
Historical Evolutionary Dynamics and Phylogeography Analysis of Transmissible Gastroenteritis Virus and Porcine Deltacoronavirus: Findings from 59 Suspected Swine Viral Samples from China

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
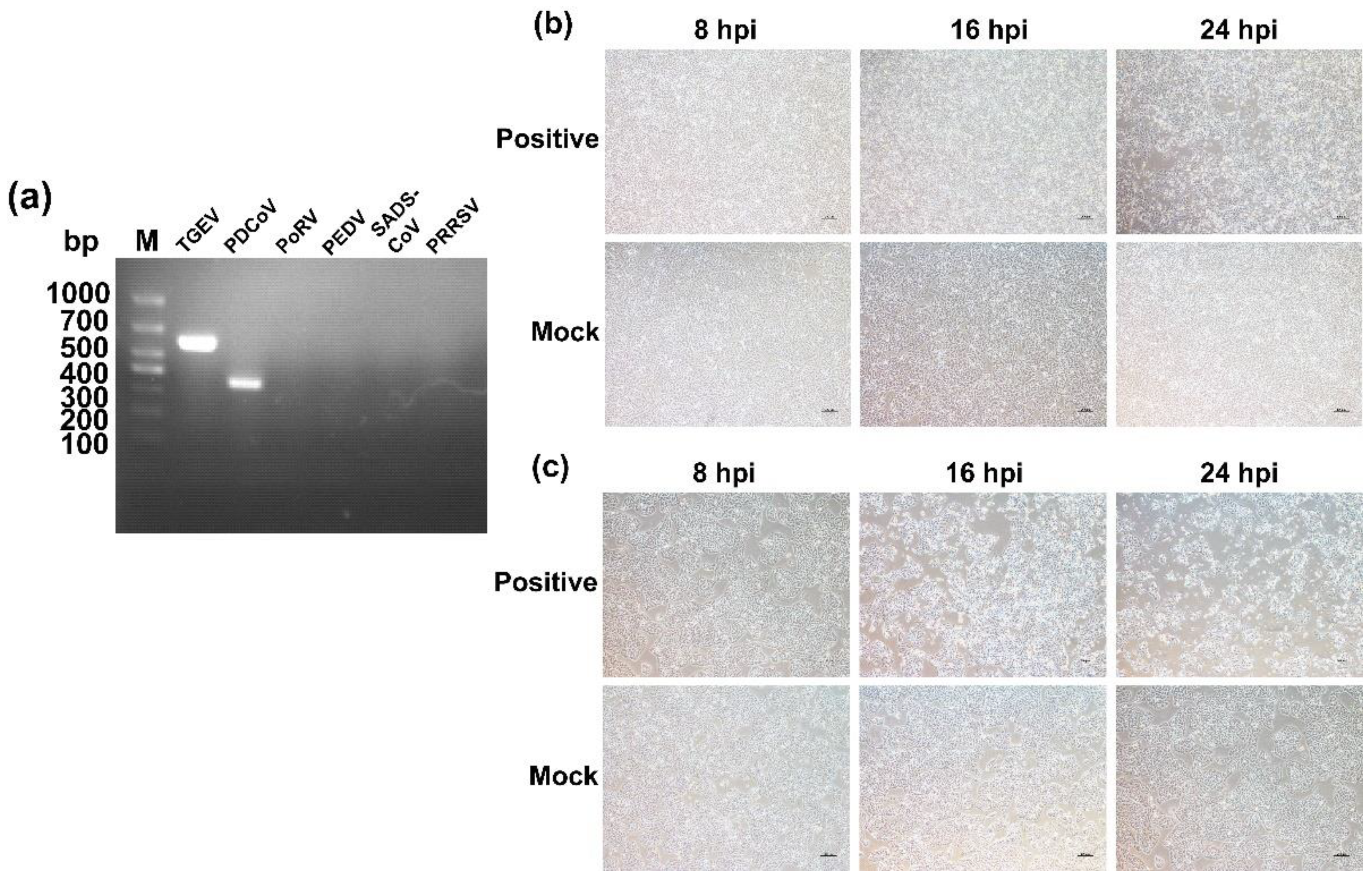
2.1. Virus Detection and Isolation
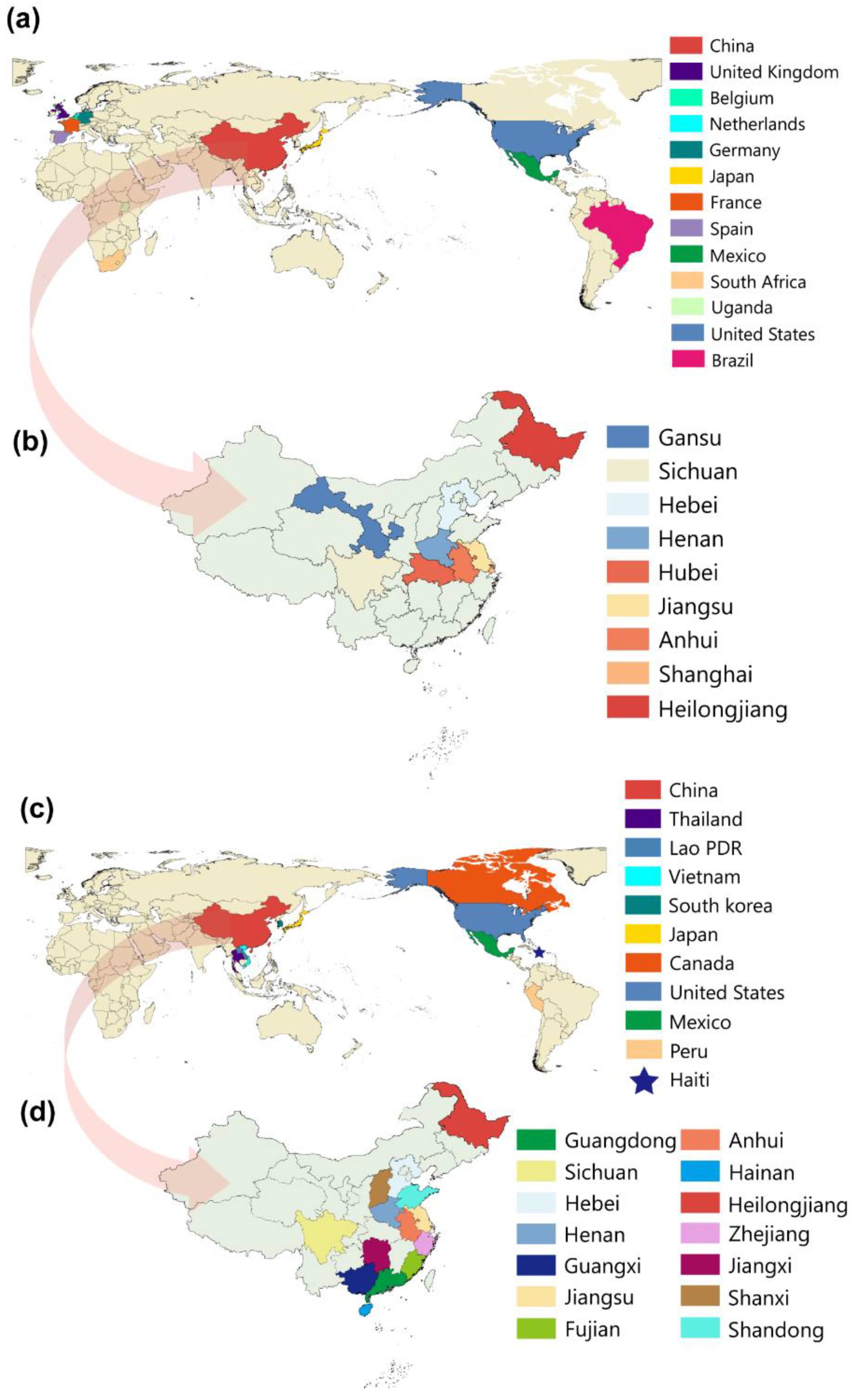
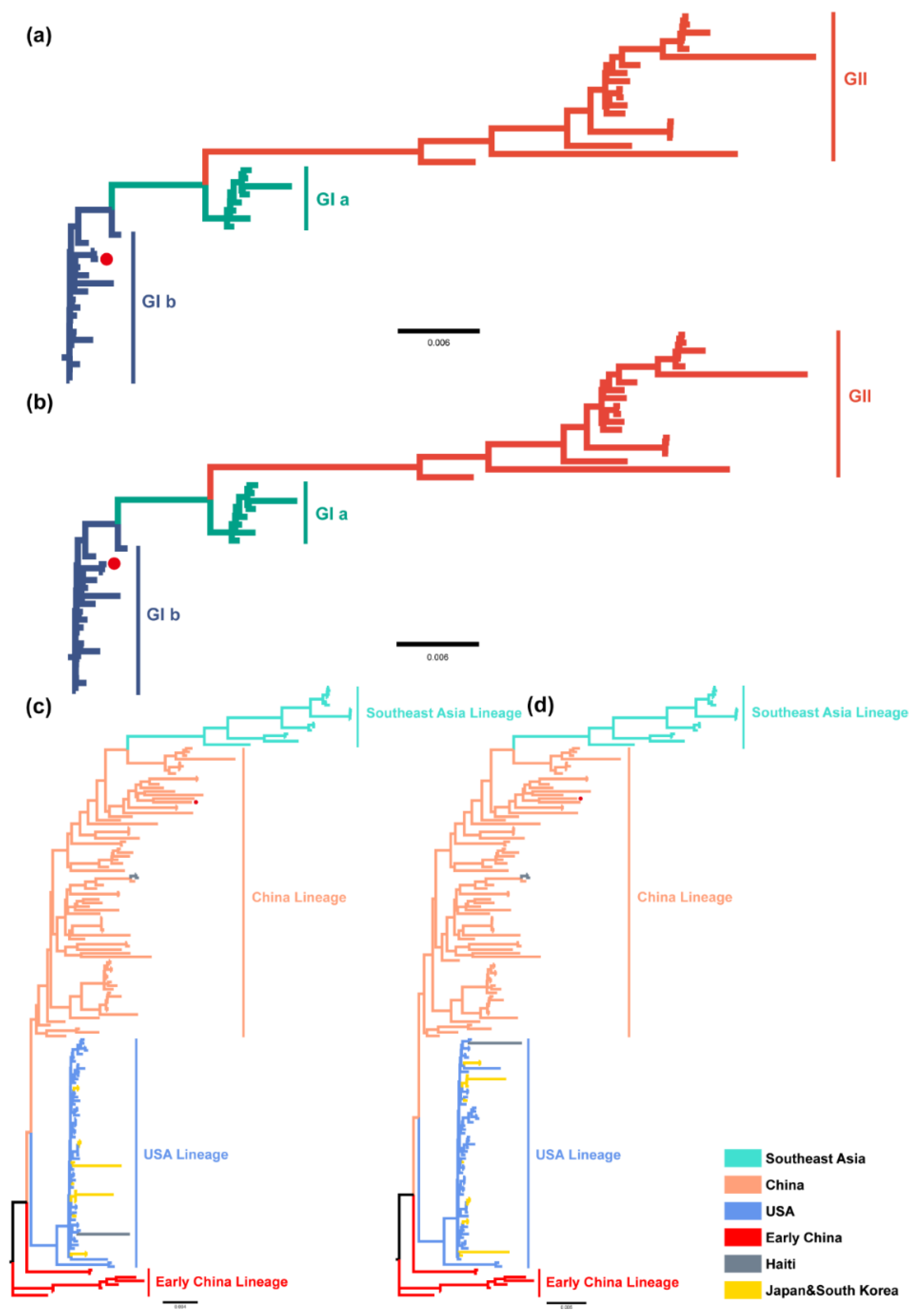
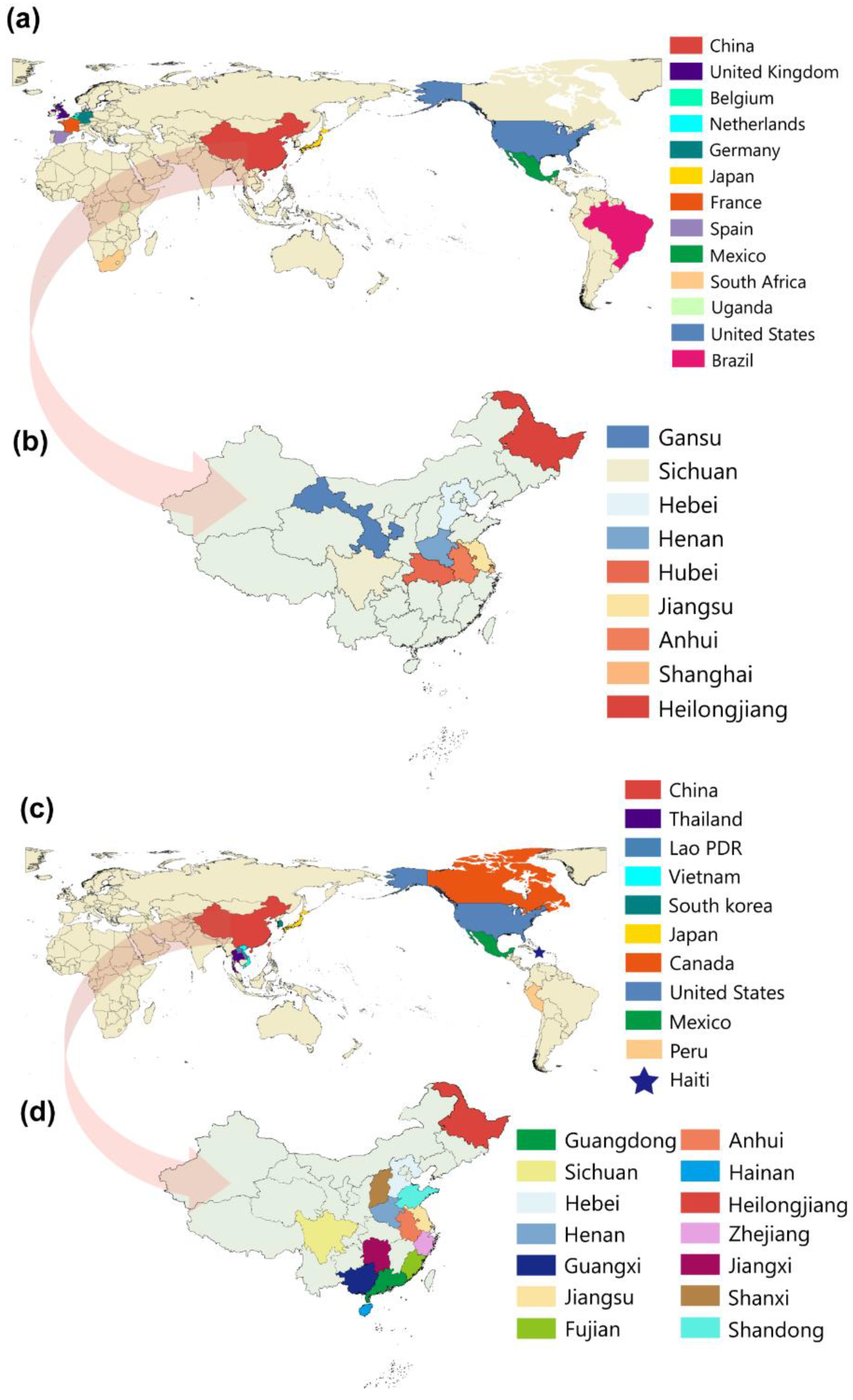
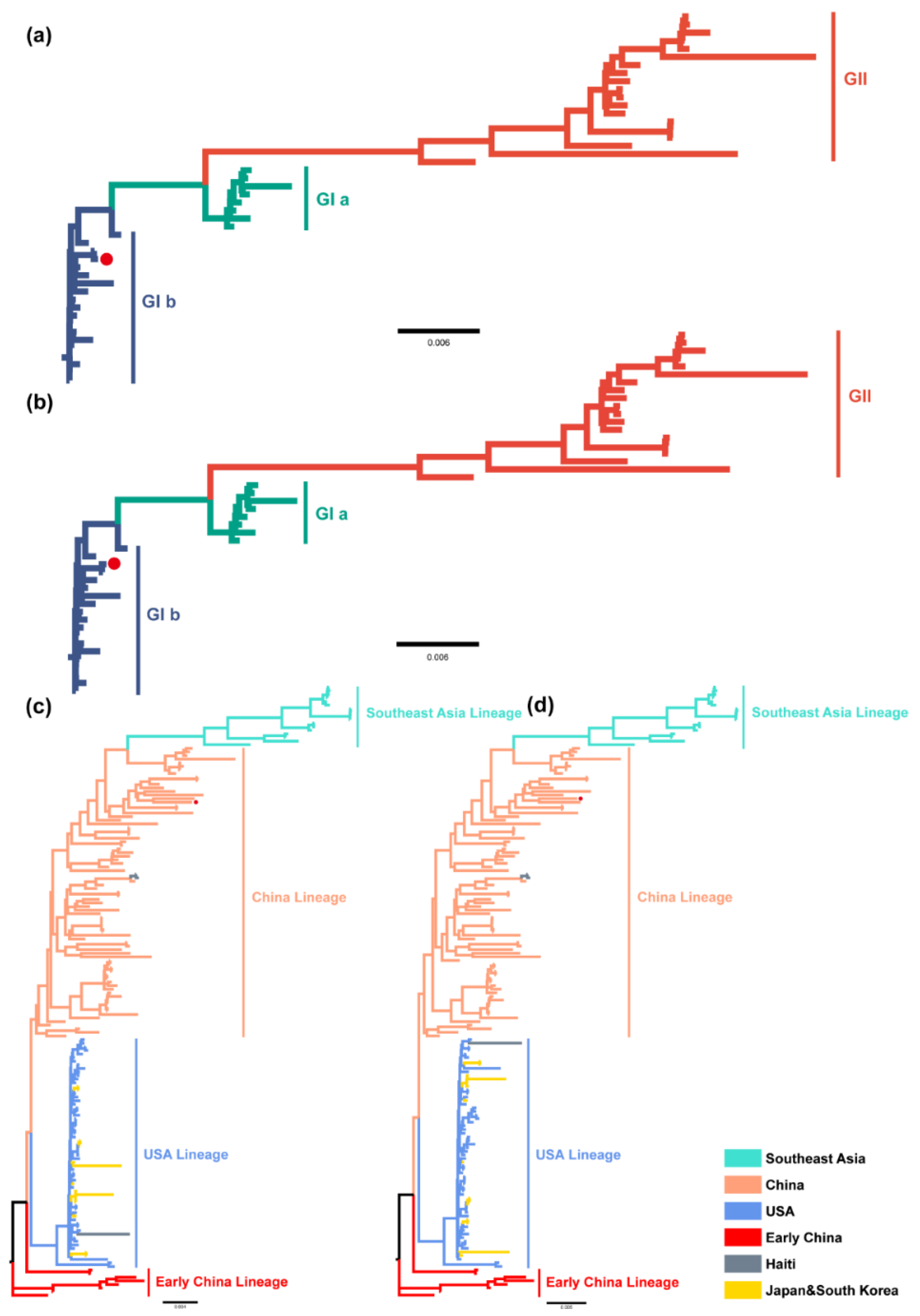
2.2. Distribution and Phylogenetic Analysis of TGEV and PDCoV
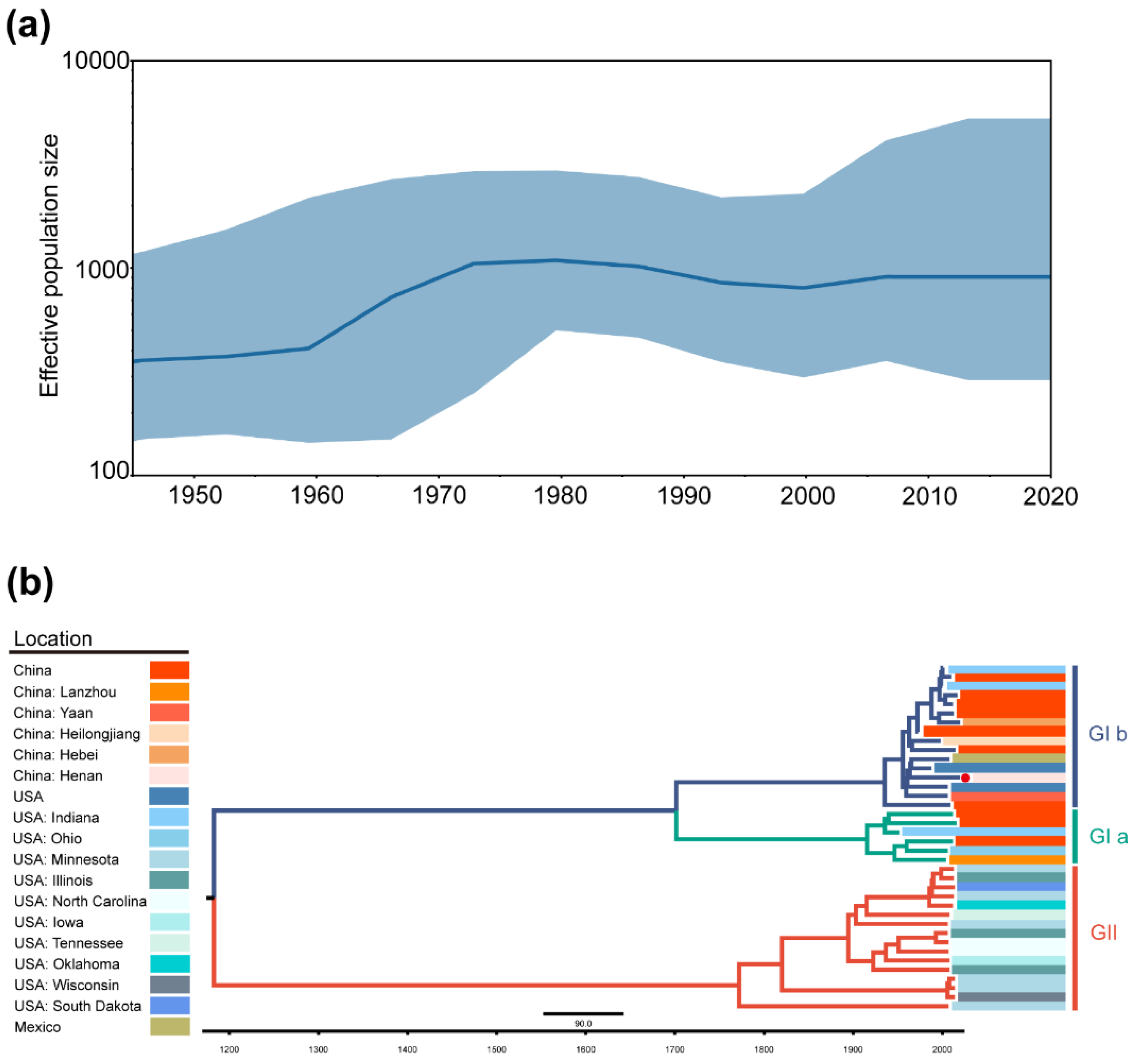
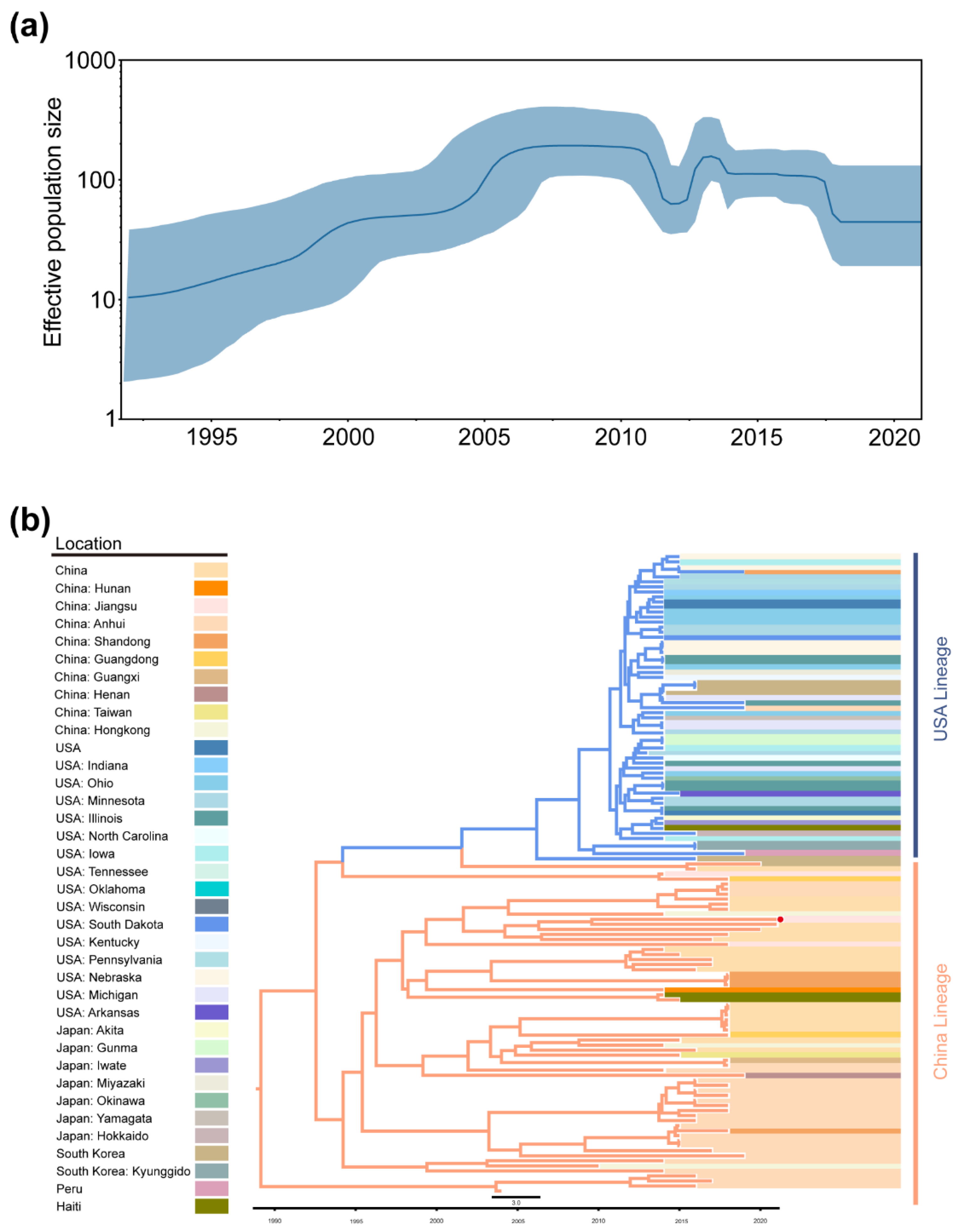
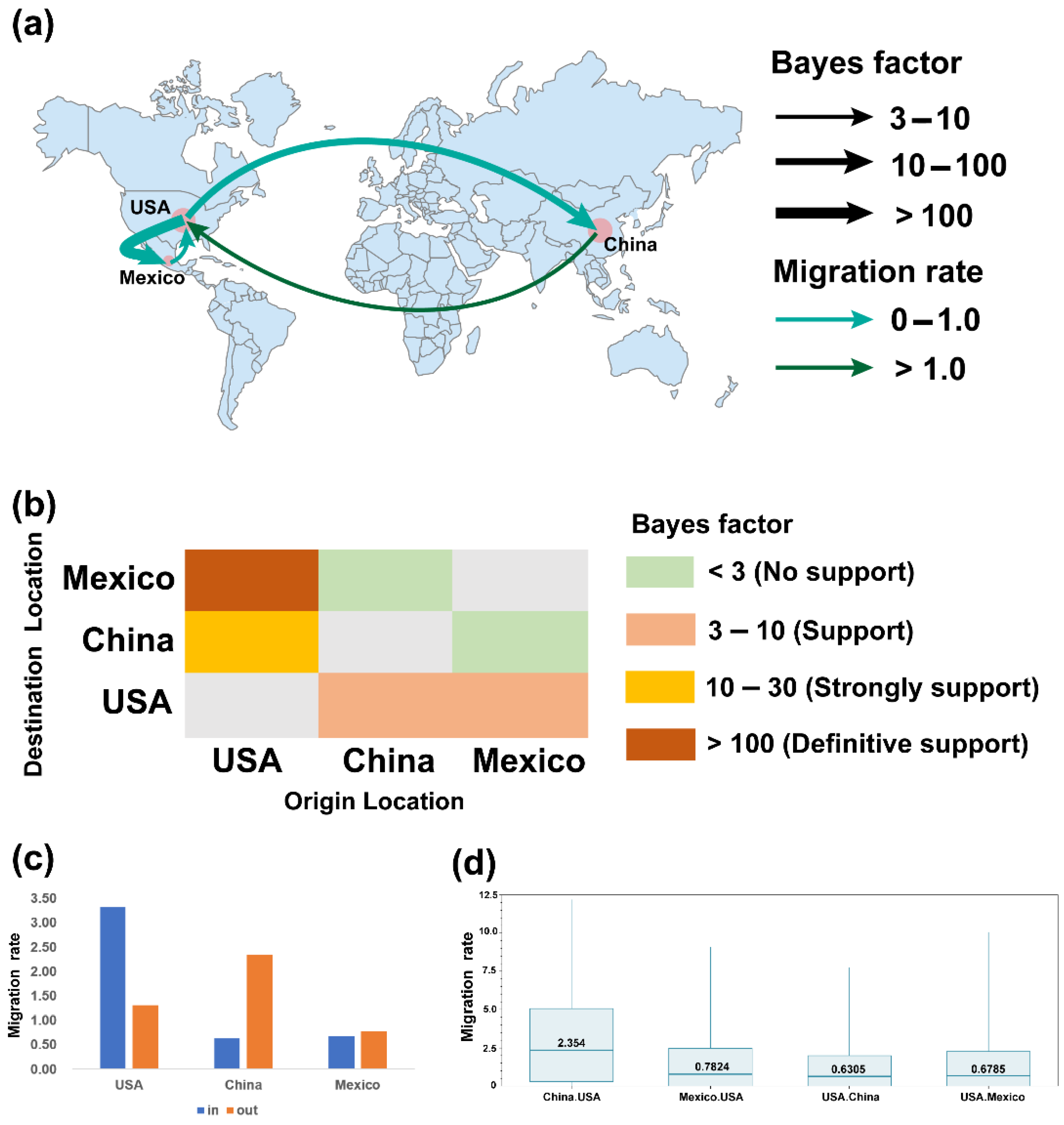
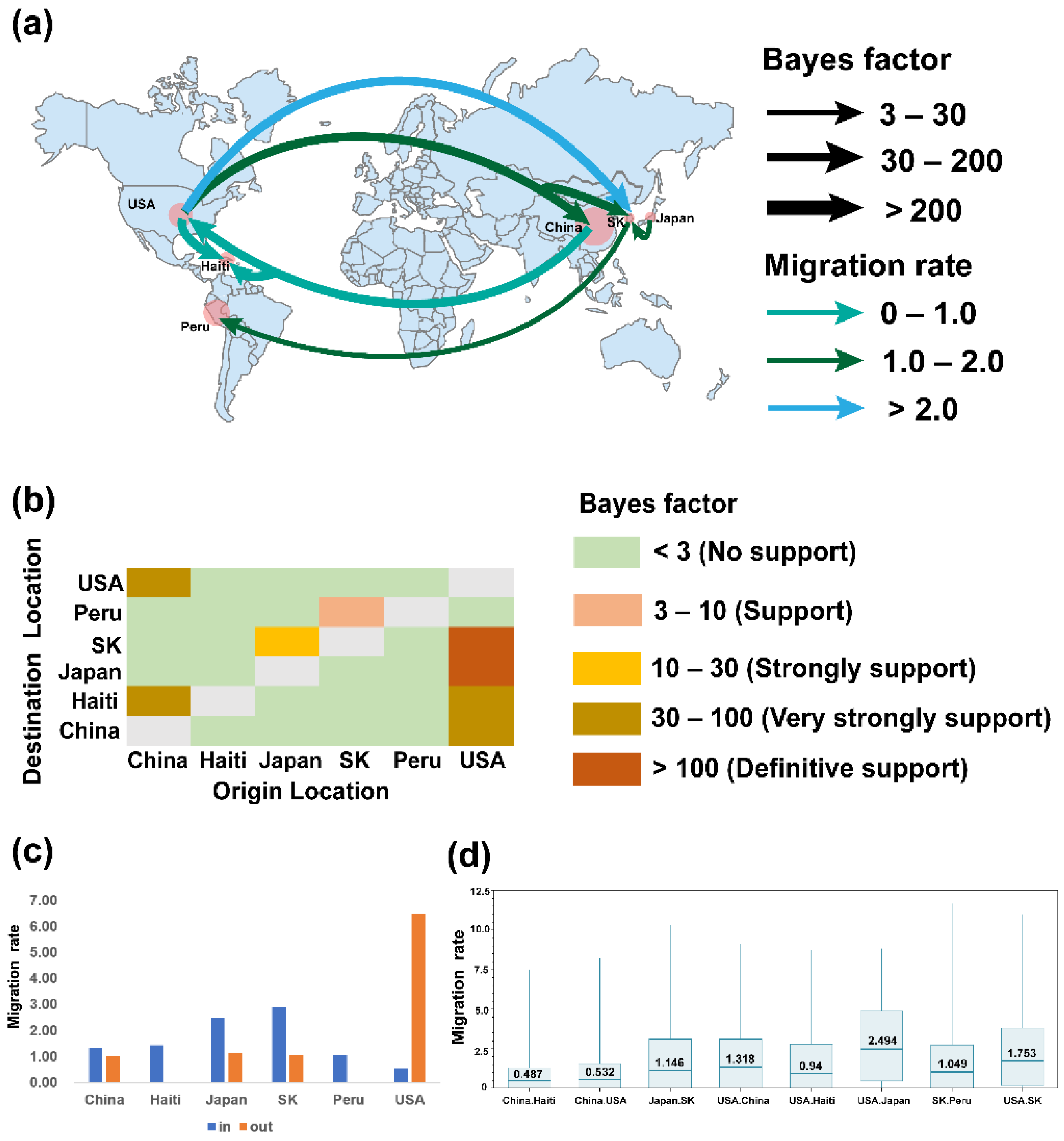
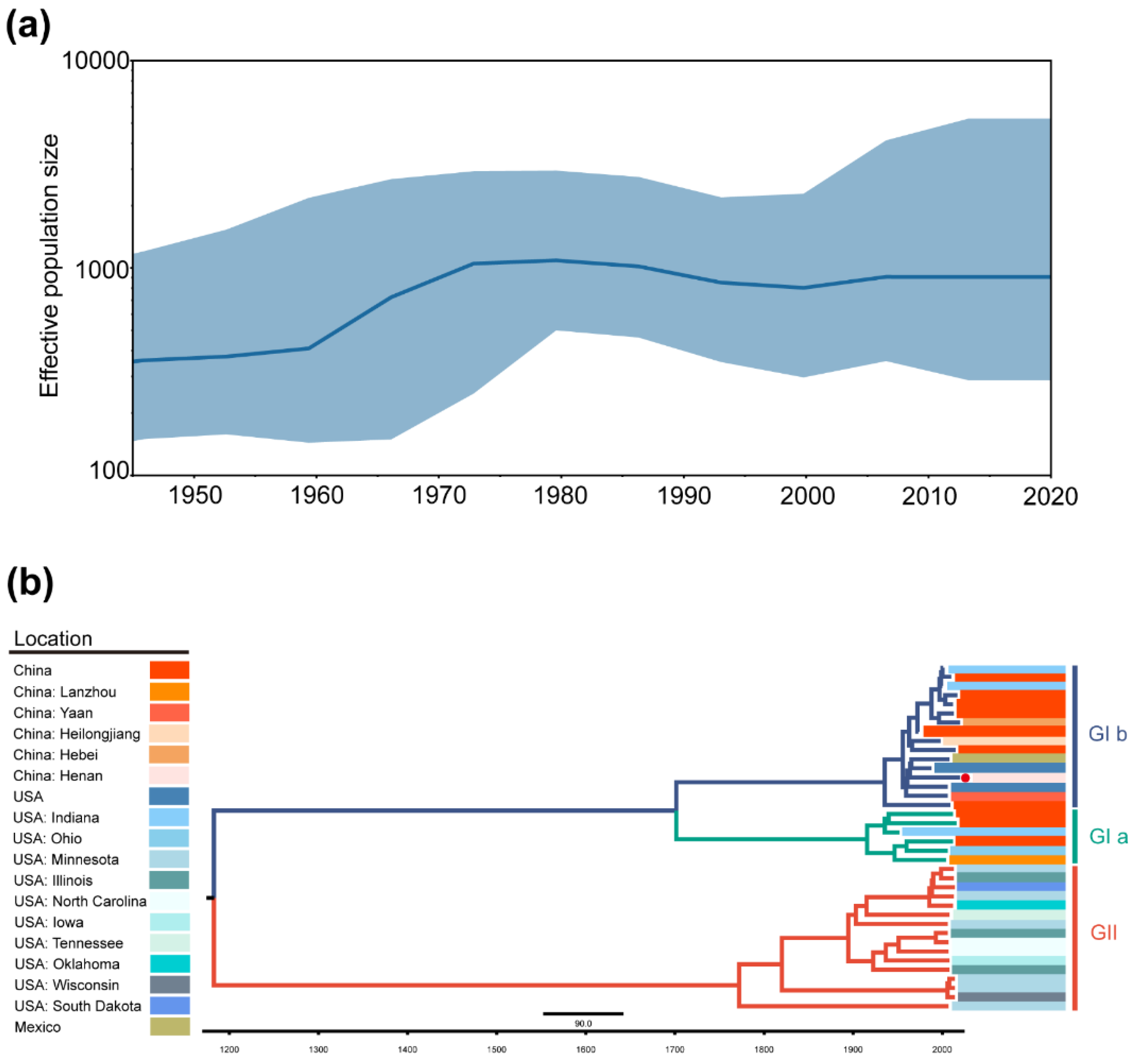
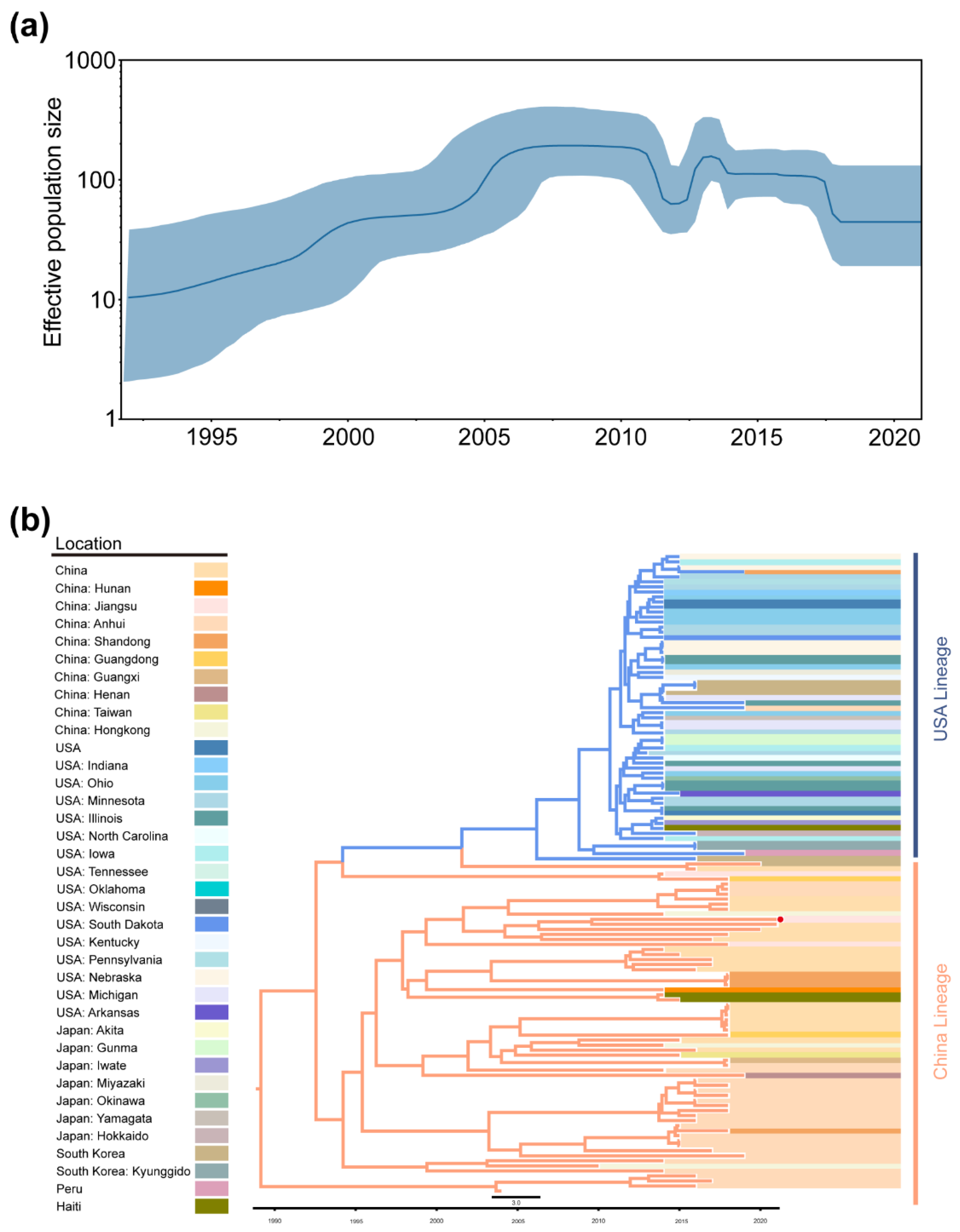
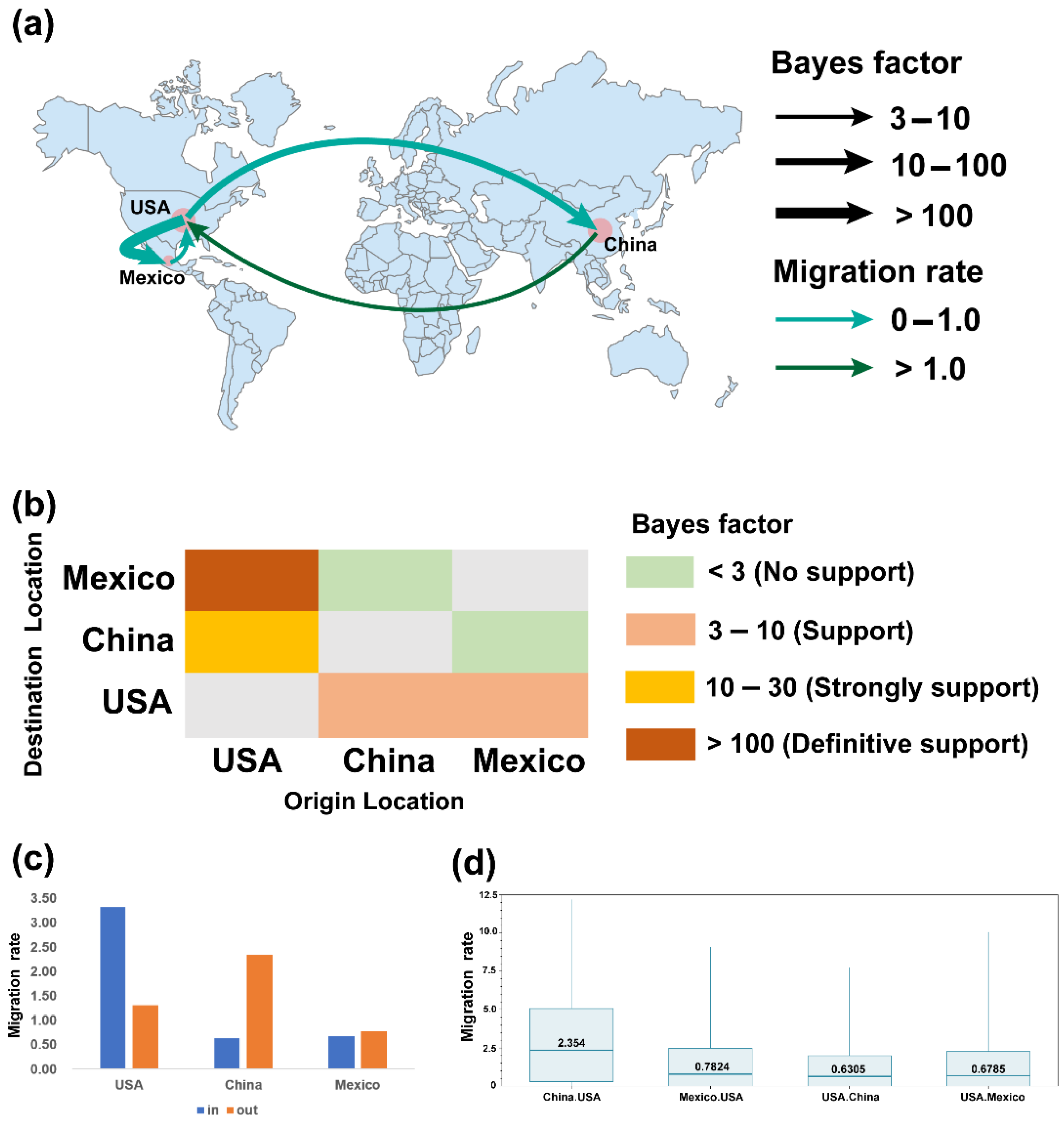
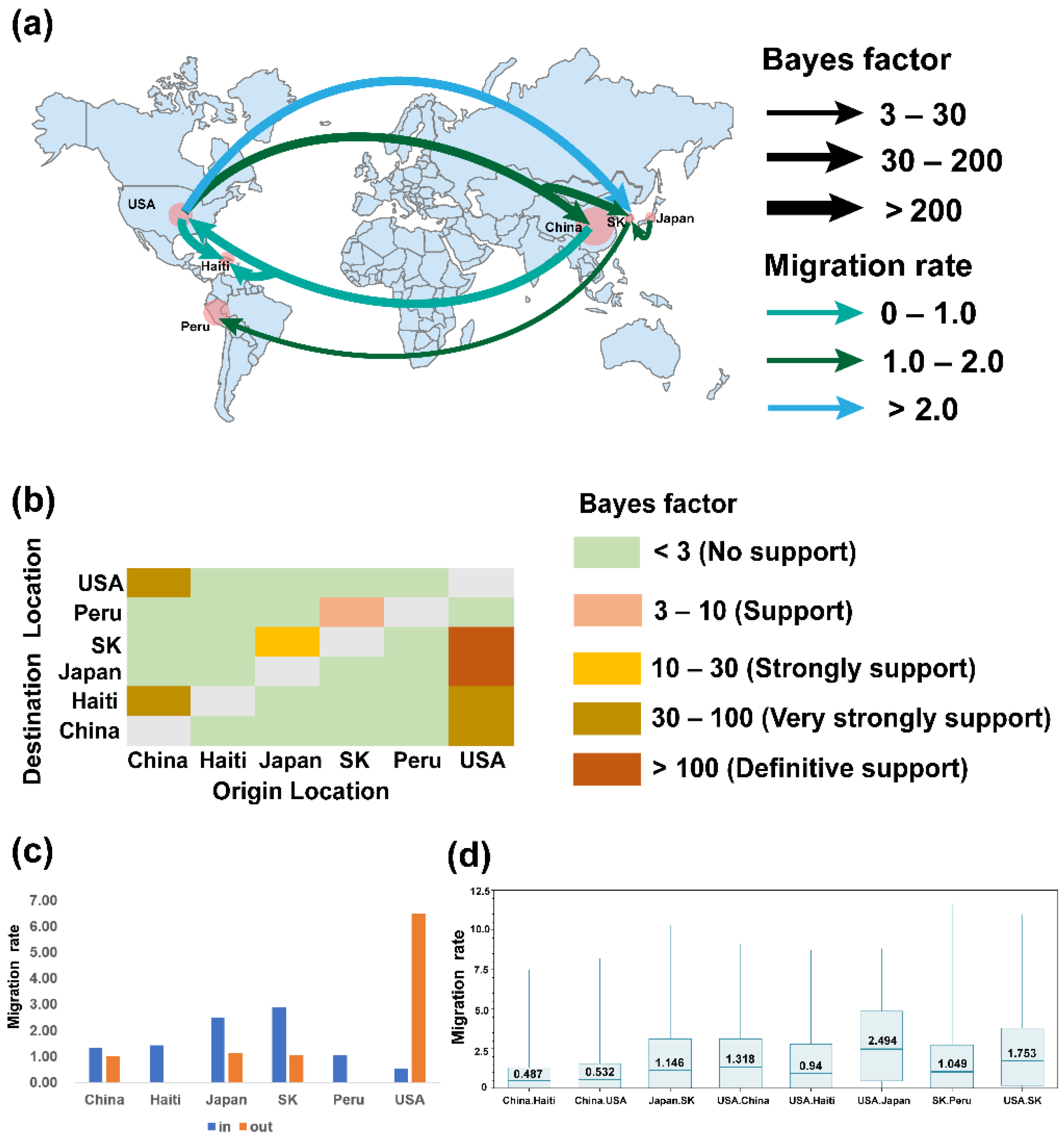
2.3. Phylogeographic Inference of TGEV and PDCoV
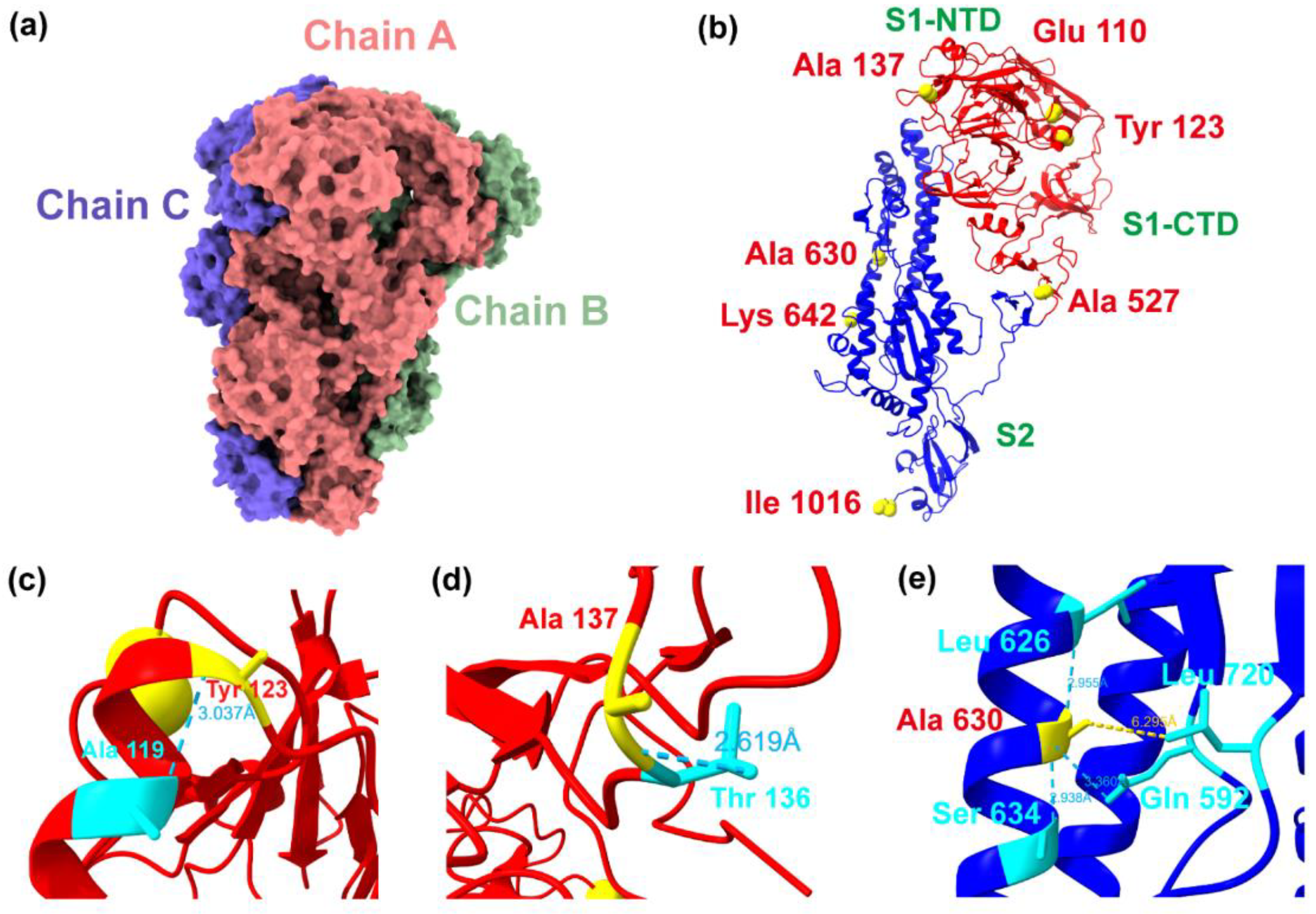
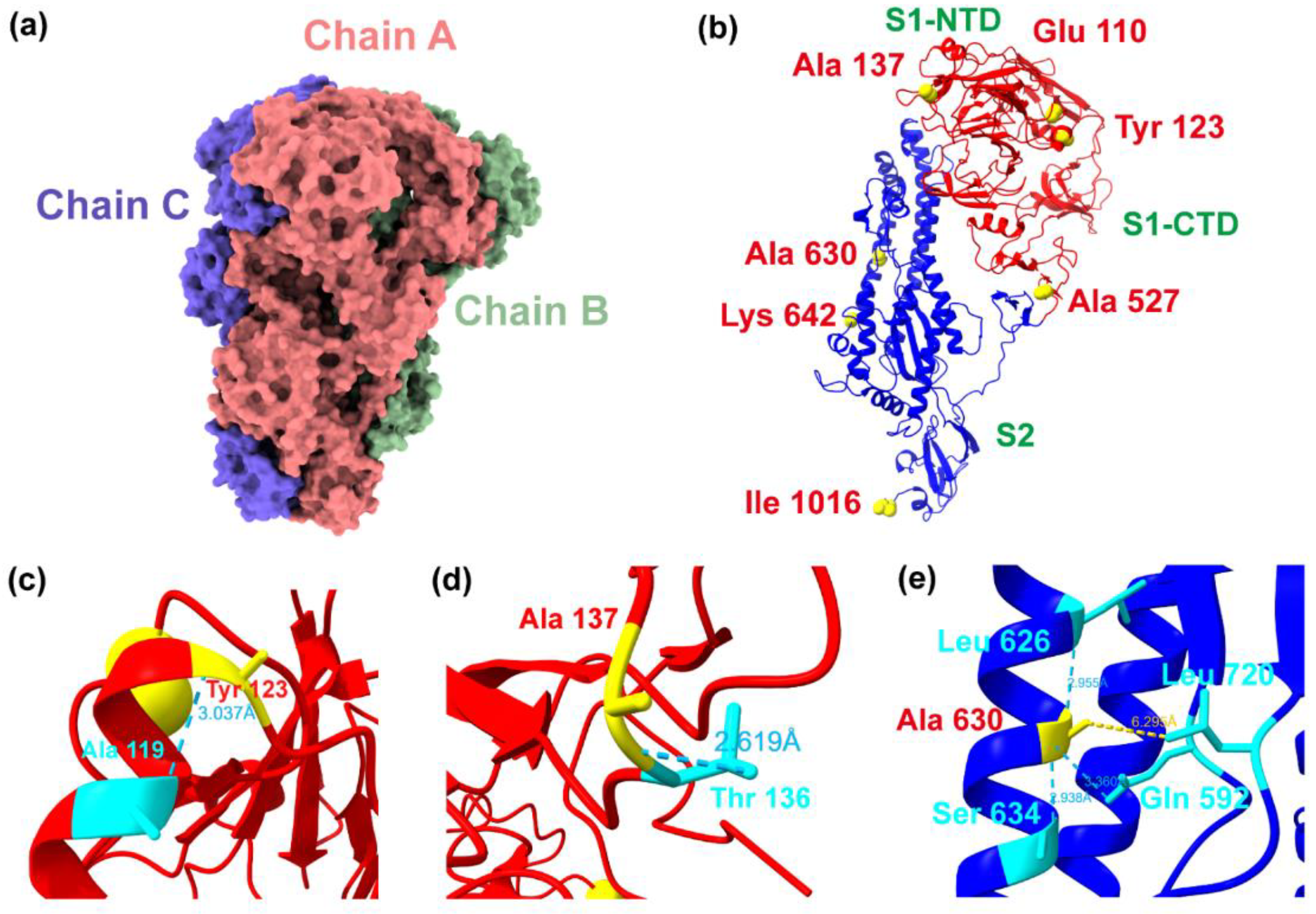
2.4. Adaptive Evolution Sites and Structural Analysis of the S Protein
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Virus Isolation
4.2. Viral RNA Extraction and Genome Sequencing
4.3. Sequence Analysis and Phylogenetic Analysis
4.4. Phylogeographic Analyses of TGEV and PDCoV
4.5. S protein Positive Selection and Functional Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef]
- Sila, T.; Sunghan, J.; Laochareonsuk, W.; Surasombatpattana, S.; Kongkamol, C.; Ingviya, T.; Siripaitoon, P.; Kositpantawong, N.; Kanchanasuwan, S.; Hortiwakul, T.; et al. Suspected Cat-to-Human Transmission of SARS-CoV-2, Thailand, July-September 2021. Emerg. Infect. Dis. 2022, 28, 1485–1488. [Google Scholar] [CrossRef]
- Tan, C.C.S.; Lam, S.D.; Richard, D.; Owen, C.J.; Berchtold, D.; Orengo, C.; Nair, M.S.; Kuchipudi, S.V.; Kapur, V.; van Dorp, L.; et al. Transmission of SARS-CoV-2 from humans to animals and potential host adaptation. Nat. Commun. 2022, 13, 2988. [Google Scholar] [CrossRef]
- Barua, A.; Grot, N.; Plawski, A. The basis of mink susceptibility to SARS-CoV-2 infection. J. Appl Genet. 2022, 63, 543–555. [Google Scholar] [CrossRef]
- Yan, Q.; Liu, X.; Sun, Y.; Zeng, W.; Li, Y.; Zhao, F.; Wu, K.; Fan, S.; Zhao, M.; Chen, J.; et al. Swine Enteric Coronavirus: Diverse Pathogen-Host Interactions. Int. J. Mol. Sci. 2022, 23, 3953. [Google Scholar] [CrossRef]
- Thakor, J.C.; Dinesh, M.; Manikandan, R.; Bindu, S.; Sahoo, M.; Sahoo, D.; Dhawan, M.; Pandey, M.K.; Tiwari, R.; Emran, T.B.; et al. Swine coronaviruses (SCoVs) and their emerging threats to swine population, inter-species transmission, exploring the susceptibility of pigs for SARS-CoV-2 and zoonotic concerns. Vet. Q. 2022, 42, 125–147. [Google Scholar] [CrossRef]
- Chen, F.; Knutson, T.P.; Rossow, S.; Saif, L.J.; Marthaler, D.G. Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Sci. Rep. 2019, 9, 3953. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Fukai, K.; Ichikawa, Y.; Okuda, Y.; Shibata, I.; Motoyama, C.; Imai, K.; Kirisawa, R. Virological and serological studies of porcine respiratory coronavirus infection on a Japanese farm. J. Vet. Med. Sci. 2008, 70, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Rasschaert, D.; Duarte, M.; Laude, H. Porcine respiratory coronavirus differs from transmissible gastroenteritis virus by a few genomic deletions. J. Gen. Virol. 1990, 71, 2599–2607. [Google Scholar] [CrossRef] [PubMed]
- Akimkin, V.; Beer, M.; Blome, S.; Hanke, D.; Höper, D.; Jenckel, M.; Pohlmann, A. New Chimeric Porcine Coronavirus in Swine Feces, Germany, 2012. Emerg. Infect. Dis. 2016, 22, 1314–1315. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg. Infect. Dis. 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Vlasova, A.N.; Chepngeno, J.; Lu, Z.; Wang, Q.; Saif, L.J. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J. Clin. Microbiol. 2015, 53, 1537–1548. [Google Scholar] [CrossRef]
- Ajayi, T.; Dara, R.; Misener, M.; Pasma, T.; Moser, L.; Poljak, Z. Herd-level prevalence and incidence of porcine epidemic diarrhoea virus (PEDV) and porcine deltacoronavirus (PDCoV) in swine herds in Ontario, Canada. Transbound. Emerg. Dis. 2018, 65, 1197–1207. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, H.C.; Nguyen, V.G.; Moon, H.J.; Kim, H.K.; Park, S.J.; Lee, C.H.; Lee, G.E.; Park, B.K. Detection and Phylogenetic Analysis of Porcine Deltacoronavirus in Korean Swine Farms, 2015. Transbound. Emerg. Dis. 2016, 63, 248–252. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Wong, E.Y.M.; Tsang, C.C.; Ahmed, S.S.; Au-Yeung, R.K.H.; Yuen, K.Y.; Wernery, U.; Woo, P.C.Y. Discovery and Sequence Analysis of Four Deltacoronaviruses from Birds in the Middle East Reveal Interspecies Jumping with Recombination as a Potential Mechanism for Avian-to-Avian and Avian-to-Mammalian Transmission. J. Virol. 2018, 92, e00265-18. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Chen, Y.; Zhu, X.; Guo, J.; Da, X.; Hou, Z.; Xu, S.; Zhou, J.; Fang, L.; Wang, D.; et al. Cross-Species Transmission of Deltacoronavirus and the Origin of Porcine Deltacoronavirus. Evol. Appl. 2020, 13, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Tsang, C.C.; Lau, C.C.; Wong, P.C.; Chow, F.W.; Fong, J.Y.; Yuen, K.Y. Coronavirus HKU15 in respiratory tract of pigs and first discovery of coronavirus quasispecies in 5’-untranslated region. Emerg. Microbes. Infect. 2017, 6, e53. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.A.; Tagliamonte, M.S.; White, S.K.; Elbadry, M.A.; Alam, M.M.; Stephenson, C.J.; Bonny, T.S.; Loeb, J.C.; Telisma, T.; Chavannes, S.; et al. Independent infections of porcine deltacoronavirus among Haitian children. Nature 2021, 600, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Tortorici, M.A.; Snijder, J.; Yoshioka, C.; Walls, A.C.; Li, W.; McGuire, A.T.; Rey, F.A.; Bosch, B.-J.; Veesler, D.; et al. Glycan Shield and Fusion Activation of a Deltacoronavirus Spike Glycoprotein Fine-Tuned for Enteric Infections. J. Virol. 2018, 92, e01628-17. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Alexander, F.C. Experiences with African swine fever in Haiti. Ann. N. Y. Acad. Sci. 1992, 653, 251–256. [Google Scholar] [CrossRef]
- Nelson, M.I.; Viboud, C.; Vincent, A.L.; Culhane, M.R.; Detmer, S.E.; Wentworth, D.E.; Rambaut, A.; Suchard, M.A.; Holmes, E.C.; Lemey, P. Global migration of influenza A viruses in swine. Nat. Commun. 2015, 6, 6696. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, X.; Lemey, P.; Suchard, M.A.; Bi, Y.; Shi, W.; Liu, D.; Qi, W.; Zhang, G.; Stenseth, N.C.; et al. Assessing the role of live poultry trade in community-structured transmission of avian influenza in China. Proc. Natl. Acad. Sci. USA 2020, 117, 5949–5954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, C.; Du, L.; Jiang, S.; Shi, Z.; Baric, R.S.; Li, F. Two Mutations Were Critical for Bat-to-Human Transmission of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2015, 89, 9119–9123. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wu, H.; Chen, Z. Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective. Int J. Mol. Sci. 2020, 21, 7898. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Shan, N.; Wang, X.; Lin, S.; Ma, K.; Li, B.; Li, H.; Liao, M.; Qi, W. Genetic diversity, phylogeography, and evolutionary dynamics of highly pathogenic avian influenza A (H5N6) viruses. Virus Evol. 2020, 6, veaa079. [Google Scholar] [CrossRef]
- Schrauwen, E.J.; Fouchier, R.A. Host adaptation and transmission of influenza A viruses in mammals. Emerg. Microbes. Infect. 2014, 3, e9. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl Acad Sci USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Li, G.; Zhai, S.L.; Zhou, X.; Chen, T.B.; Niu, J.W.; Xie, Y.S.; Si, G.B.; Cong, F.; Chen, R.A.; He, D.S. Phylogeography and evolutionary dynamics analysis of porcine delta-coronavirus with host expansion to humans. Transbound. Emerg. Dis. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic. Acids. Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Wu, K.; Zeng, W.; Yu, S.; Li, Y.; Sun, Y.; Liu, X.; Ruan, Y.; Huang, J.; Ding, H.; et al. Historical Evolutionary Dynamics and Phylogeography Analysis of Transmissible Gastroenteritis Virus and Porcine Deltacoronavirus: Findings from 59 Suspected Swine Viral Samples from China. Int. J. Mol. Sci. 2022, 23, 9786. https://doi.org/10.3390/ijms23179786
Yan Q, Wu K, Zeng W, Yu S, Li Y, Sun Y, Liu X, Ruan Y, Huang J, Ding H, et al. Historical Evolutionary Dynamics and Phylogeography Analysis of Transmissible Gastroenteritis Virus and Porcine Deltacoronavirus: Findings from 59 Suspected Swine Viral Samples from China. International Journal of Molecular Sciences. 2022; 23(17):9786. https://doi.org/10.3390/ijms23179786
Chicago/Turabian StyleYan, Quanhui, Keke Wu, Weijun Zeng, Shu Yu, Yuwan Li, Yawei Sun, Xiaodi Liu, Yang Ruan, Juncong Huang, Hongxing Ding, and et al. 2022. "Historical Evolutionary Dynamics and Phylogeography Analysis of Transmissible Gastroenteritis Virus and Porcine Deltacoronavirus: Findings from 59 Suspected Swine Viral Samples from China" International Journal of Molecular Sciences 23, no. 17: 9786. https://doi.org/10.3390/ijms23179786
APA StyleYan, Q., Wu, K., Zeng, W., Yu, S., Li, Y., Sun, Y., Liu, X., Ruan, Y., Huang, J., Ding, H., Yi, L., Zhao, M., Chen, J., & Fan, S. (2022). Historical Evolutionary Dynamics and Phylogeography Analysis of Transmissible Gastroenteritis Virus and Porcine Deltacoronavirus: Findings from 59 Suspected Swine Viral Samples from China. International Journal of Molecular Sciences, 23(17), 9786. https://doi.org/10.3390/ijms23179786