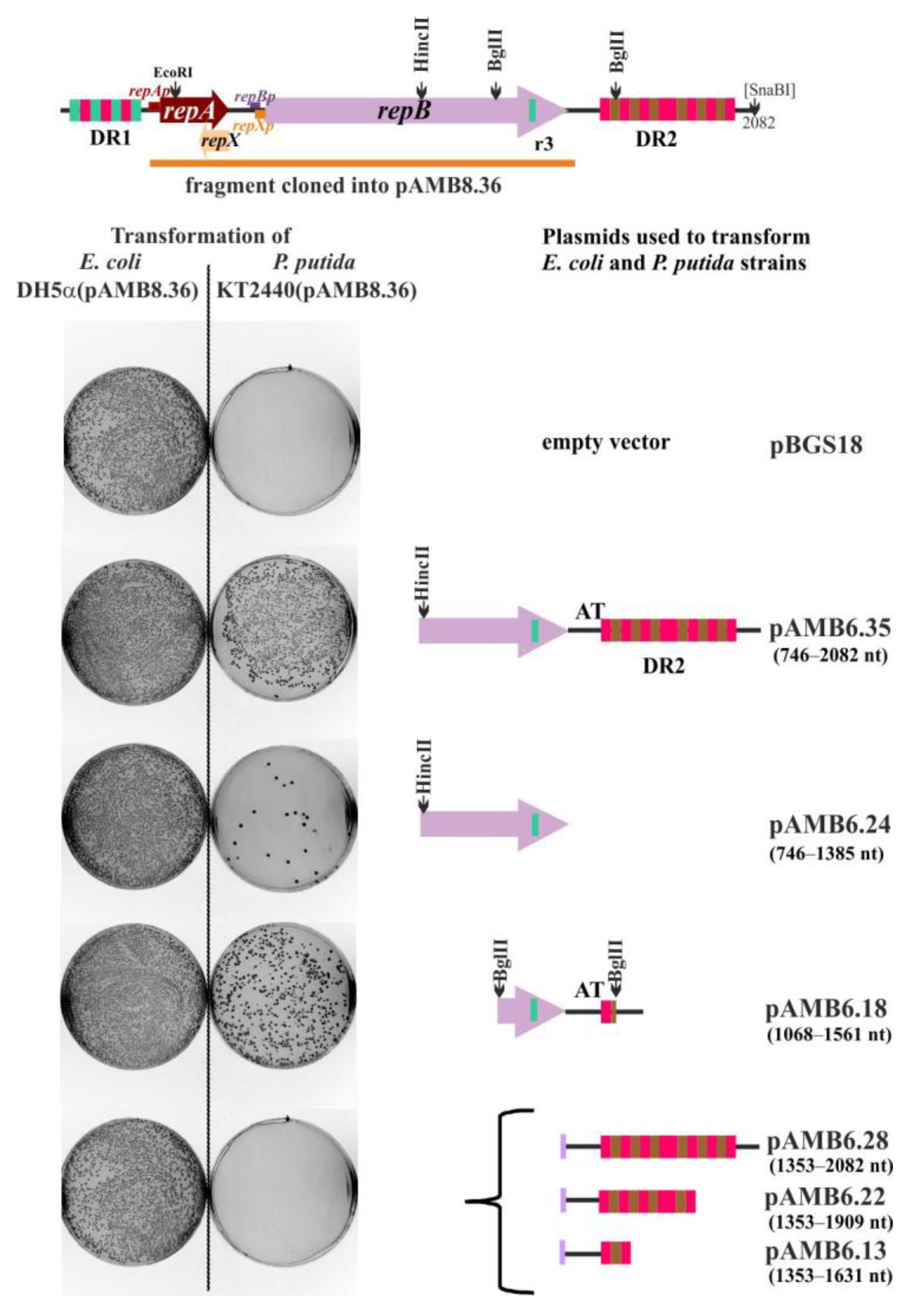
2.3. Defining oriV by Deletion and Complementation Analysis
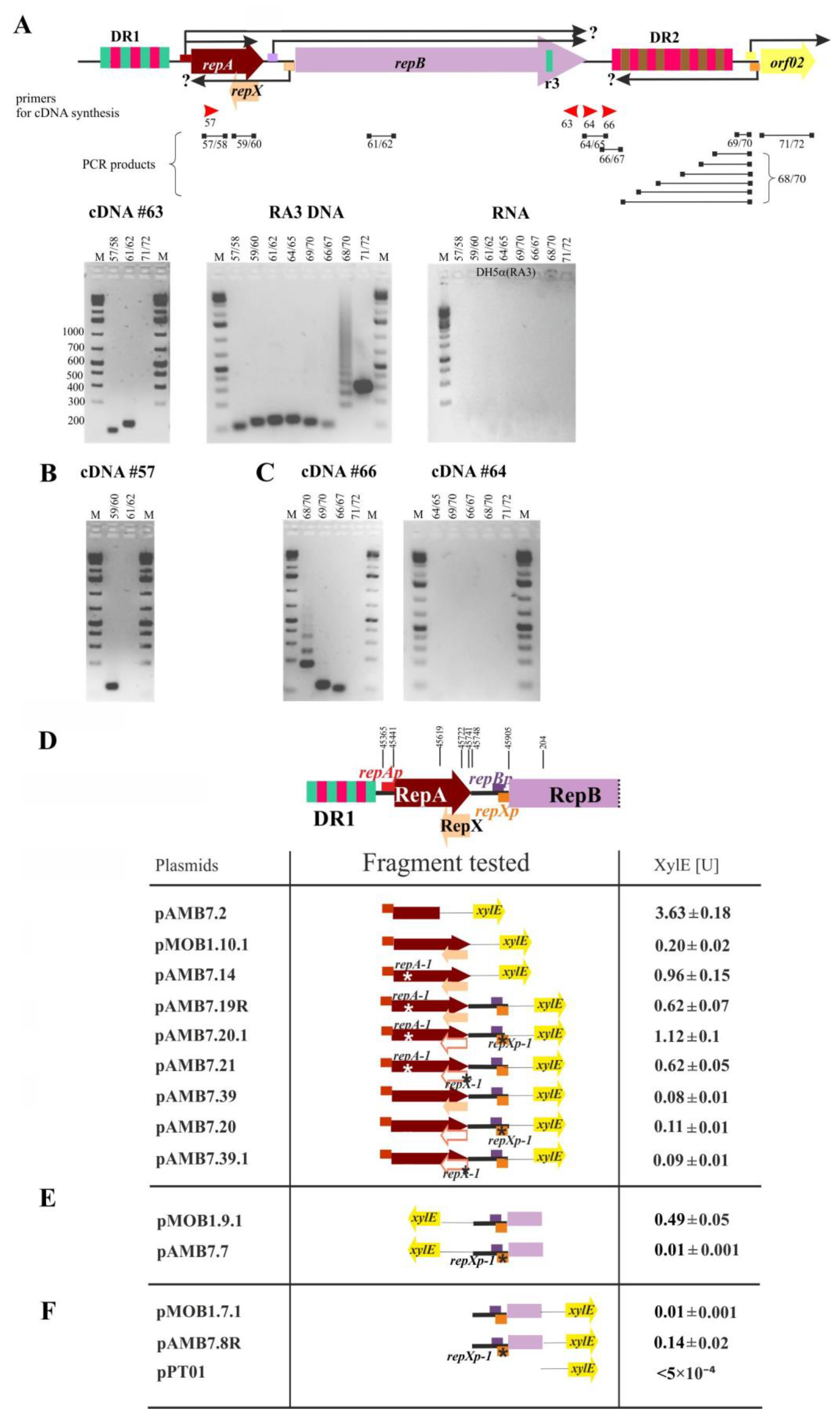
2.4. The repA and repB form an Operon and the repXmRNA Is Complementary to repAmRNA over Its Full Length
2.5. The Extent of mRNA Synthesized from orf02prev toward oriV
2.6. Transcriptional Analysis of the repA, repB, and repX Promoters
2.7. Properties of RepA and Its Variants
2.8. The Copy Number of miniRA3 Depends on repAp, repBp, and repXp Transcription and Presence of RepX
2.9. Northern Analysis of the Various Transcripts Produced by the Minireplicon
3. Discussion
4. Materials and Methods
- Bacterial strains and growth conditions
- Plasmid DNA isolation, analysis, DNA amplification, and manipulation
- PCR based site-directed mutagenesis–mutagenesis in situ (Thermo Fisher Sci.PL)
- Determination of catechol 2,3-oxygenase activity (XylE)
- Transformation procedures
- Overproduction and purification of His6-tagged RepAs by affinity chromatography
- Crosslinking with glutaraldehyde
- Plasmid copy number

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Description | Additional Information |
|---|---|---|
| Plasmids provided by others | ||
| pABB21 | oriRA3, CmR, transcriptional terminator Ttnp513from RA3, vector based on the miniRA3-1 | [37] |
| pABB21.1 | derivative of pABB21 with truncated DR1 [r3r1] and DR2 [r1r2r1] in the miniRA3-1 | A. Bartosik |
| pABB21.2 | derivative of pABB21 with truncated DR1 [(r3r1)2] and DR2 [(r1r2)3r1(r1r2)r1] in the miniRA3-1 | A. Bartosik |
| pAKB1.102 | pGEM_T-Easy derivative, 1174 bp RA3 fragment DR1 repAp | A. Kulinska |
| pBBR1MCS-3 | IncA/C, CmR, broad-host-range (BHR) vector | [38] |
| pBGS18 | oriMB1, KmR, high copy | [39] |
| pET28a | oriMB1, KmR, T7p, lacO, His6-tag, T7 tag, medium copy | Novagen |
| pGBT30 | oriMB1, ApR, lacIq tacp, expression vector, high copy | [19] |
| pGEM-T-Easy | oriMB1, ApR, cloning vector, high copy | Promega |
| pJSB18 | WT miniRA3, TcR (RA3 coordinates 1–2300;43327–45909) | J. Godziszewska |
| pKRP11 | oriMB1, ApR, KmR, high copy | [16] |
| pKRP12 | oriMB1, ApR, TcR, high copy | [16] |
| pPT01 | oriSC101, KmR, medium copy | [21] |
| pUC18 | oriMB1, ApR, high copy | [40] |
| RA3 | IncU, CmR, SmR, SuR, 45.9 kb BHR, conjugative, low copy number plasmid | F. Hayes |
| Plasmids constructed during this work | ||
| RA3 derivatives | ||
| pAMB1.1 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates 1–2082; 38989–45909) | repA mutant, (RepAD76V) mutation A→T at position 45667 |
| pAMB1.2 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | WT |
| pAMB1.3 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, (RepAD52N), mutation G→A at position 45594 in repA; deletion of 1 nt at position 45661 causing frame-shift after 74th codon of RepA and 25th of RepX |
| pAMB1.4 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, (RepAA29G), mutation C→G at position 45526 |
| pAMB1.5 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, (RepAL57Q), mutation T→A at position 45610 |
| pAMB1.6 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, (RepAP9L), mutation C→T at position 45466 |
| pAMB1.7 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repXp-3 mutant, mutation A→C at position 45890, change in a putative -35 motif for repXp |
| pAMB1.8 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, mutation C→A at position 45451 introducing stop codon in repA after 3th codon |
| pAMB1.9 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repXp-4 mutant, 7 nt duplication between putative -35 and -10 motifs of repXp (duplication starts at position 45875) |
| pAMB1.10 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant, (RepAA29V) mutation C→T at position 45526 |
| pAMB1.11 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repA mutant (RepAT30P), mutation A→C at position 45528 |
| pAMB1.14 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates as above) | repXp-1, mutation A→G in a putative -10 motif for repXp (position 45869) |
| pAMB1.14.1 | pAMB1.14 derivative, KmR cassette from pKRP11 inserted as the HincII fragment into SnaBI site; SmR, KmR | repXp-1, mutation A→G in a putative -10 motif for repXp (position 45869) |
| pAMB2.2 | miniRA3-2 repA-1, repAp-1 (IR1*), KmR, PCR based- site directed mutagenesis of pMOB1.3.2 (miniRA3-1) with primers #25 and #26 | repA-1 mutation A→C at position 45534; repAp-1(IR1*), 3 nt modified in the IR1 |
| pAMB2.3 | miniRA3-7 repAp-2 (IR2*); KmR, PCR based- site directed mutagenesis of pMOB1.3.2 with primers #27 and #28 | repA-1, repAp-2 (IR2*), 5 nt modified in the IR2; repA-1 allele mutation A→C at position 45534 |
| pAMB2.4 | miniRA3-4, repA-1, repX-1; KmR, PCR based- site directed mutagenesis of pMOB1.3.2 with primers #29 and #30 | repA-1 allele, mutation A→C at position 45534, repX-1, mutation A→G at position 45734 eliminating codon ATG for RepX; |
| pAMB2.7 | miniRA3-5, repXp-1; KmR, pAMB1.14.1 digested with PvuII and self-ligated; (RA3 coordinates 1–2082; 38989–39371; 44942–45909) | repXp-1, mutation A→G in a putative -10 motif of repXp (position 45869) |
| pAMB2.11 | miniRA3-5, repXp-1, EcoRI*; KmR, PCR based-site directed mutagenesis of pAMB2.7 with primers #37 and #38 | repXp-1–mutation A→G in a putative -10 motif ofrepXp (position 45869); inactivation of EcoRI site within repA gene without a change in RepA amino acid sequence; |
| pMOB1.3 | 9 kb self-replicating SnaBI restriction fragment from RA3, SmR (RA3 coordinates 1–2082; 38989–45909) | repA-1 mutant, (RepAT32P), mutation A→C at position 45534 |
| pMOB1.3.1 | pMOB1.3 derivative, KmR cassette from pKRP11 inserted as the HincII fragment into SnaBI site, SmR KmR | repA-1 mutant, (RepAT32P), mutation A→C at position 45534 |
| pMOB1.3.2 | miniRA3-1, repA-1; pMOB1.3.1 digested with PvuII and self-ligated, KmR; (RA3 coordinates 1–2082; 38989–39371; 44942–45909) | repA-1 mutant, (RepAT32P), mutation A→C at position 45534 |
| pMOB1.16 | pMOB1.3.2 derivative; PCR based site-directed mutagenesis with primers #73 and #74 to inactivate EcoRI site in the repB | WT repB, inactivated EcoRI site within repB gene without a change in the amino acid sequence |
| pGBT30 derivatives | ||
| pAMB3.33 | tacp-repB; 1385 bp PCR amplified fragment on pMOB1.16 with primers #10 and #16 cloned between EcoRI-SalI sites (RA3 coordinates 1–1385) | |
| pAMB3.36 | repAp repA-1 repBp [repXp repX] repB; 538 bp PCR fragment amplified on miniRA3-1 with primers #1 and #17 cloned as the BamHI-EcoRI fragment into pAMB3.33 | repA-1 allele; mutation A→C at position 45534 (RepAT32P); (RA3 coordinates 45365–45903; 1–1385) |
| pAMB3.40 | tacp-repA-1; 300 bp PCR fragment amplified on miniRA3-1 with primers #19 and #3 cloned as the SacI-SalI fragment (RA3 coordinates 45441–45741) | repA-1 allele; mutation A→C at position 45534 (RepAT32P) |
| pAMB3.42 | tacp-repA-3; 300 bp PCR fragment amplified on pAMB1.1 with primers #19 and #3 cloned as the SacI-SalI fragment (RA3 coordinates as above) | repA-3 allele; mutation A→T at position 45667 (RepAD76V) |
| pAMB3.43 | tacp-repA-4; 300 bp PCR fragment amplified on pAMB1.5 with primers #19 and #3 cloned as the SacI-SalI fragment (RA3 coordinates as above) | repA-4 allele; mutation T→A at position 45610 (RepAL57Q) |
| pAMB3.44 | tacp-repA-5; 300 bp PCR fragment amplified on pAMB1.6 with primers #19 and #3 cloned as the SacI-SalI fragment (RA3 coordinates as above) | repA-5 allele; mutation C→T at position 45466 (RepAP9L) |
| pAMB3.45 | tacp-repA; 300 bp PCR fragment amplified on pAMB2.11 with primers #6 and #3 cloned as the EcoRI-SalI fragment (RA3 coordinates as above) | WT repA; inactivated EcoRI site within repA gene without a change in the amino acid sequence |
| pGEM-T Easy derivatives | ||
| pAMB4.25 | repA-1 repBp [repXp repX]; 684 bp PCR fragment amplified on miniRA3-1 with primers #5 and #13 (RA3 coordinates 1–204; 45429–45909) | repA-1 allele without repAp; intergenic repA-repB region |
| pUC18 derivatives | ||
| pAMB5.2 | repAp, 256 bp PCR fragment amplified on miniRA3-1 with primers #1 and #4; cloned as the BamHI fragment (RA3 coordinates 45365–45621) | |
| pAMB5.3 | repAp-1 (IR1*), 256 bp PCR fragment amplified on miniRA3-2 with primers #1 and #4; cloned as the BamHI fragment (RA3 coordinates 45365–45621) | repAp-1 (IR1*); 3 nt modified in the one arm of the IR1 |
| pAMB5.3.1 | repAp-3 (IR1*), PCR based site-directed mutagenesis of pAMB5.3 with primers #31 and #32 | repAp-3 (IR1*); 6 nt modified in the IR1 |
| pAMB5.2.2 | repAp-2 (IR2*), 256 bp PCR fragment amplified on miniRA3-7 with primers #1 and #4; cloned as the BamHI fragment (RA3 coordinates 45365–45621) | repAp-2 (IR2*); 5 nt modified in the IR2 |
| pAMB5.19 | repAp repA-1 repBp [repXp repX]; 540 bp PCR fragment amplified on miniRA3-1 with primers #1 and #15; cloned as the BamHI fragment (RA3 coordinates 45365–45905) | repA-1; mutation A→C at position 45534 |
| pAMB5.25 | repA -repA-1 repBp [repXp repX] repB’; 748 bp PCR fragment amplified on miniRA3-1 with primers #1 and #9 cloned; as the BamHI-SphI fragment (RA3 coordinates 1–204; 45365–45909) | repA-1; mutation A→C at position 45534 |
| pAMB5.29 | ‘repB; 848 bp EcoRI-SalI fragment from pMOB1.6.1 | part of repB encoding amino acids from 180 to 459 |
| pAMB5.30 | ‘repA repBp [repXp repX] repB’, 1kb EcoRI fragment from miniRA3-1 (RA3 coordinates 1–537; 45486–45909) | |
| pAMB5.30.1 | ‘repA repBp [repXp-1 repX] repB’; PCR based site- directed mutagenesis of pAMB5.30 with primers #23 and #24 to introduce repXp-1 mutation | repXp-1; A→G in a putative -10 motif of repXp (position 45869) |
| pAMB5.31 | ‘repA repBp [repXp repX-1] repB’;960 bp EcoRI fragment from pAMB2.4 (RA3 coordinates 1–537; 45486–45909) | repX-1; codon ATG for RepX eliminated (mutation A→G at position 45734) |
| pAMB5.34 | repB DR2; 1545 bp fragment from miniRA3-1 cloned as the EcoRI-HindIII fragment (RA3 coordinates 537–2082) | |
| pAMB5.38 | pUC18–miniRA3-1 hybrid plasmid; pMOB1.16 digested with EcoRI-PvuII and ligated with pUC18 digested EcoRI-HincII; ApR, KmR (RA3 coordinates 1–2082; 38989–39371; 45486–45909) | repA-1; mutation A→C at position 45534 |
| pMOB1.9 | repBp [repXp];365 bp PCR fragment amplified on RA3 with #8 and #9 primers, cloned as the SphI-BamHI fragment (RA3 coordinates 1–204; 45748–45909) | |
| pMOB1.10 | repAp repA;376 bp PCR fragment amplified on RA3 with #1 and #2 primers, cloned as BamHI fragment (RA3 coordinates 45365–45741) | |
| pMOB1.13 | r2; 42 nt oligonucleotides #39 and #40 after annealing were cloned between BamHI-SalI sites | |
| pMOB1.14 | r1; 38 nt oligonucleotides #41 and #42 after annealing were cloned between SalI-PstI sites with inactivation of SalI site | |
| pMOB1.15 | r2 r1; 38 nt oligonucleotides #41 and #42 after annealing were cloned between SalI-PstI sites of pMOB1.13 with inactivation of SalI site | |
| pBGS18 derivatives | ||
| pAMB6.13 | truncated DR2; 278 bp PCR fragment amplified on pABB21.1 with primers #22 and #76; cloned as the EcoRI-SacI fragment (RA3 coordinates 1353–1631) | |
| pAMB6.18 | ‘repB r1; 493 bp fragment from miniRA3-1 inserted as the BglII fragment into BamHI site (RA3 coordinates 1068–1561) | |
| pAMB6.22 | truncated DR2; 556 bp PCR fragment amplified on pABB21.2 with primers #22 and #76; cloned as the EcoRI-SacI fragment (RA3 coordinates 1353–1909) | |
| pAMB6.24 | ‘repB; 639 bp fragment from pAMB5.29; cloned as the SmaI-HincII fragment (RA3 coordinates 746–1385) | |
| pAMB6.28 | DR2; 729 bp PCR fragment amplified on pABB21 with primers #22 and #76; cloned as the EcoRI-SacI fragment (RA3 coordinates 1353–2082) | |
| pAMB6.35 | ‘repB DR2; 1336 bp fragment from pAMB5.34 inserted as the HincII-HindIII fragment between SmaI-HindIII sites | |
| pMOB1.6 | repB; 1407 bp PCR fragment amplified on RA3 template with the use of #75 and #10 primers; cloned as the SacI-SalI fragment (RA3 coordinates 1–1385; 45887–45909) | |
| pPT01 derivatives | ||
| pAMB7.2 | repAp; 256 bp BamHI fragment from pAMB5.2 | repAp-xylE transcriptional fusion |
| pAMB7.3.1 | repAp-3 (IR1*); 256 bp BamHI fragment from pAMB5.3.1 | repAp-3 (IR1*)-xylE transcriptional fusion; 6 nt modified in the IR1 |
| pAMB7.4 | repAp-2 (IR2*); 256 bp BamHI fragment from pAMB5.4 | repAp-2 (IR2*)-xylE transcriptional fusion; 5 nt modified in the IR2 |
| pAMB7.7 | repBp [repXp-1]; 365 bp fragment PCR amplified on pAMB5.30.1 with primers #8 and #9; cloned as the BamHI fragment | repXp-1-xylE transcriptional fusion (RA3 coordinates 1–204; 45748–45909) |
| pAMB7.8R | repBp [repXp-2]; 365 bp PCR fragment amplified on pAMB2.5 with primers #11 and #13; cloned as the BamHI-SphI fragment | repB-xylE transcriptional fusion (RA3 coordinates 1–204; 45748–45909) |
| pAMB7.11 | repBp [repXp]; 391 bp PCR fragment amplified on pJSB18 with primers #18 and #9; cloned as the BamHI-SphI fragment | repXp-xylE transcriptional fusion (RA3 coordinates 1–204; 45722–45909) |
| pAMB7.12 | repBp [repXp-1]; 365 bp PCR fragment amplified on pAMB1.12 with primers #11 and #13, cloned as the BamHI-SphI fragment | repBp-xylE transcriptional fusion (RA3 coordinates 1–204; 45748–45909) |
| pAMB7.14 | repAp repA-1; 376 bp PCR fragment amplified on miniRA3-1 with primers #1 and #2; cloned as the BamHI fragment | repAp repA-1-xylE transcriptional fusion (RA3 coordinates 45365–45741) |
| pAMB7.17 | repA repBp [repXp repX]; 494 bp PCR fragment amplified on miniRA3-1 with primers #7 and #9; cloned as the BamHI-SphI fragment | repXp repX-xylE transcriptional fusion (RA3 coordinates 1–204; 45619–45909) |
| pAMB7.19R | repAp repA-1 repBp [repXp repX]; 540 bp fragment from pAMB5.19 cloned as the BamHI fragment | repAp repA-1 repBp [repXp repX]-xylE transcriptional fusion (RA3 coordinates 45365–45905) |
| pAMB7.20 | repAp repA repBp [repXp-1 repX]; 540 bp PCR fragment amplified on pAMB1.14 with primers #1 and #15; cloned as the BamHI fragment; | repAp repA repBp [repXp-1 repX]-xylE transcriptional fusion (RA3 coordinates 45365–45905), in the background repXp-1 mutation (position 45869) |
| pAMB7.20.1 | repAp repA-1 repBp [repXp-1 repX]; PCR-based site-directed mutagenesis of pAMB7.20 with primers #33 and #34 to introduce repA-1 mutation | repAp repA-1 repBp [repXp-1 repX]-xylE transcriptional fusion, in the background repXp-1 mutation (position 45869) |
| pAMB7.21 | repAp repA-1 repBp [repXp repX-1]; 540 bp PCR fragment amplified on miniRA3-4 with primers #1 and #15, cloned as the BamHI fragment | repAp repA-1 repBp [repXp repX-1]-xylE transcriptional fusion, in the background repX-1 mutation (position 45734) |
| pAMB7.23 | repA-1 repBp [repXp repX]; 672 bp PCR fragment amplified on miniRA3-1 with primers #5 and #9; cloned as the BamHI-SphI fragment | repXp repX [repA-1 repBp]-xylE transcriptional fusion (RA3 coordinates 1–204; 45441–45909), in the background repA-1 mutation (position 45534) |
| pAMB7.39 | repAp repA repBp [repXp repX]; 540 bp PCR fragment amplified on pJSB18 (WT mini RA3) with primers #1 and #15, cloned as the BamHI fragment | repAp repA repBp [repXp repX]-xylE transcriptional fusion |
| pAMB7.39R | repAp repA repBp [repXp repX]; 540 bp BamHI fragment of pAMB7.39 in the reversed orientation | repXp repX [repAp repA repBp]-xylE transcriptional fusion |
| pAMB7.39.1 | repAp repA repBp [repXp repX-1]; PCR-based site-directed mutagenesis of pAMB7.39 with primers #29 and #30 to introduce repX-1 mutation | repAp repA repBp [repXp repX-1]-xylE transcriptional fusion, in the background repX-1 mutation (position 45734) |
| pMOB1.7.1 | repBp [repXp];365 bp PCR fragment amplified on RA3 with #11 and #13 primers, cloned as the SphI-BamHI fragment (RA3 coordinates 1–204; 45748–45909) | repBp [repXp]-xylE transcriptional fusion |
| pMOB1.9.1 | repBp [repXp];365 bp SphI-BamHI fragment from pMOB1.9 (RA3 coordinates 1–204; 45748–45909) | repXp [repBp] -xylE transcriptional fusion |
| pMOB1.10.1 | repAp repA;376 bp BamHI fragment from pMOB1.10 (RA3 coordinates 1–204; 45365–45741) | repAp repA-xylE transcriptional fusion |
| pBBR1MCS-3 derivatives | ||
| pAMB8.0 | pBBR1MCS-3 modified in tetM to remove EcoRI site without a change in amino acid sequence of TetM; PCR based site-directed mutagenesis with primers #35 and #36 | |
| pAMB8.36 | repAp repA-1 repBp [repXp repX] repB; 2829 bp fragment from pAMB3.36 inserted as the SalI-PstI fragment between XhoI-PstI sites | repA-1 mutation at position 45534 |
| pET28a derivatives | ||
| pAMB11.41 | T7p-His6-repA-1; 296 bp PCR fragment amplified on miniRA3-1 with primers #12 and #3, cloned as the BamHI-SalI fragment (RA3 coordinates 45441–45737) | repA-1 allele encodes RepAT32P (mutation A→C at position 45534) |
| pAMB11.42 | T7p-His6-repA-3; 300 bp BamHI-SalI fragment from pAMB3.42 | repA-3 allele encodes RepAD76V (mutation A→T at position 45667) |
| pAMB11.43 | T7p-His6-repA-4; 300 bp BamHI-SalI fragment from pAMB3.43 | repA-4 allele encodes RepAL57Q (mutation T→A at position 45610) |
| pAMB11.44 | T7p-His6-repA-5; 300 bp BamHI-SalI fragment from pAMB3.44 | repA-5 allele encodes RepAP9L (mutation C→T at position 45466) |
| pAMB11.47 | T7p-His6-repA; 296 bp PCR fragment amplified on RA3 with primers #12 and #3, cloned as the BamHI-SalI fragment (RA3 coordinates 45441–45737) | WT RepA |
| No(#) | Designation | Sequence |
|---|---|---|
| 1 | ant1 | CGGATCCGCGGGCCTGATCTATTGTTG |
| 2 | ant2 | CGGATCCGCATGCTTTCTATGCCGCTAACGGC |
| 3 | ant2Sal | CGTCGACTATGCCGCTAACGGCCTCAC |
| 4 | ant5 | CGGGATCCTAGCTGCTGCCAGGATAAAC |
| 5 | ant6 | CGGATCCATGAACCAATCACGACCGGC |
| 6 | ant6Eco | CGAATTCATGAACCAATCACGACCGGC |
| 7 | ant7 | CGGATCCCTACACGAACAGAGCCGGAA |
| 8 | rep1 | CGGATCCCCGGAAACCAACTTGGCG |
| 9 | rep2 | CGGATCCGCATGCCGCATAAACTCGGCCTGT |
| 10 | rep4 | CCGTCGACGCCATCTAAACGGCTTTACA |
| 11 | rep5 | CGCATGCCCCCGGAAAACCAACTTGGCG |
| 12 | rep6Bam | CGGATCCATGAACCAATCACGACCGGC |
| 13 | rep7 | CGGATCCGCCGCATAAACTCGGCCTGT |
| 14 | rep8 | CATGAGCCGGGCTAAATG |
| 15 | rep9 | CGGATCCGCATGCGATGCACCCCTAACTTGCC |
| 16 | rep11 | GCGAATTCATGGCGCAAGCTCAGTTGTC |
| 17 | rep12 | GAATTCTGCACCCCTAACTTGCCAAGG |
| 18 | endrepAF | CGGATCCGCCGTTAGCGGCATAGAAAG |
| 19 | repAmodF | CTGAGCTCGAGGGAGGATCCATGAACCAATCACGACCG |
| 20 | 1527R2 | CACCTTCAGCGGTCGTCAAC |
| 21 | orf02pR | GCGCATGCCGATCACGCTCCCAGGTCAA |
| 22 | terrepBFEco | CCGAATTCGGTACCACAGGCGGCTAGGTGTAAAG |
| 23 | mutRep1 | GAGCCTGGATAAGCTTAAGGGTTGCACCTCCTATTATGGCGGGAGTGTA |
| 24 | mutRep2 | GGTACACTCCCGCCATAATAGGAGGTGCAACCCTTAAGCTTATCCAGGC |
| 25 | opA1 | CTAGGTTACACTCTAGAACACATCATTCTG |
| 26 | opA2 | GAATGATGTGTTCTAGAGTGTAACCTAGTTG |
| 27 | opB1 | CTTCAAAACAGTCGACTCTGATGAGGGCTT |
| 28 | opB2 | GCCCTCATCAGAGTCGACTGTTTTGAAGTG |
| 29 | repXm1 | GTTAGCGGCGTAGAAAGGGAGCTCCCCGGAAACC |
| 30 | repXm2 | TTCCGGGGAGCTCCCTTTCTACGCCGCTAACGG |
| 31 | cdMtopA1 | CTATTGTTGAAGTTCGCGAACTAGGTTACAC |
| 32 | cdMtopA2 | GTAACCTAGTTCGCGAACTTCAACAATAGATC |
| 33 | wpMt605L | GGCCACCCATCCCGGGTGGACAAGAAAG |
| 34 | wpMt605R | CTTTCTTGTCCACCCGGGATGGGTGGCC |
| 35 | modEcTcF | CATGAGAATTGTTGAAGACG |
| 36 | modEcTcR | CGTCTTCAACAATTCTCATG |
| 37 | mRA3ecoa | GATACTTGAAAGGGAGTTCTTGGCCCCG |
| 38 | mRA3ecob | GTACGGGGCCAAGAACTCCCTTTCAAG |
| 39 | dr2BamHI | GATCCGCCAAGTTCAGATCTGGACGCCAGAAGGAAATCAACCAGGTGG |
| 40 | dr2SalI | TCGACCACCTGGTTGATTTCCTTCTGGCGTCCAGATCTGAACTTGGCG |
| 41 | dr1SalI | TCGAAGCCAACTCACCAGGCACCGGCAGCAGCTCGACCAGGTGCTGCA |
| 42 | dr1PstI | GCACCTGGTCGAGCTGCTGCCGGTGCCTGGTGAGTTGGCT |
| 43 | sphmob | GCGCATGCTTTTCTCGTTGGAGGGTGAT |
| 44 | inc230P | GCGGATCCGATAGCTCTTTGCCATTAAC |
| 45 | wt1palG | TTGAAGTTTACCAACTAGGTTACACTTCAA |
| 46 | wt1palD | TTGAAGTGTAACCTAGTTGGTAAACTTCAA |
| 47 | wt2palG | AAACACATCATTCTGATGAGGGC |
| 48 | wt2palD | GCCCTCATCAGAATGATGTGTTT |
| 49 | mutAll1palG | TTGAAGTTCGCGAACTAGGTTACACTCTAG |
| 50 | mutAll1palD | CTAGAGTGTAACCTAGTTCGCGAACTTCAA |
| 51 | mutAll2palG | AAACAGTCGACTCTGATGAGGGC |
| 52 | mutAll2palD | GCCCTCATCAGAGTCGACTGTTT |
| 53 | repB2F | CATCGAGAAGCAAAAGGCG |
| 54 | repB2R | CCAACTTGCGTAGGTCTTCCAG |
| 55 | galK-F | ATGATCTTTCTTGCCGAGCG |
| 56 | galK-R | AGCAGCTTTATCATCTGCCGC |
| 57 | repAF | CAAACAGACTTGGCCACCC |
| 58 | repAR | GACTGTAACAGGCACTCGCC |
| 59 | repAF1 | GATAGCGCGTTTATCCTGGC |
| 60 | repAR1 | CTCGTCATTCTCTGCGTCCC |
| 61 | repBF | CTGGAATGCTTGCCAAACCC |
| 62 | repBR | TTCACGGTATTGACCAGGCG |
| 63 | repBR1 | TCACTTTGAAATAGCCATCTAAACGG |
| 64 | 02prevUF | TGTAAAGCCGTTTAGATGGC |
| 65 | 02prevUR | CAGCATGGCTATACGCCTGC |
| 66 | 02prevUF1 | GTTAGCAGGCGTATAGCCATG |
| 67 | 02prevUR1 | CTGGCAAGTTGATCTAAAGG |
| 68 | r2F | GTTCAGATCTGGACGCCAGAAG |
| 69 | 02prevDF | GTACGAAATCAGGCGACGCTATGC |
| 70 | 02prevDR | GGCAATAAAAAGCGCGCTCTAC |
| 71 | EcoOrf2F | CGGAATTCATGATCCACACAGCTAACCG |
| 72 | SalOrf2R | CGCGTCGACATAGGCCAAATCGGCCTACT |
| 73 | EcrepB1 | ACCGTCAACTCAAAGAGCGG |
| 74 | EcrepB2 | CCGCTCTTTGAGTTGACGGT |
| 75 | rep3 | CCGAGCTCTTGGCAAGTTAGGGGTGCAT |
| 76 | CmR | GAGCTCGCCCGGTAGTGATCTTATTTC |
| 77 | NorRepBF | TAACTTTCTCCTTCTCTCTGG |
| 78 | NorRepB7 | GAATTAATACGACTCACTATAGGGTAGTAATAGGGGAGGGACTTG |
- RT reaction followed by PCR
- Northern analysis
- (a)
- Hybridization with the radioactive single stranded DNA labelled at 5′ end as a probe
- (b)
- Hybridization with [α– 32P] UTP labelled mRNA
- In vitro analysis of protein-DNA interactions by electrophoretic mobility shift assay (EMSA)
- Immunodetection of proteins–western blot analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hülter, N.; Ilhan, J.; Wein, T.; Kadibalban, A.S.; Hammerschmidt, K.; Dagan, T. An Evolutionary Perspective on Plasmid Lifestyle Modes. Curr. Opin. Microbiol. 2017, 38, 74–80. [Google Scholar] [CrossRef]
- Aoki, T.; Egusa, S.; Ogata, Y.; Watanabe, T. Detection of Resistance Factors in Fish Pathogen Aeromonas Liquefaciens. J. Gen. Microbiol. 1971, 65, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, G.; Huys, G.; Swings, J.; McGann, P.; Hiney, M.; Smith, P.; Pickup, R.W. Distribution of Oxytetracycline Resistance Plasmids between Aeromonads in Hospital and Aquaculture Environments: Implication of Tn1721 in Dissemination of the Tetracycline Resistance Determinant Tet A. Appl. Environ. Microbiol. 2000, 66, 3883–3890. [Google Scholar] [CrossRef] [PubMed]
- Tschäpe, H.; Tietze, E.; Koch, C. Characterization of Conjugative R Plasmids Belonging to the New Incompatibility Group IncU. J. Gen. Microbiol. 1981, 127, 155–160. [Google Scholar] [CrossRef]
- Cattoir, V.; Poirel, L.; Aubert, C.; Soussy, C.-J.; Nordmann, P. Unexpected Occurrence of Plasmid-Mediated Quinolone Resistance Determinants in Environmental Aeromonas spp. Emerg. Infect. Dis. 2008, 14, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Dang, B.; Xu, Y.; Mao, D.; Luo, Y. Complete Nucleotide Sequence of Plasmid PNA6 Reveals the High Plasticity of IncU Family Plasmids. Gene 2016, 591, 74–79. [Google Scholar] [CrossRef]
- Brown, C.J.; Sen, D.; Yano, H.; Bauer, M.L.; Rogers, L.M.; der Auwera, G.A.V.; Top, E.M. Diverse Broad-Host-Range Plasmids from Freshwater Carry Few Accessory Genes. Appl. Environ. Microbiol. 2013, 79, 7684–7695. [Google Scholar] [CrossRef]
- Rahube, T.O.; Viana, L.S.; Koraimann, G.; Yost, C.K. Characterization and Comparative Analysis of Antibiotic Resistance Plasmids Isolated from a Wastewater Treatment Plant. Front. Microbiol. 2014, 5, 558. [Google Scholar] [CrossRef]
- Kulinska, A.; Czeredys, M.; Hayes, F.; Jagura-Burdzy, G. Genomic and Functional Characterization of the Modular Broad-Host-Range RA3 Plasmid, the Archetype of the IncU Group. Appl. Environ. Microbiol. 2008, 74, 4119–4132. [Google Scholar] [CrossRef]
- Mitura, M.; Lewicka, E.; Godziszewska, J.; Adamczyk, M.; Jagura-Burdzy, G. Alpha-Helical Protein KfrC Acts as a Switch between the Lateral and Vertical Modes of Dissemination of Broad-Host-Range RA3 Plasmid from IncU (IncP-6) Incompatibility Group. Int. J. Mol. Sci. 2021, 22, 4880. [Google Scholar] [CrossRef]
- Lewicka, E.; Dolowy, P.; Godziszewska, J.; Litwin, E.; Ludwiczak, M.; Jagura-Burdzy, G. Transcriptional Organization of the Stability Module of Broad-Host-Range Plasmid RA3, from the IncU Group. Appl. Environ. Microbiol. 2020, 86, e00847-20. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Król, J.E.; Suzuki, H.; Foster, B.; Van Houdt, R.; Brown, C.J.; Mergeay, M.; Top, E.M. Plasmids Captured in C. Metallidurans CH34: Defining the PromA Family of Broad-Host-Range Plasmids. Antonie Van Leeuwenhoek 2009, 96, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Haines, A.S.; Cheung, M.; Thomas, C.M. Evidence That IncG (IncP-6) and IncU Plasmids Form a Single Incompatibility Group. Plasmid 2006, 55, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, K.; Kehrenberg, C.; Schwarz, S. Tet(A)-Mediated Tetracycline Resistance in Porcine Bordetella Bronchiseptica Isolates Is Based on Plasmid-Borne Tn1721 Relics. J. Antimicrob. Chemother. 2006, 58, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, A.; Szczepanowski, R.; Pühler, A.; Top, E.M. Genomics of IncP-1 Antibiotic Resistance Plasmids Isolated from Wastewater Treatment Plants Provides Evidence for a Widely Accessible Drug Resistance Gene Pool. FEMS Microbiol. Rev. 2007, 31, 449–477. [Google Scholar] [CrossRef] [PubMed]
- Reece, K.S.; Phillips, G.J. New Plasmids Carrying Antibiotic-Resistance Cassettes. Gene 1995, 165, 141–142. [Google Scholar] [CrossRef]
- Ludwiczak, M.; Dolowy, P.; Markowska, A.; Szarlak, J.; Kulinska, A.; Jagura-Burdzy, G. Global Transcriptional Regulator KorC Coordinates Expression of Three Backbone Modules of the Broad-Host-Range RA3 Plasmid from IncU Incompatibility Group. Plasmid 2013, 70, 131–145. [Google Scholar] [CrossRef]
- Kulinska, A.; Godziszewska, J.; Wojciechowska, A.; Ludwiczak, M.; Jagura-Burdzy, G. Global Transcriptional Regulation of Backbone Genes in Broad-Host-Range Plasmid RA3 from the IncU Group Involves Segregation Protein KorB (ParB Family). Appl. Environ. Microbiol. 2016, 82, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Jagura-Burdzy, G.; Ibbotson, J.P.; Thomas, C.M. The KorF Region of Broad-Host-Range Plasmid RK2 Encodes Two Polypeptides with Transcriptional Repressor Activity. J. Bacteriol. 1991, 173, 826–833. [Google Scholar] [CrossRef][Green Version]
- Kovach, M.E.; Phillips, R.W.; Elzer, P.H.; Roop, R.M.; Peterson, K.M. PBBR1MCS: A Broad-Host-Range Cloning Vector. BioTechniques 1994, 16, 800–802. [Google Scholar]
- Macartney, D.P.; Williams, D.R.; Stafford, T.; Thomas, C.M. Divergence and Conservation of the Partitioning and Global Regulation Functions in the Central Control Region of the IncP Plasmids RK2 and R751. Microbiol. Read. Engl. 1997, 143 Pt 7, 2167–2177. [Google Scholar] [CrossRef]
- Kulinska, A.; Cao, Y.; Macioszek, M.; Hayes, F.; Jagura-Burdzy, G. The Centromere Site of the Segregation Cassette of Broad-Host-Range Plasmid RA3 Is Located at the Border of the Maintenance and Conjugative Transfer Modules. Appl. Environ. Microbiol. 2011, 77, 2414–2427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dobiasova, H.; Videnska, P.; Dolejska, M. Complete Sequences of IncU Plasmids Harboring Quinolone Resistance Genes QnrS2 and Aac(6’)-Ib-Cr in Aeromonas Spp. from Ornamental Fish. Antimicrob. Agents Chemother. 2016, 60, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Marti, E.; Balcázar, J.L. Multidrug Resistance-Encoding Plasmid from Aeromonas sp. Strain P2G1. Clin. Microbiol. Infect. 2012, 18, E366–E368. [Google Scholar] [CrossRef]
- Godziszewska, J.; Kulińska, A.; Jagura-Burdzy, G. MobC of Conjugative RA3 Plasmid from IncU Group Autoregulates the Expression of Bicistronic MobC-Nic Operon and Stimulates Conjugative Transfer. BMC Microbiol. 2014, 14, 235. [Google Scholar] [CrossRef]
- Godziszewska, J.; Moncalián, G.; Cabezas, M.; Bartosik, A.A.; de la Cruz, F.; Jagura-Burdzy, G. Concerted Action of NIC Relaxase and Auxiliary Protein MobC in RA3 Plasmid Conjugation. Mol. Microbiol. 2016, 101, 439–456. [Google Scholar] [CrossRef]
- Lewicka, E.; Mitura, M.; Steczkiewicz, K.; Kieracinska, J.; Skrzynska, K.; Adamczyk, M.; Jagura-Burdzy, G. Unique Properties of the Alpha-Helical DNA-Binding Protein KfrA Encoded by the IncU Incompatibility Group Plasmid RA3 and Its Host-Dependent Role in Plasmid Maintenance. Appl. Environ. Microbiol. 2021, 87, e01771-20. [Google Scholar] [CrossRef]
- Xu, B.-S.; Liu, M.; Zhou, K.; Geng, Z.; Gao, Z.-Q.; Dong, Y.-H.; She, Z.; Liu, Q.-S. Conformational Changes of Antitoxin HigA from Escherichia coli Str. K-12 upon Binding of Its Cognate Toxin HigB Reveal a New Regulation Mechanism in Toxin-Antitoxin Systems. Biochem. Biophys. Res. Commun. 2019, 514, 37–43. [Google Scholar] [CrossRef]
- Hanahan, D. Studies on Transformation of Escherichia Coli with Plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef] [PubMed]
- McKenney, K.; Shimatake, H.; Court, D.; Schmeissner, U.; Brady, C.; Rosenberg, M. A System to Study Promoter and Terminator Signals Recognized by Escherichia coli RNA Polymerase. Gene Amplif. Anal. 1981, 2, 383–415. [Google Scholar]
- Sambrook, J.; Maniatis, T.; Fritsch, E.F.; Laboratory, C.S.H. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1987; ISBN 978-0-87969-309-1. [Google Scholar]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 1, 263–273. [Google Scholar] [CrossRef]
- Zukowski, M.M.; Gaffney, D.F.; Speck, D.; Kauffmann, M.; Findeli, A.; Wisecup, A.; Lecocq, J.P. Chromogenic Identification of Genetic Regulatory Signals in Bacillus Subtilis Based on Expression of a Cloned Pseudomonas Gene. Proc. Natl. Acad. Sci. USA 1983, 80, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Jagura-Burdzy, G.; Thomas, C.M. Purification of KorA Protein from Broad Host Range Plasmid RK2: Definition of a Hierarchy of KorA Operators. J. Mol. Biol. 1995, 253, 39–50. [Google Scholar] [CrossRef]
- Škulj, M.; Okršlar, V.; Jalen, Š.; Jevševar, S.; Slanc, P.; Štrukelj, B.; Menart, V. Improved Determination of Plasmid Copy Number Using Quantitative Real-Time PCR for Monitoring Fermentation Processes. Microb. Cell Factories 2008, 7, 6. [Google Scholar] [CrossRef]
- Bartosik, A.A.; Markowska, A.; Szarlak, J.; Kulińska, A.; Jagura-Burdzy, G. Novel Broad-Host-Range Vehicles for Cloning and Shuffling of Gene Cassettes. J. Microbiol. Methods 2012, 88, 53–62. [Google Scholar] [CrossRef]
- Kovach, M.E.; Elzer, P.H.; Hill, D.S.; Robertson, G.T.; Farris, M.A.; Roop, R.M.; Peterson, K.M. Four New Derivatives of the Broad-Host-Range Cloning Vector PBBR1MCS, Carrying Different Antibiotic-Resistance Cassettes. Gene 1995, 166, 175–176. [Google Scholar] [CrossRef]
- Spratt, B.G.; Hedge, P.J.; te Heesen, S.; Edelman, A.; Broome-Smith, J.K. Kanamycin-Resistant Vectors That Are Analogues of Plasmids PUC8, PUC9, PEMBL8 and PEMBL9. Gene 1986, 41, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 Phage Cloning Vectors and Host Strains: Nucleotide Sequences of the M13mp18 and PUC19 Vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]







| Plasmid | Mutations in the Minireplicon | Stability (%) after 60 Generations of Growth without Selection | Plasmid Copy Number per Chromosome (Real-Time qPCR) |
|---|---|---|---|
| in the E. coli DH5α strain | |||
| RA3 | 100% | 1 | |
| WT miniRA3 | 100% | 1 | |
| miniRA3-1 | repA-1 (RepAT32P) | 100% | 2.1 |
| miniRA3-2 | repAp-1 (IR1*) repA-1 | 100% | 74.6 |
| miniRA3-7 | repAp-2 (IR2*) repA-1 | 100% | 43.3 |
| miniRA3-4 | repX-1, repA-1 | 100% | 13.8 |
| miniRA3-5 | repXp-1 | 100% | 2.2 |
| in the P. putida KT2440 strain | |||
| RA3 | 10% (±3) | 1 | |
| WT miniRA3 | <3% * | 1 | |
| miniRA3-1 | repA-1 (RepAT32P) | 31% (±4) | 4.6 |
| miniRA3-2 | repAp-1 (IR1*) repA-1 | 12% (±8) | 7.6 |
| miniRA3-7 | repAp-2 (IR2*) repA-1 | 18% (±3) | 9.8 |
| miniRA3-4 | repX-1, repA-1 | 27% (±4) | 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowska-Barkic, A.; Lewicka, E.; Czeredys, M.; Mitura, M.; Jagura-Burdzy, G. Deciphering the Regulatory Circuits of RA3 Replication Module - Mechanisms of the Copy Number Control. Int. J. Mol. Sci. 2022, 23, 9964. https://doi.org/10.3390/ijms23179964
Markowska-Barkic A, Lewicka E, Czeredys M, Mitura M, Jagura-Burdzy G. Deciphering the Regulatory Circuits of RA3 Replication Module - Mechanisms of the Copy Number Control. International Journal of Molecular Sciences. 2022; 23(17):9964. https://doi.org/10.3390/ijms23179964
Chicago/Turabian StyleMarkowska-Barkic, Aleksandra, Ewa Lewicka, Magdalena Czeredys, Monika Mitura, and Grazyna Jagura-Burdzy. 2022. "Deciphering the Regulatory Circuits of RA3 Replication Module - Mechanisms of the Copy Number Control" International Journal of Molecular Sciences 23, no. 17: 9964. https://doi.org/10.3390/ijms23179964
APA StyleMarkowska-Barkic, A., Lewicka, E., Czeredys, M., Mitura, M., & Jagura-Burdzy, G. (2022). Deciphering the Regulatory Circuits of RA3 Replication Module - Mechanisms of the Copy Number Control. International Journal of Molecular Sciences, 23(17), 9964. https://doi.org/10.3390/ijms23179964