
Bioinformatic Analysis of Ixodes ricinus Long Non-Coding RNAs Predicts Their Binding Ability of Host miRNAs

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
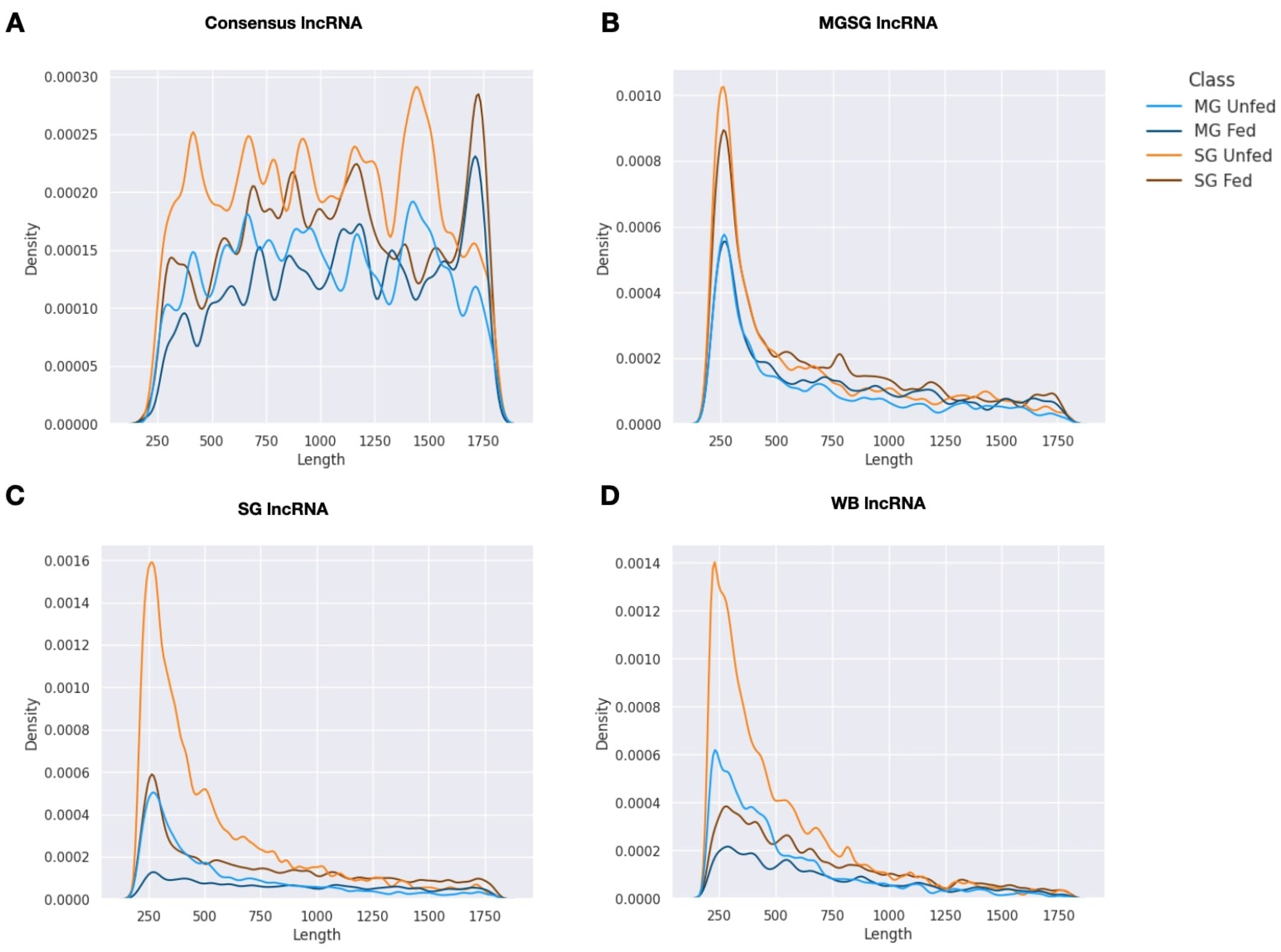
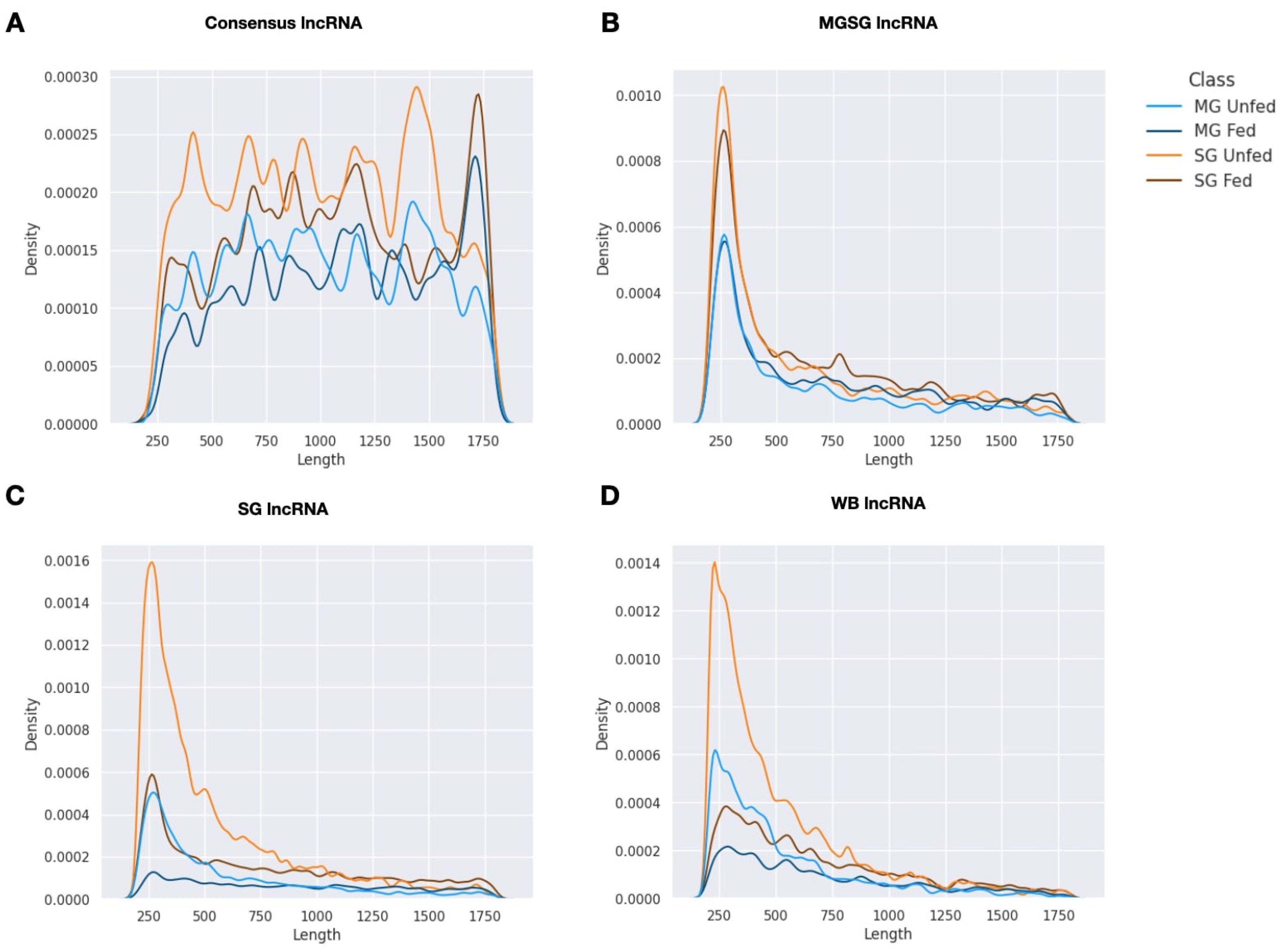
2.1. Summary of Transcriptome Assembly and lncRNA Isolation
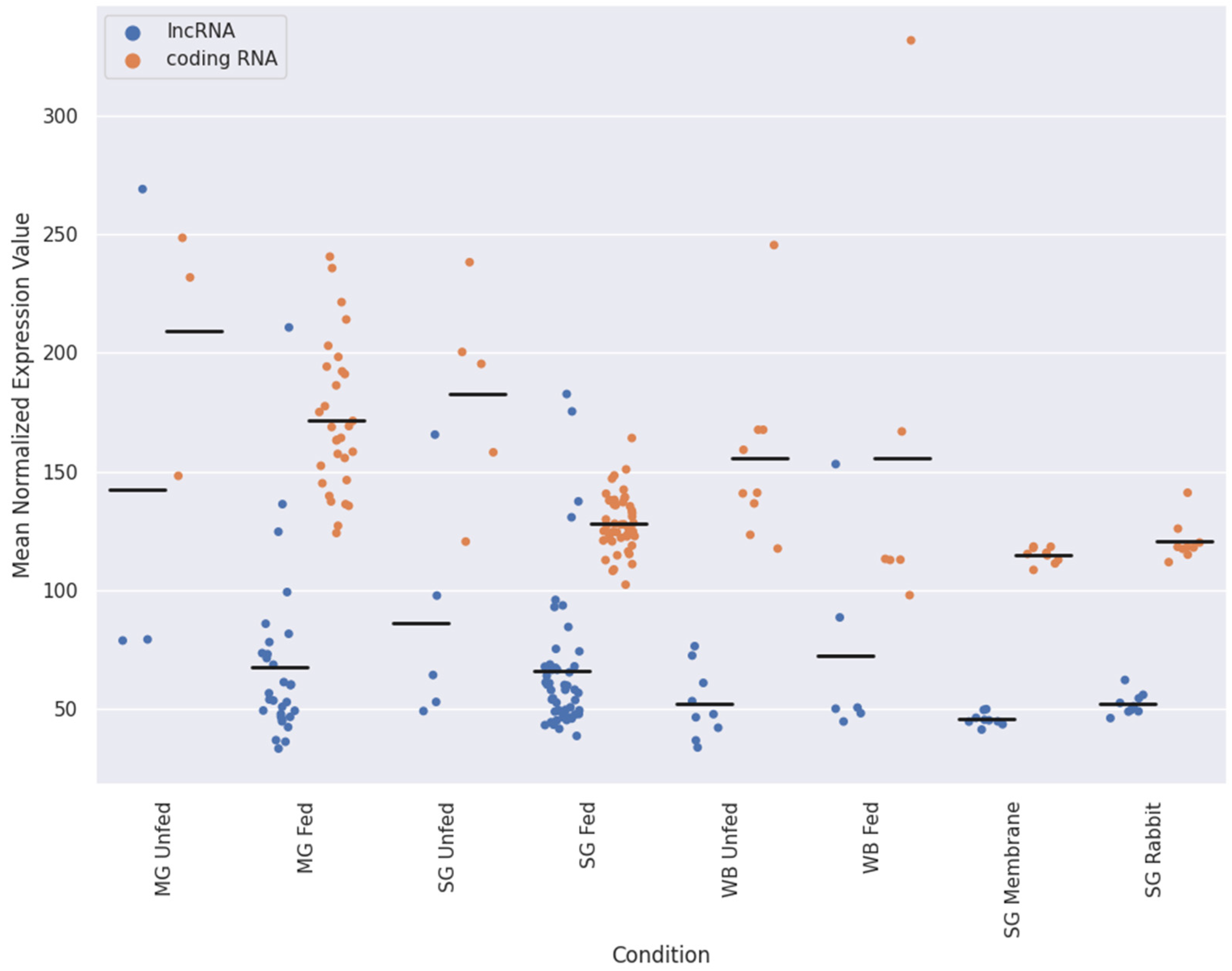
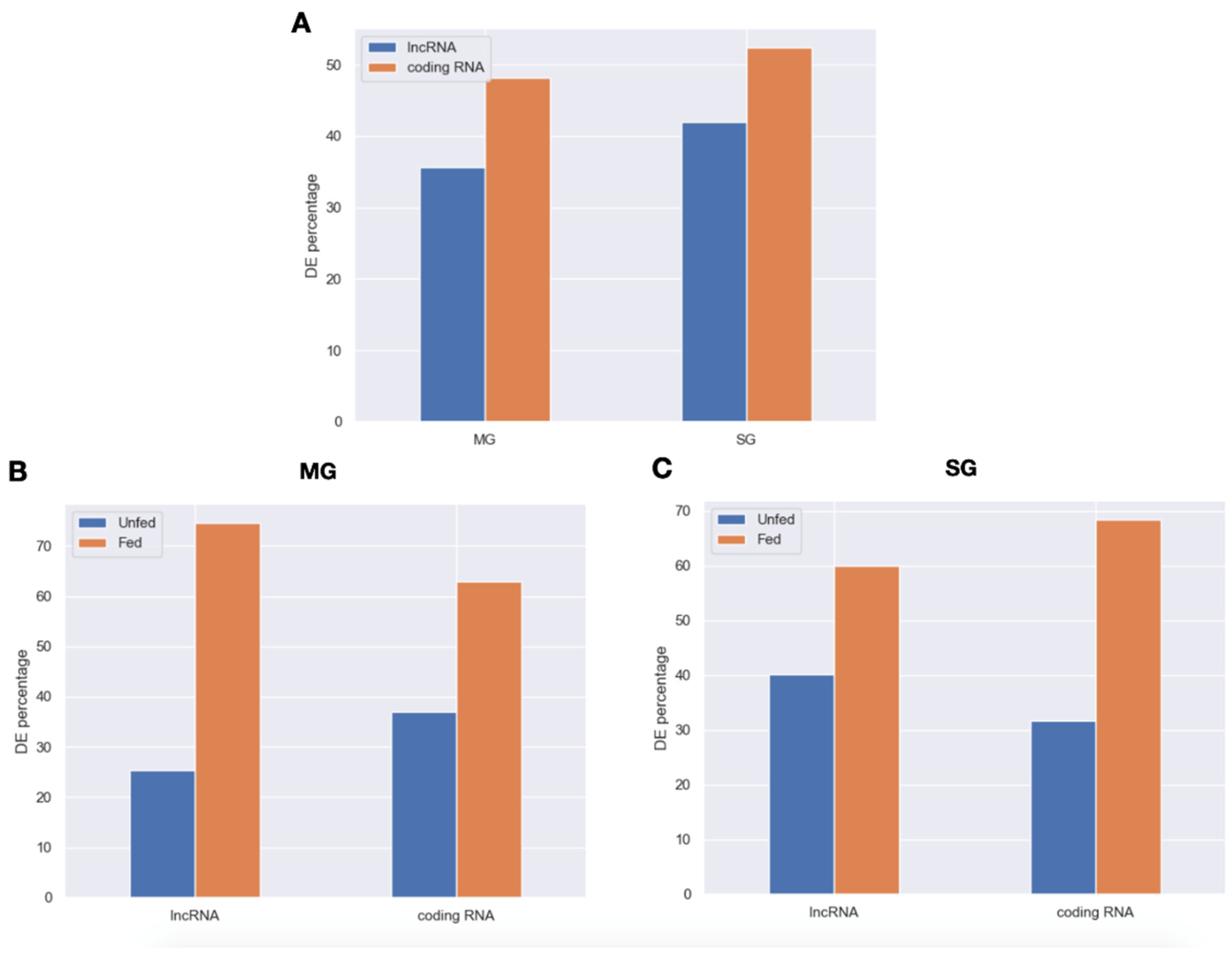
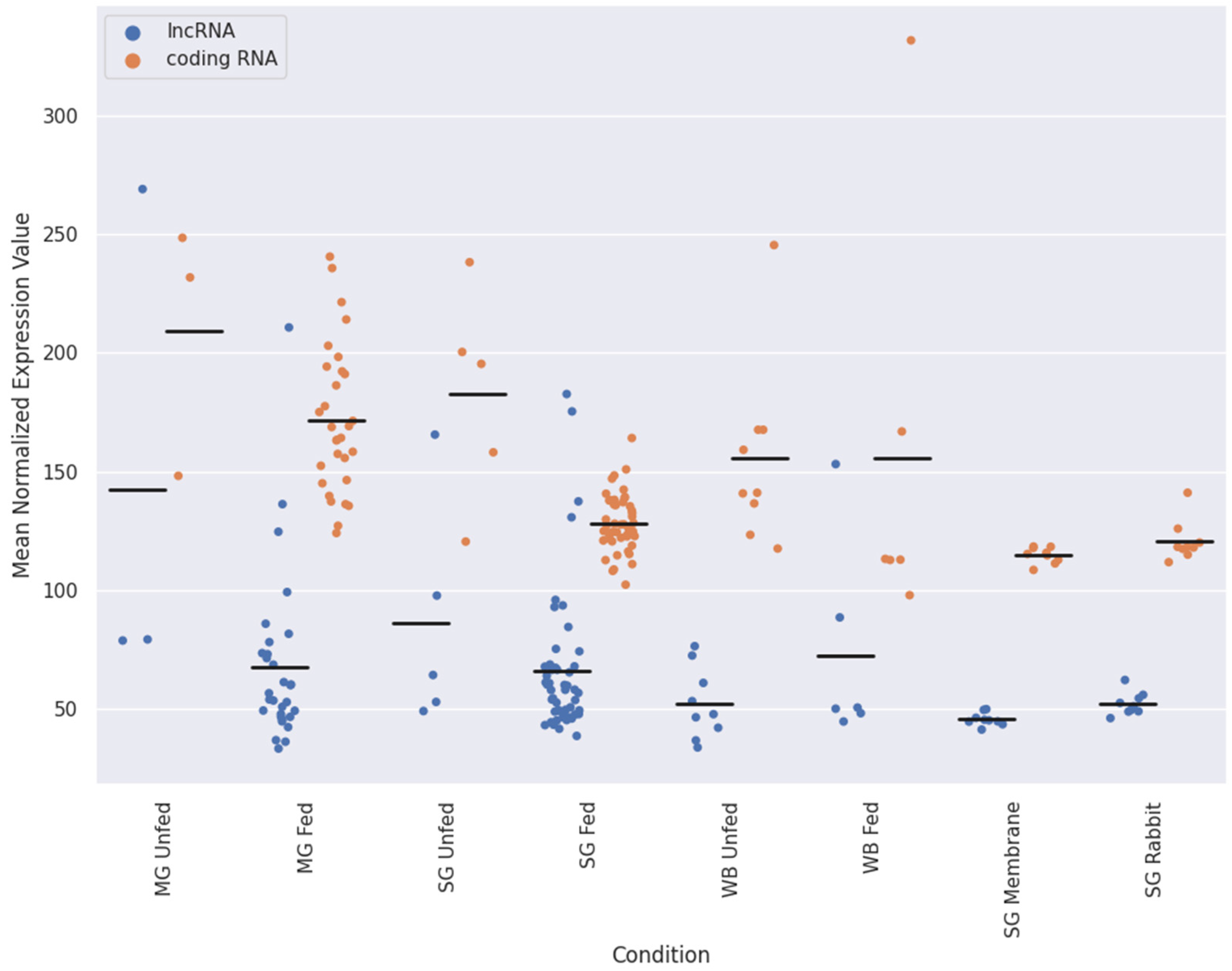
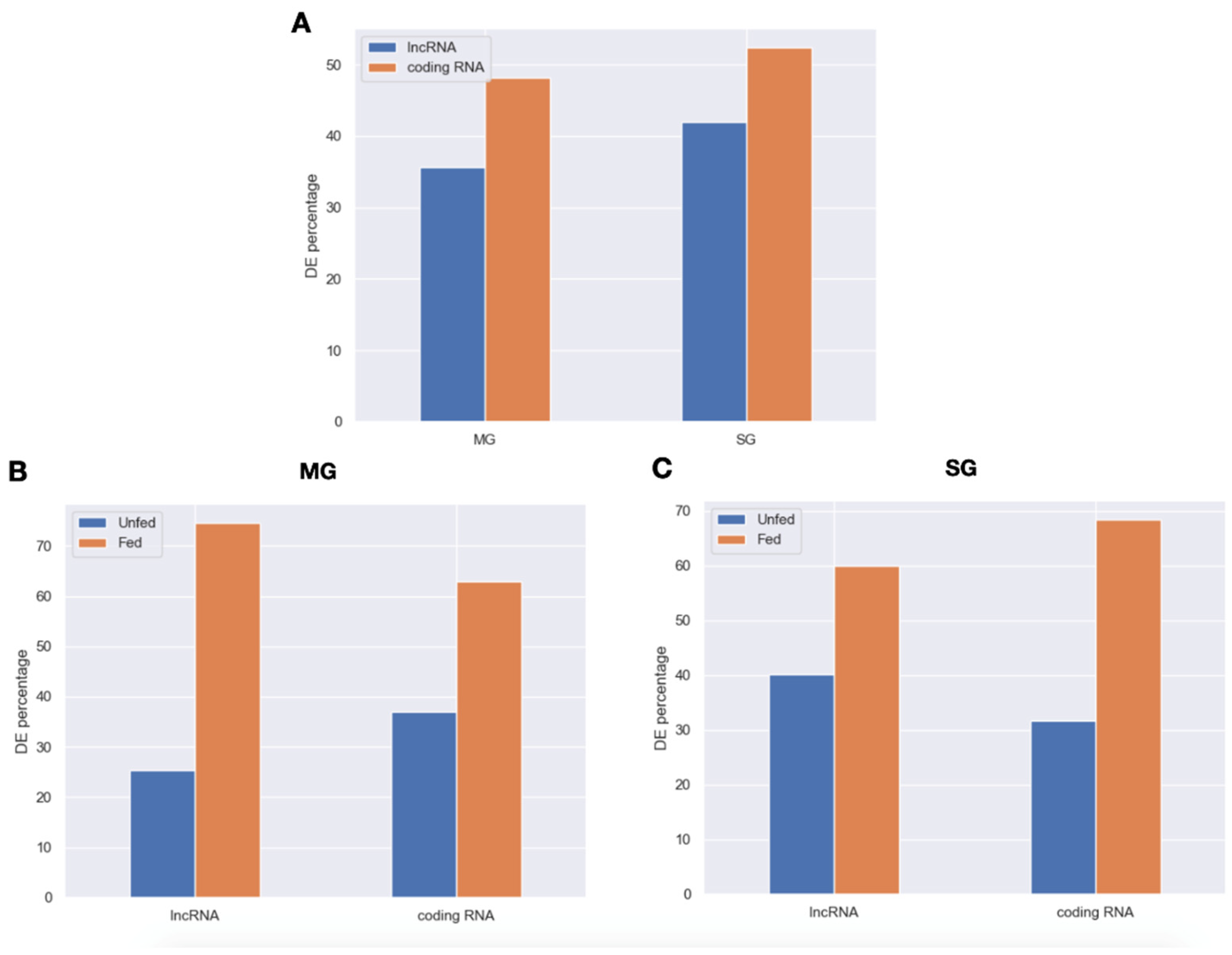
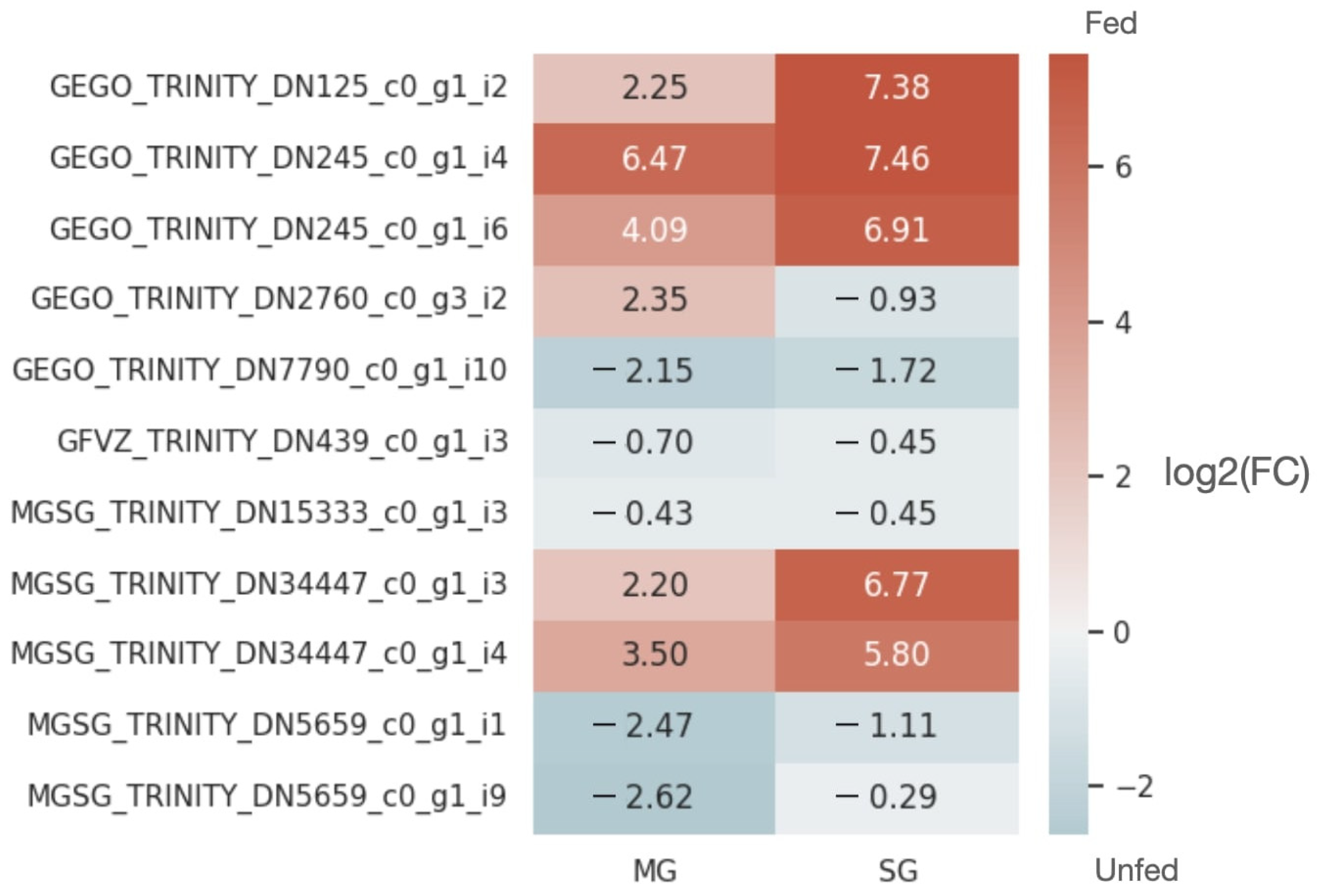
2.2. Expression Patterns of lncRNAs in the Midgut and Salivary Glands under Different Feeding Treatments
2.3. Target Prediction and KEGG Analysis of the Consensus Differentially Expressed lncRNAs
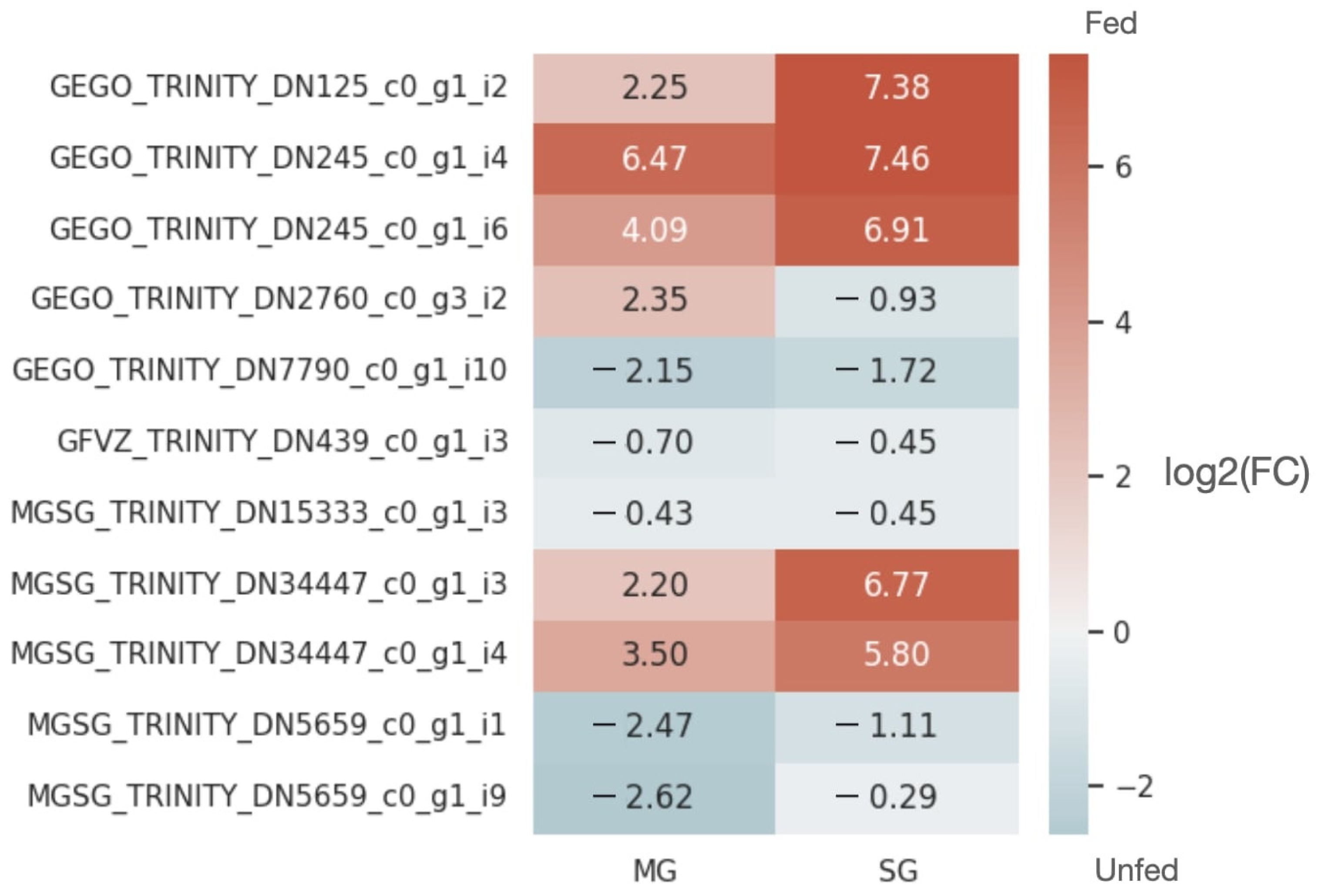
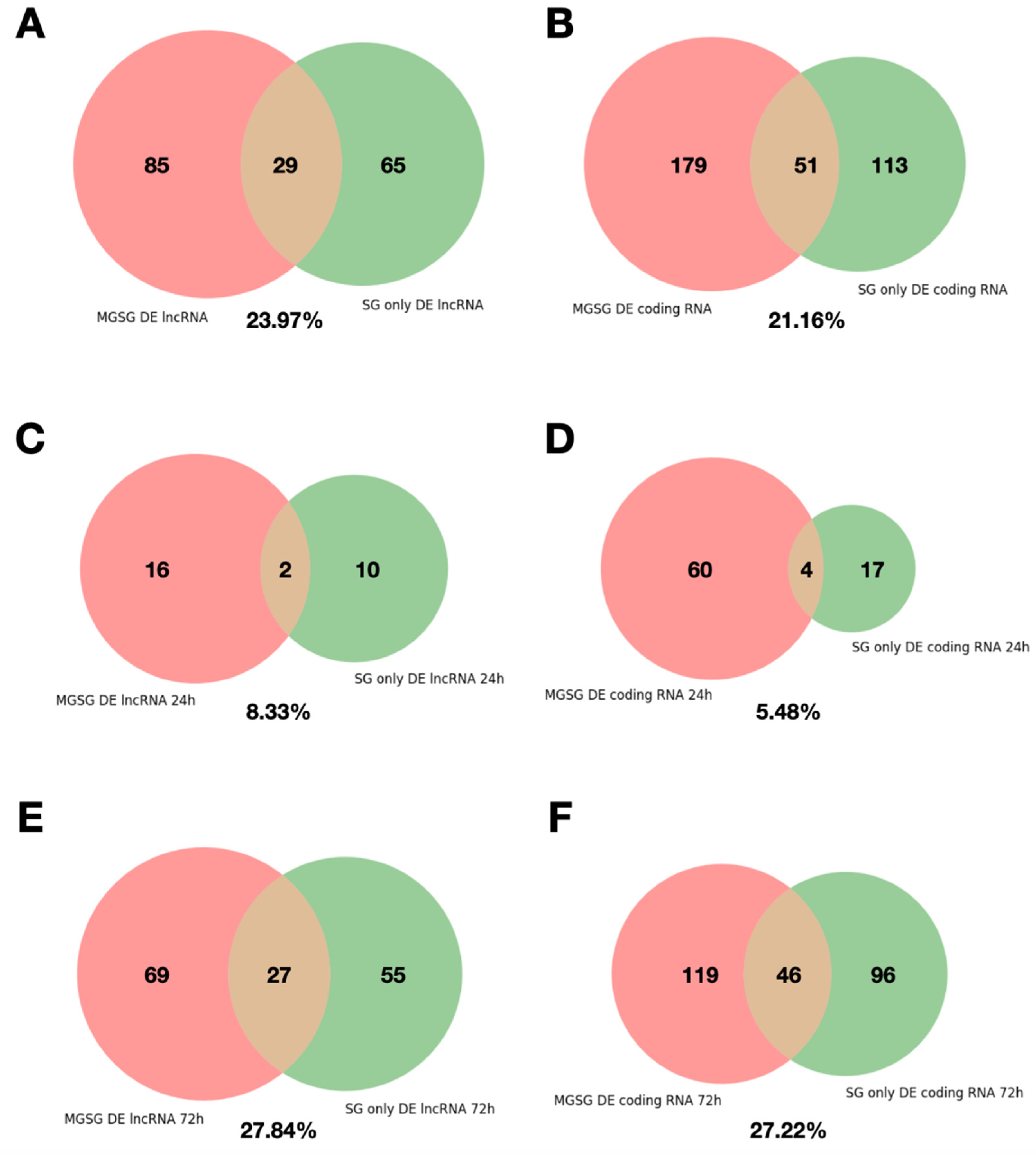
2.4. Analysis of the Reproducibility of the Differential Expression of lncRNAs
2.5. Prediction of Sponge Candidates and Functional Analysis
3. Discussion
4. Materials and Methods
4.1. Transcriptome Assembly, Expression Analysis and Consensus lncRNA Isolation
4.2. Differential Expression Analysis
4.3. Target Prediction
4.4. Functional In Silico Analysis of Differentially Expressed lncRNAs
4.5. Sponge Candidates and Functional In Silico Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chmelar, J.; Calvo, E.; Pedra, J.H.F.; Francischetti, I.M.B.; Kotsyfakis, M. Tick Salivary Secretion as a Source of Antihemostatics. J. Proteom. 2012, 75, 3842. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.N.; Chlastáková, A.; Jmel, M.A.; Iliaki-Giannakoudaki, E.; Chmelař, J.; Kotsyfakis, M. Serpins in Tick Physiology and Tick-Host Interaction. Front. Cell. Infect. Microbiol. 2022, 12, 892770. [Google Scholar] [CrossRef] [PubMed]
- Kotál, J.; Langhansová, H.; Lieskovská, J.; Andersen, J.F.; Francischetti, I.M.B.; Chavakis, T.; Kopecký, J.; Pedra, J.H.F.; Kotsyfakis, M.; Chmelař, J. Modulation of Host Immunity by Tick Saliva. J. Proteom. 2015, 128, 58. [Google Scholar] [CrossRef] [PubMed]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef]
- Chmelař, J.; Kotál, J.; Karim, S.; Kopacek, P.; Francischetti, I.M.B.; Pedra, J.H.F.; Kotsyfakis, M. Sialomes and Mialomes: A Systems-Biology View of Tick Tissues and Tick–Host Interactions. Trends Parasitol. 2016, 32, 242–254. [Google Scholar] [CrossRef]
- De La Fuente, J.; Villar, M.; Estrada-Peña, A.; Olivas, J.A. High Throughput Discovery and Characterization of Tick and Pathogen Vaccine Protective Antigens Using Vaccinomics with Intelligent Big Data Analytic Techniques. Expert Rev. Vaccines 2018, 17, 569–576. [Google Scholar] [CrossRef]
- Wang, H.L.V.; Chekanova, J.A. An Overview of Methodologies in Studying LncRNAs in the High-Throughput Era: When Acronyms ATTACK! Methods Mol. Biol. 2019, 1933, 1–30. [Google Scholar] [CrossRef]
- Dempsey, J.L.; Cui, J.Y. Long Non-Coding RNAs: A Novel Paradigm for Toxicology. Toxicol. Sci. 2017, 155, 3–21. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Mariner, P.D.; Walters, R.D.; Espinoza, C.A.; Drullinger, L.F.; Wagner, S.D.; Kugel, J.F.; Goodrich, J.A. Human Alu RNA Is a Modular Transacting Repressor of MRNA Transcription during Heat Shock. Mol. Cell 2008, 29, 499–509. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA-LncRNA Interactions. Methods Mol. Biol. 2016, 1402, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, P.; Bensaoud, C.; Mekki, I.; Rehman, M.U.; Kotsyfakis, M. Long Non-Coding RNAs and Their Potential Roles in the Vector-Host-Pathogen Triad. Life 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-Wide Identification of Tissue-Specific Long Non-Coding RNA in Three Farm Animal Species 06 Biological Sciences 0604 Genetics. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef]
- Bayer-Santos, E.; Marini, M.M.; da Silveira, J.F. Non-Coding RNAs in Host–Pathogen Interactions: Subversion of Mammalian Cell Functions by Protozoan Parasites. Front. Microbiol. 2017, 8, 474. [Google Scholar] [CrossRef]
- Bensaoud, C.; Hackenberg, M.; Kotsyfakis, M. Noncoding RNAs in Parasite–Vector–Host Interactions. Trends Parasitol. 2019, 35, 715–724. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. SRNAbench and SRNAtoolbox 2019: Intuitive Fast Small RNA Profiling and Differential Expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef]
- Fromm, B.; Høye, E.; Domanska, D.; Zhong, X.; Aparicio-Puerta, E.; Ovchinnikov, V.; Umu, S.U.; Chabot, P.J.; Kang, W.; Aslanzadeh, M.; et al. MirGeneDB 2.1: Toward a Complete Sampling of All Major Animal Phyla. Nucleic Acids Res. 2021, 50, D204–D210. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-MiRPath v3.0: Deciphering MicroRNA Function with Experimental Support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Preusse, M.; Theis, F.J.; Mueller, N.S. MiTALOS v2: Analyzing Tissue Specific MicroRNA Function. PLoS ONE 2016, 11, e0151771. [Google Scholar] [CrossRef]
- Sonenshine, E.D. Biology of Ticks; Oxford University Press: Oxford, NY, USA, 1991. [Google Scholar]
- Sauer, J.R.; McSwain, J.L.; Bowman, A.S.; Essenberg, R.C. Tick Salivary Gland Physiology. Annu. Rev. Entomol. 1995, 40, 245–267. [Google Scholar] [CrossRef]
- Narasimhan, S.; DePonte, K.; Marcantonio, N.; Liang, X.; Royce, T.E.; Nelson, K.F.; Booth, C.J.; Koski, B.; Anderson, J.F.; Kantor, F.; et al. Immunity against Ixodes Scapularis Salivary Proteins Expressed within 24 Hours of Attachment Thwarts Tick Feeding and Impairs Borrelia Transmission. PLoS ONE 2007, 2, e451. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.B.; Mather, T.N.; Ribeiro, J.M.C. Tick Saliva Is a Potent Inhibitor of Endothelial Cell Proliferation and Angiogenesis. Thromb. Haemost. 2005, 94, 167–174. [Google Scholar] [CrossRef]
- Díaz-Martín, V.; Manzano-Román, R.; Oleaga, A.; Encinas-Grandes, A.; Pérez-Sánchez, R. Cloning and Characterization of a Plasminogen-Binding Enolase from the Saliva of the Argasid Tick Ornithodoros Moubata. Vet. Parasitol. 2013, 191, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Basson, M.D.; Sanders, M.A.; Gomez, R.; Hatfield, J.; VanderHeide, R.; Thamilselvan, V.; Zhang, J.; Walsh, M.F. Focal Adhesion Kinase Protein Levels in Gut Epithelial Motility. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G491–G499. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Shao, J.; Townsend, C.M.; Evers, B.M. Phosphatidylinositol 3-Kinase Mediates Proliferative Signals in Intestinal Epithelial Cells. Gut 2003, 52, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chi, H.; Kinashi, T. Editorial: Hippo Signaling in the Immune System. Front. Immunol. 2020, 11, 587514. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Malik, A.B.; Sun, Y.; Hu, S.; Reynolds, A.B.; Minshall, R.D.; Hu, G. Innate Immune Function of the Adherens Junction Protein P120-Catenin in Endothelial Response to Endotoxin. J. Immunol. 2011, 186, 3180. [Google Scholar] [CrossRef]
- Tian, E.; ten Hagen, K.G. Recent Insights into the Biological Roles of Mucin-Type O-Glycosylation. Glycoconj J. 2009, 26, 325–334. [Google Scholar] [CrossRef]
- Gray, E.; Hogwood, J.; Mulloy, B. The Anticoagulant and Antithrombotic Mechanisms of Heparin. Handb. Exp. Pharm. 2012, 207, 43–61. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.L.; Flavell, R.A. Transforming Growth Factor-Beta Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Martínez, V.G.; Hernández-López, C.; Valencia, J.; Hidalgo, L.; Entrena, A.; Zapata, A.G.; Vicente, A.; Sacedón, R.; Varas, A. The Canonical BMP Signaling Pathway Is Involved in Human Monocyte-Derived Dendritic Cell Maturation. Immunol. Cell Biol. 2011, 89, 610–618. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, L.A.; Cohen, A.; Baron, S.; Burchfiel, C.M. Respond to “Search for Preventable Causes of Cardiovascular Disease”. Am. J. Epidemiol. 2009, 169, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Choi, S.H.; Jin, Y.B.; Lee, H.J.; Ji, Y.H.; Kim, J.; Lee, Y.S.; Lee, Y.J. The Effect of Oxidized Low-Density Lipoprotein (Ox-LDL) on Radiation-Induced Endothelial-to-Mesenchymal Transition. Int. J. Radiat. Biol. 2013, 89, 356–363. [Google Scholar] [CrossRef]
- Etebari, K.; Asad, S.; Zhang, G.; Asgari, S. Identification of Aedes Aegypti Long Intergenic Non-Coding RNAs and Their Association with Wolbachia and Dengue Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0005069. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Das, K.T.; Yunus, M.A.; Azzam, G. Genome-Wide Identification of Aedes Albopictus Long Noncoding RNAs and Their Association with Dengue and Zika Virus Infection. PLoS Negl. Trop. Dis. 2021, 15, e0008351. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.M.; Jmel, M.A.; Cuveele, B.; Gómez-Martín, C.; Aparicio-Puerta, E.; Mekki, I.; Kotál, J.; Martins, L.A.; Hackenberg, M.; Bensaoud, C.; et al. Transcriptomic Analysis of the Tick Midgut and Salivary Gland Responses upon Repeated Blood-Feeding on a Vertebrate Host. Front. Cell. Infect. Microbiol. 2022, 12, 919786. [Google Scholar] [CrossRef] [PubMed]
- Perner, J.; Kropáčková, S.; Kopáček, P.; Ribeiro, J.M.C. Sialome Diversity of Ticks Revealed by RNAseq of Single Tick Salivary Glands. PLoS Negl. Trop. Dis. 2018, 12, e0006410. [Google Scholar] [CrossRef] [PubMed]
- Charrier, N.P.; Couton, M.; Voordouw, M.J.; Rais, O.; Durand-Hermouet, A.; Hervet, C.; Plantard, O.; Rispe, C. Whole Body Transcriptomes and New Insights into the Biology of the Tick Ixodes Ricinus. Parasites Vectors 2018, 11, 364. [Google Scholar] [CrossRef]
- Babraham Bioinformatics. Babraham Bioinformatics—Trim Galore. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 4 July 2018).
- Babraham Bioinformatics. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 4 July 2018).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a Full-Length Transcriptome without a Genome from RNA-Seq Data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. UniProtKB/Swiss-Prot. Methods Mol. Biol. 2007, 406, 89–112. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Medina, J.M.; Jmel, M.A.; Cuveele, B.; Mekki, I.; Kotal, J.; Almeida Martins, L.; Hackenberg, M.; Kotsyfakis, M.; Bensaoud, C. IxoriDB. Available online: https://arn.ugr.es/IxoriDB (accessed on 20 August 2022).
- Schmid-Hempel, P. Immune Defence, Parasite Evasion Strategies and Their Relevance for ‘Macroscopic Phenomena’ Such as Virulence. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 85. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina, J.M.; Abbas, M.N.; Bensaoud, C.; Hackenberg, M.; Kotsyfakis, M. Bioinformatic Analysis of Ixodes ricinus Long Non-Coding RNAs Predicts Their Binding Ability of Host miRNAs. Int. J. Mol. Sci. 2022, 23, 9761. https://doi.org/10.3390/ijms23179761
Medina JM, Abbas MN, Bensaoud C, Hackenberg M, Kotsyfakis M. Bioinformatic Analysis of Ixodes ricinus Long Non-Coding RNAs Predicts Their Binding Ability of Host miRNAs. International Journal of Molecular Sciences. 2022; 23(17):9761. https://doi.org/10.3390/ijms23179761
Chicago/Turabian StyleMedina, José María, Muhammad Nadeem Abbas, Chaima Bensaoud, Michael Hackenberg, and Michail Kotsyfakis. 2022. "Bioinformatic Analysis of Ixodes ricinus Long Non-Coding RNAs Predicts Their Binding Ability of Host miRNAs" International Journal of Molecular Sciences 23, no. 17: 9761. https://doi.org/10.3390/ijms23179761
APA StyleMedina, J. M., Abbas, M. N., Bensaoud, C., Hackenberg, M., & Kotsyfakis, M. (2022). Bioinformatic Analysis of Ixodes ricinus Long Non-Coding RNAs Predicts Their Binding Ability of Host miRNAs. International Journal of Molecular Sciences, 23(17), 9761. https://doi.org/10.3390/ijms23179761