
Rhodococcus equi-Derived Extracellular Vesicles Promoting Inflammatory Response in Macrophage through TLR2-NF-κB/MAPK Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
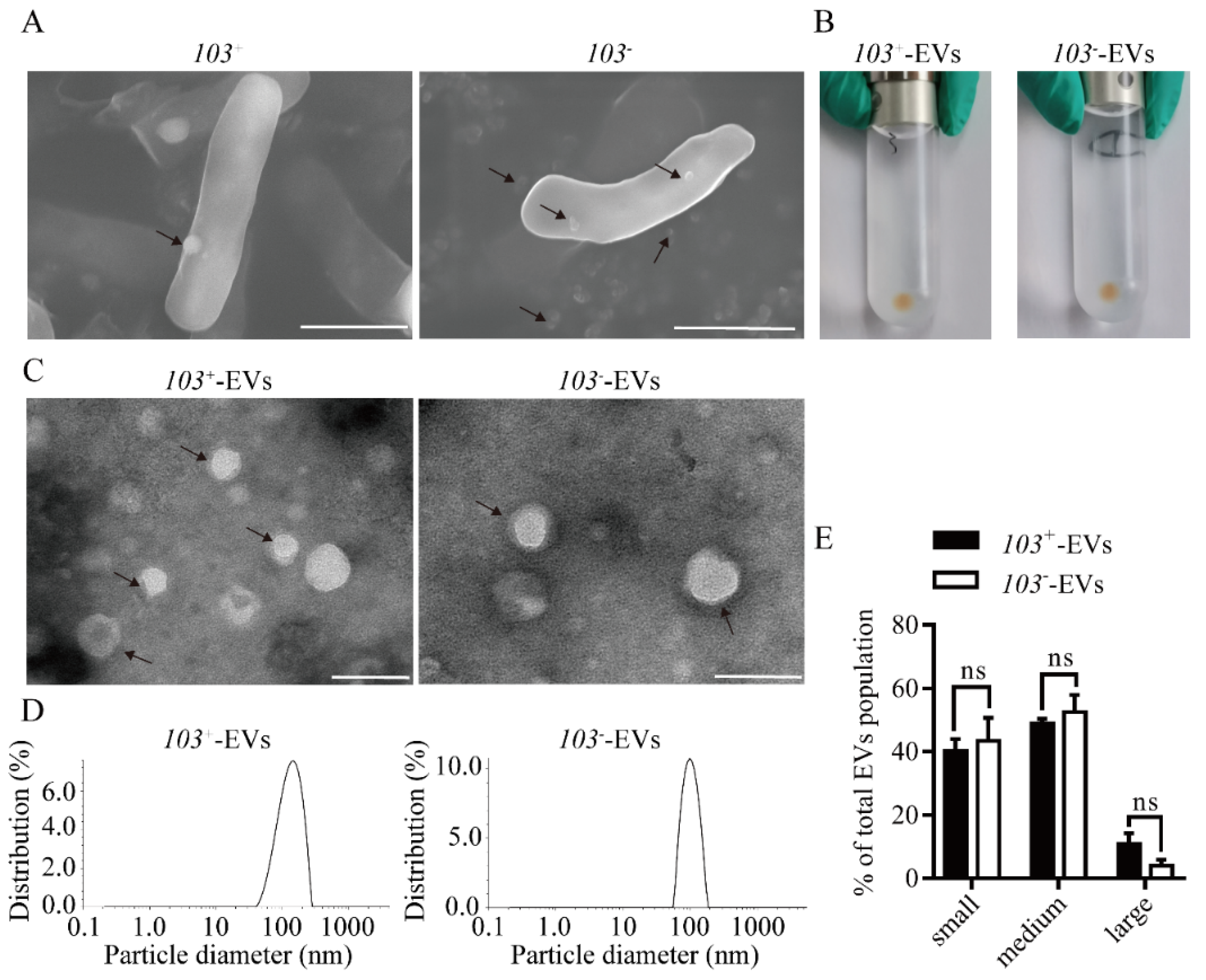
2.1. Characterization of EVs from R. equi (R. equi-EVs)
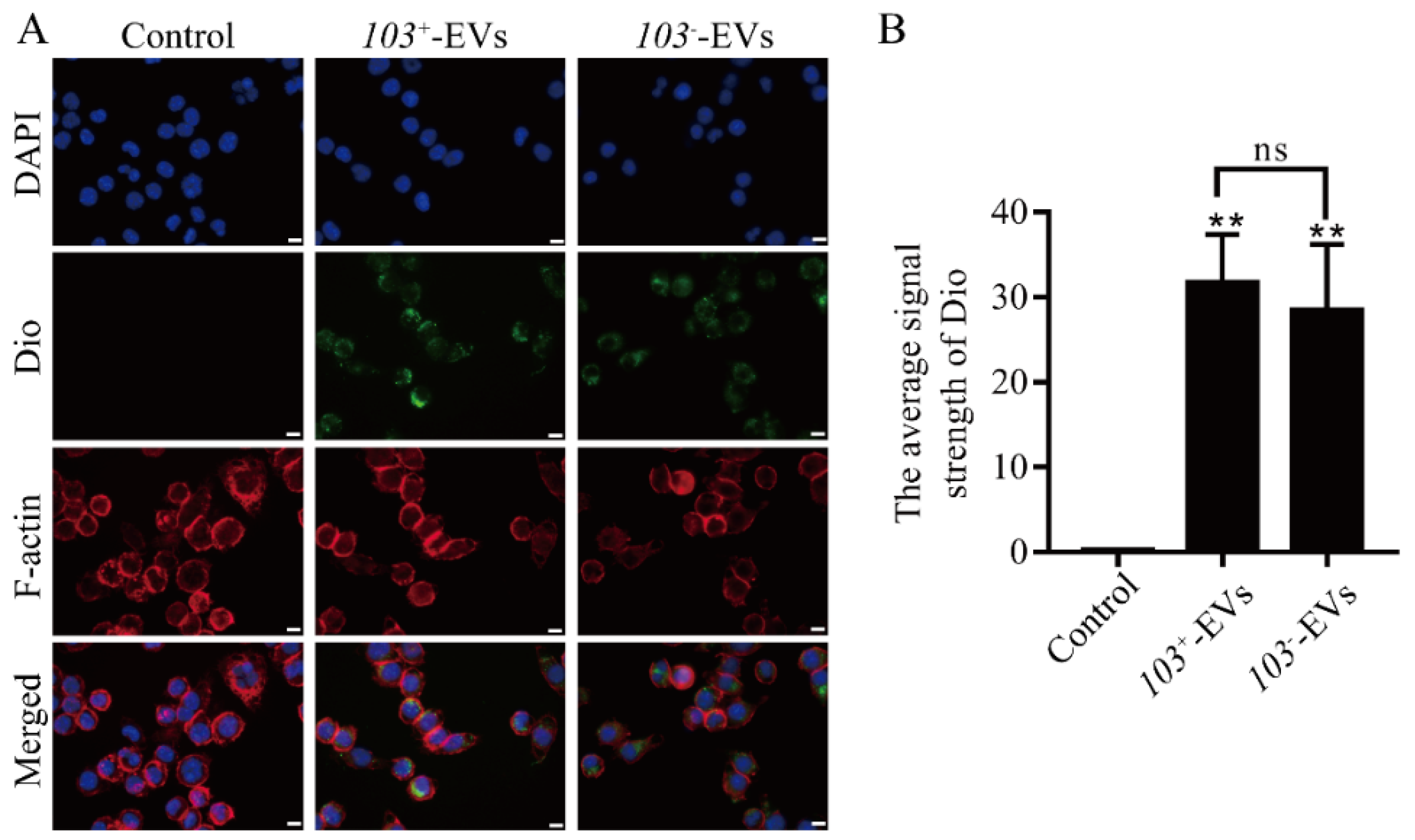
2.2. R. equi EVs Were Taken up by Macrophage
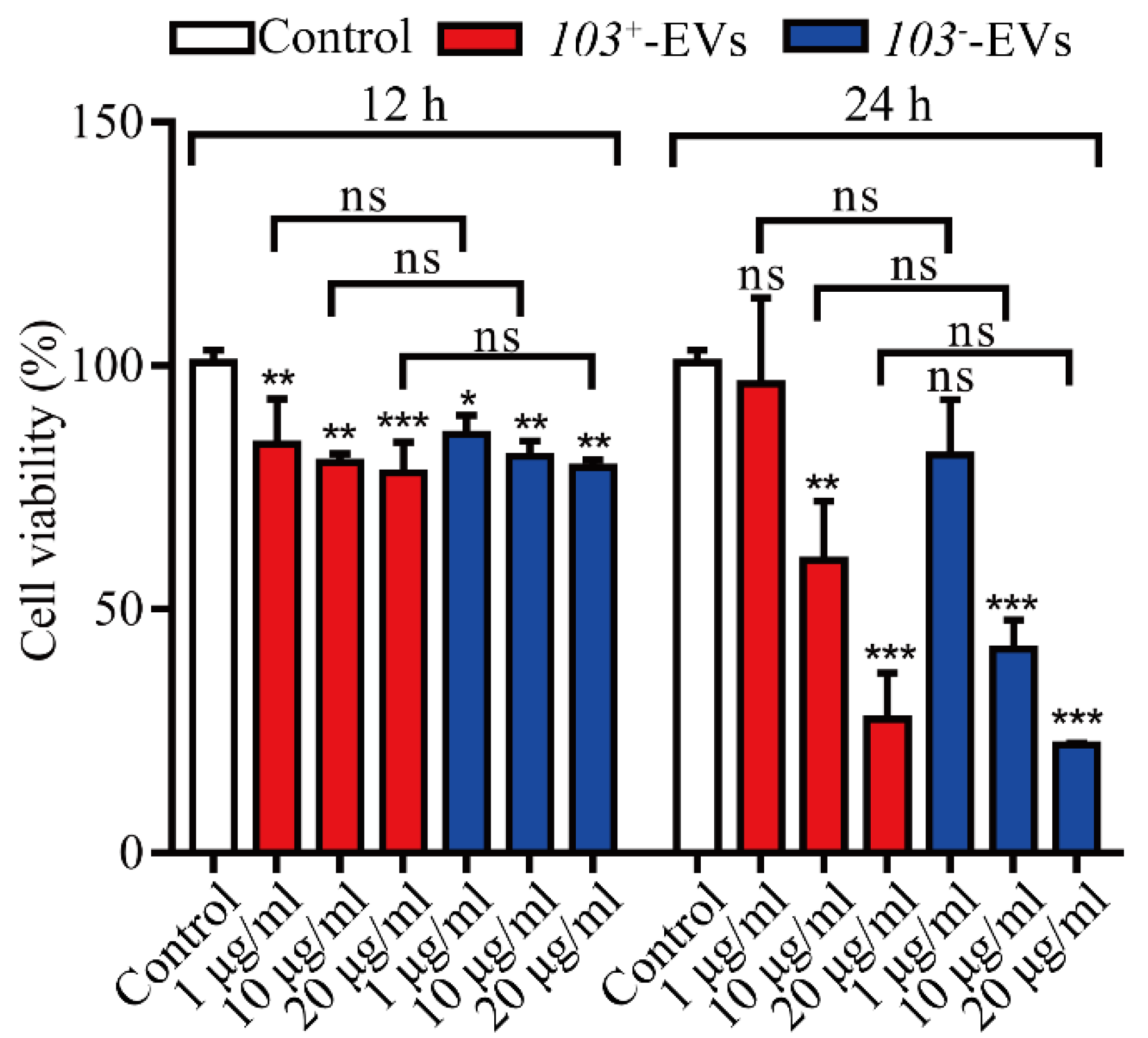
2.3. R. equi-EVs Induced Cytotoxicity in J774A.1 Cells
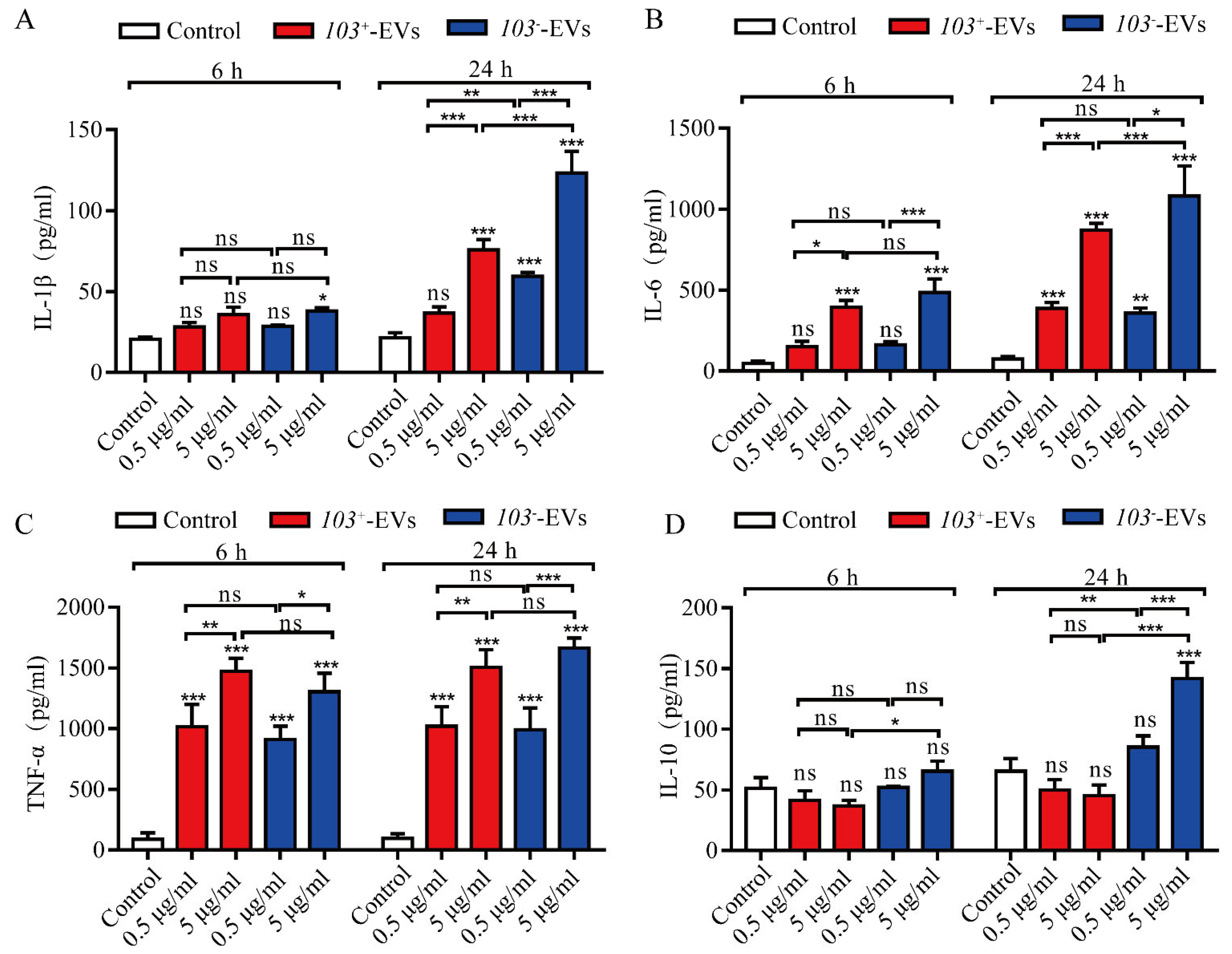
2.4. R. equi-EVs Activate Inflammatory Response in J774A.1 Cells
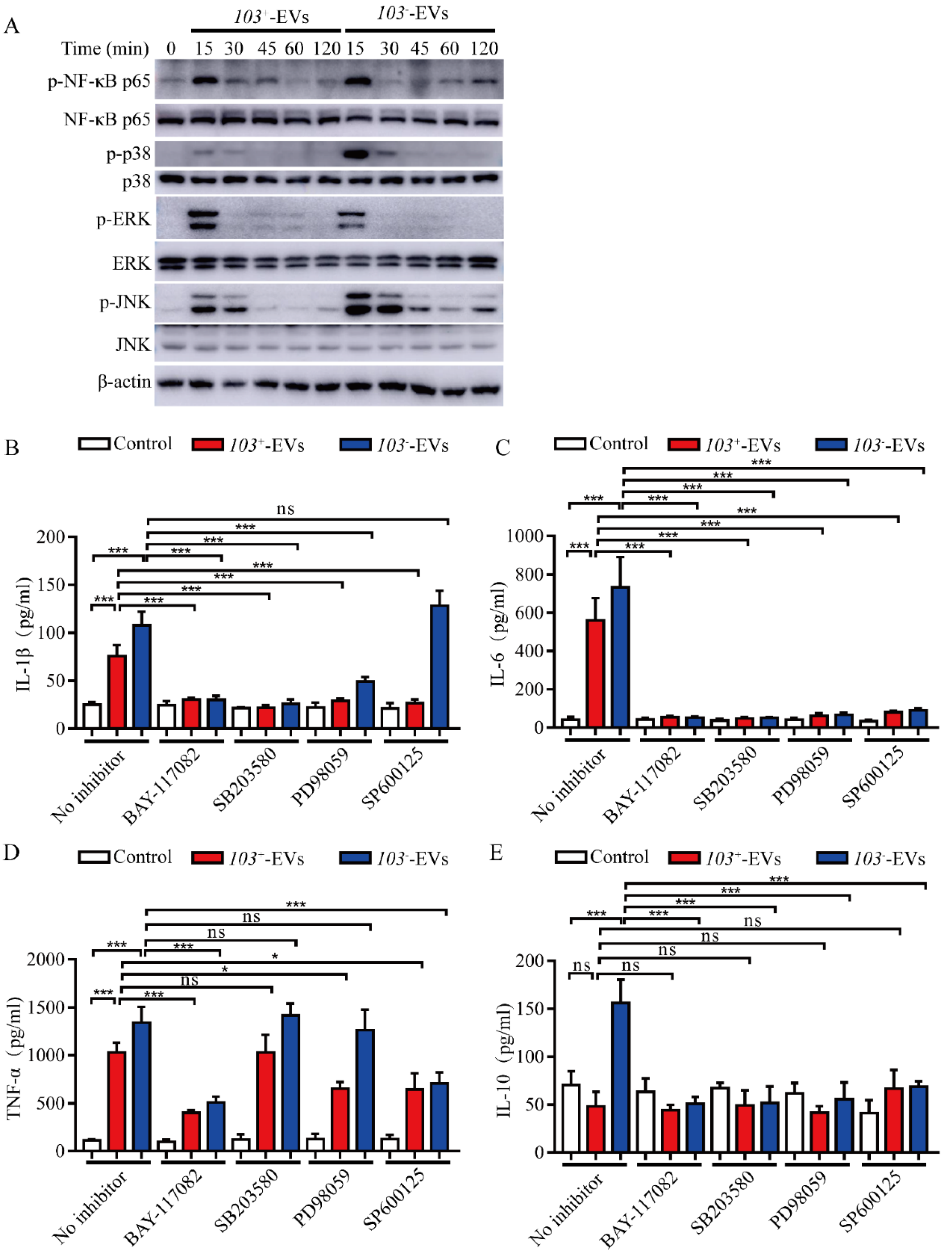
2.5. R. equi-EVs Induced Inflammation Mediated by NF-κB/MAPK Pathways
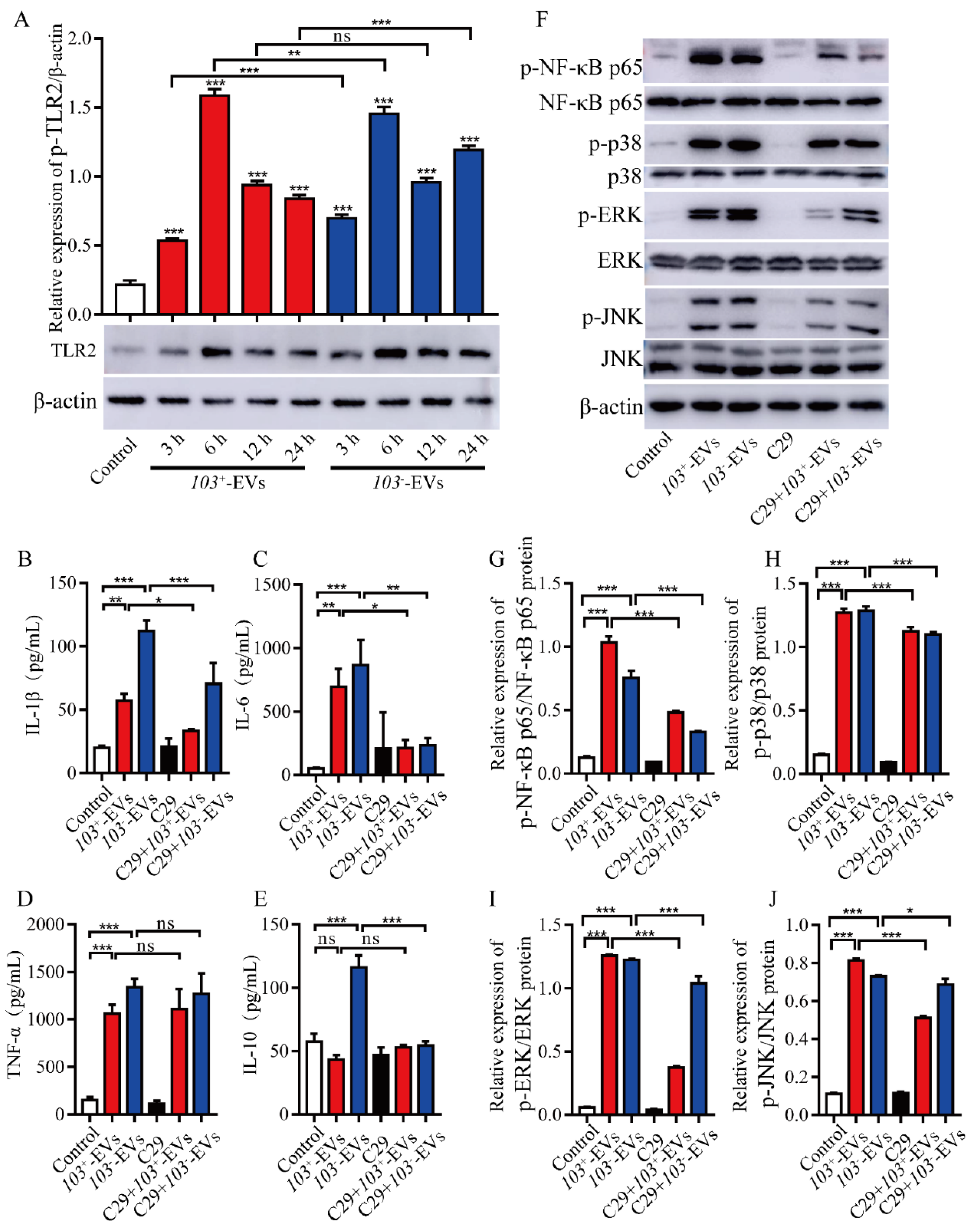
2.6. TLR2-NF-κB/MAPK Is Involved in R. equi-EVs Induced Macrophages Inflammatory Response
2.7. The Proteins Component of R. eui-EVs Is Essential for Inducing Inflammatory Response
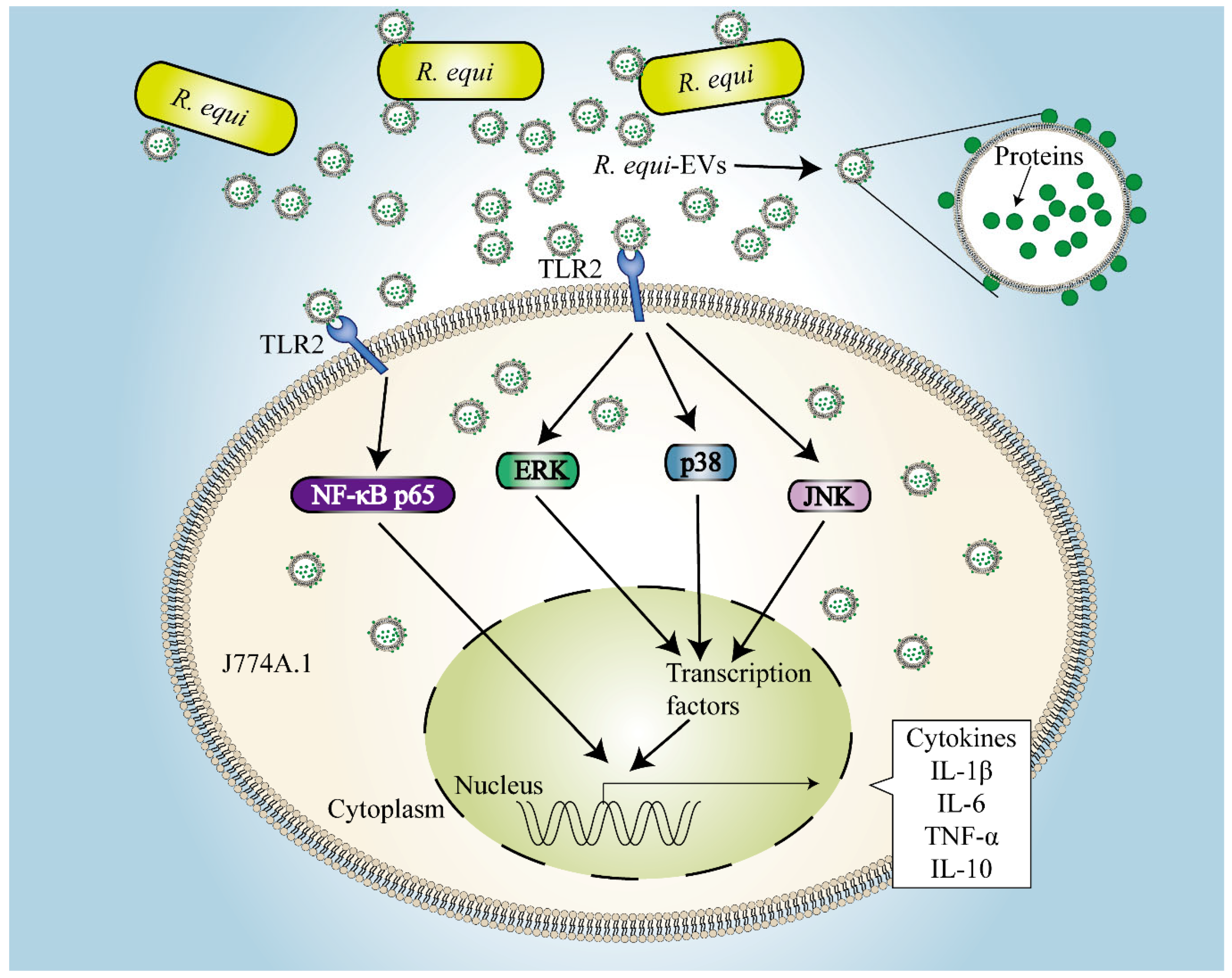
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Culture Conditions
4.2. Scanning Electron Microscope Detection of R. equi-EVs
4.3. Isolation of R. equi-EVs
4.4. Transmission Electron Microscopy Detection of R. equi-EVs
4.5. Dynamic Light Scattering Analysis
4.6. Culture Conditions for J774A.1 Cells and R. equi-EVs Internalization Studies
4.7. Cytotoxicity Assay
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. SDS–PAGE Analysis of EVs and Total Bacteria Proteins
4.10. Western Blotting
4.11. The Enzymatic Hydrolysis of EVs
4.12. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muscatello, G. Rhodococcus equi pneumonia in the foal—Part 1: Pathogenesis and epidemiology. Vet. J. 2012, 192, 20–26. [Google Scholar] [CrossRef]
- Muscatello, G. Rhodococcus equi pneumonia in the foal—Part 2: Diagnostics, treatment and disease management. Vet. J. 2012, 192, 27–33. [Google Scholar] [CrossRef]
- Hondalus, M.K.; Diamond, M.S.; Rosenthal, L.A.; Springer, T.A.; Mosser, D.M. The intracellular bacterium Rhodococcus equi requires Mac-1 to bind to mammalian cells. Infect. Immun. 1993, 61, 2919–2929. [Google Scholar] [CrossRef]
- Hondalus, M.K.; Mosser, D.M. Survival and replication of Rhodococcus equi in macrophages. Infect. Immun. 1994, 62, 4167–4175. [Google Scholar] [CrossRef]
- Galeas-Pena, M.; McLaughlin, N.; Pociask, D. The role of the innate immune system on pulmonary infections. Biol. Chem. 2019, 400, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Salgame, P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007, 27, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed]
- von Bargen, K.; Haas, A. Molecular and infection biology of the horse pathogen Rhodococcus equi. FEMS Microbiol. Rev. 2009, 33, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Bloom, B.R.; Hondalus, M.K. Deletion of vapA encoding Virulence Associated Protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol. Microbiol. 2003, 50, 115–128. [Google Scholar] [CrossRef]
- Sangkanjanavanich, N.; Kawai, M.; Kakuda, T.; Takai, S. Rescue of an intracellular avirulent Rhodococcus equi replication defect by the extracellular addition of virulence-associated protein A. J. Vet. Med. Sci. 2017, 79, 1323–1326. [Google Scholar] [CrossRef] [Green Version]
- von Bargen, K.; Scraba, M.; Krämer, I.; Ketterer, M.; Nehls, C.; Krokowski, S.; Repnik, U.; Wittlich, M.; Maaser, A.; Zapka, P.; et al. Virulence-associated protein A from Rhodococcus equi is an intercompartmental pH-neutralising virulence factor. Cell. Microbiol. 2019, 21, e12958. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Darrah, P.A.; Monaco, M.C.; Jain, S.; Hondalus, M.K.; Golenbock, D.T.; Mosser, D.M. Innate immune responses to Rhodococcus equi. J. Immunol. 2004, 173, 1914–1924. [Google Scholar] [CrossRef]
- Giguère, S.; Prescott, J.F. Cytokine induction in murine macrophages infected with virulent and avirulent Rhodococcus equi. Infect. Immun. 1998, 66, 1848–1854. [Google Scholar] [CrossRef]
- Liu, M.; Bordin, A.; Liu, T.; Russell, K.; Cohen, N. Gene expression of innate Th1-, Th2-, and Th17-type cytokines during early life of neonatal foals in response to Rhodococcus equi. Cytokine 2011, 56, 356–364. [Google Scholar] [CrossRef]
- Giguère, S.; Wilkie, B.N.; Prescott, J.F. Modulation of cytokine response of pneumonic foals by virulent Rhodococcus equi. Infect. Immun. 1999, 67, 5041–5047. [Google Scholar] [CrossRef]
- Vail, K.J.; da Silveira, B.P.; Bell, S.L.; Cohen, N.D.; Bordin, A.I.; Patrick, K.L.; Watson, R.O. The opportunistic intracellular bacterial pathogen Rhodococcus equi elicits type I interferon by engaging cytosolic DNA sensing in macrophages. PLoS Pathog. 2021, 17, e1009888. [Google Scholar] [CrossRef]
- Berghaus, L.J.; Giguère, S.; Bordin, A.I.; Cohen, N.D. Effects of priming with cytokines on intracellular survival and replication of Rhodococcus equi in equine macrophages. Cytokine 2018, 102, 7–11. [Google Scholar] [CrossRef]
- Kasuga-Aoki, H.; Takai, S.; Sasaki, Y.; Tsubaki, S.; Madarame, H.; Nakane, A. Tumour necrosis factor and interferon-gamma are required in host resistance against virulent Rhodococcus equi infection in mice: Cytokine production depends on the virulence levels of R. equi. Immunology 1999, 96, 122–127. [Google Scholar] [CrossRef]
- Darrah, P.A.; Hondalus, M.K.; Chen, Q.; Ischiropoulos, H.; Mosser, D.M. Cooperation between reactive oxygen and nitrogen intermediates in killing of Rhodococcus equi by activated macrophages. Infect. Immun. 2000, 68, 3587–3593. [Google Scholar] [CrossRef] [PubMed]
- Kanaly, S.T.; Hines, S.A.; Palmer, G.H. Cytokine modulation alters pulmonary clearance of Rhodococcus equi and development of granulomatous pneumonia. Infect. Immun. 1995, 63, 3037–3041. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Gill, S.; Catchpole, R.; Forterre, P. Extracellular membrane vesicles in the three domains of life and beyond. FEMS Microbiol. Rev. 2019, 43, 273–303. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.T. Outer Membrane Vesicles (OMVs) of Gram-negative Bacteria: A Perspective Update. Front. Microbiol. 2017, 8, 1053. [Google Scholar] [CrossRef]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef]
- Nagakubo, T.; Tahara, Y.O.; Miyata, M.; Nomura, N.; Toyofuku, M. Mycolic acid-containing bacteria trigger distinct types of membrane vesicles through different routes. iScience 2021, 24, 102015. [Google Scholar] [CrossRef]
- Hu, R.; Lin, H.; Wang, M.; Zhao, Y.; Liu, H.; Min, Y.; Yang, X.; Gao, Y.; Yang, M. Lactobacillus reuteri-derived extracellular vesicles maintain intestinal immune homeostasis against lipopolysaccharide-induced inflammatory responses in broilers. J. Anim. Sci. Biotechnol. 2021, 12, 25. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Bello, B.K.; Yu, G.; Yang, Q.; Yang, H.; Zhang, W.; Wang, L.; Dong, J.; Liu, G.; et al. Activation of TLR2 heterodimers-mediated NF-κB, MAPK, AKT signaling pathways is responsible for Vibrio alginolyticus triggered inflammatory response in vitro. Microb. Pathog. 2022, 162, 105219. [Google Scholar] [CrossRef]
- Chandler, C.E.; Ernst, R.K. Bacterial lipids: Powerful modifiers of the innate immune response. F1000Res 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Shishpal, P.; Kasarpalkar, N.; Singh, D.; Bhor, V.M. Characterization of Gardnerella vaginalis membrane vesicles reveals a role in inducing cytotoxicity in vaginal epithelial cells. Anaerobe 2020, 61, 102090. [Google Scholar] [CrossRef] [PubMed]
- Echeverría-Bugueño, M.; Balada, C.; Irgang, R.; Avendaño-Herrera, R. Evidence for the existence of extracellular vesicles in Renibacterium salmoninarum and related cytotoxic effects on SHK-1 cells. J. Fish Dis. 2021, 44, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Yerneni, S.S.; Werner, S.; Azambuja, J.H.; Ludwig, N.; Eutsey, R.; Aggarwal, S.D.; Lucas, P.C.; Bailey, N.; Whiteside, T.L.; Campbell, P.G.; et al. Pneumococcal Extracellular Vesicles Modulate Host Immunity. mBio 2021, 12, e0165721. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, N.R.; Nicolas, A.; Rodovalho, V.R.; Luz, B.; Briard-Bion, V.; Krupova, Z.; Thierry, A.; Coste, F.; Burel, A.; Martin, P.; et al. Extracellular vesicles produced by human and animal Staphylococcus aureus strains share a highly conserved core proteome. Sci. Rep. 2020, 10, 8467. [Google Scholar] [CrossRef] [PubMed]
- Bitto, N.J.; Cheng, L.; Johnston, E.L.; Pathirana, R.; Phan, T.K.; Poon, I.K.H.; O’Brien-Simpson, N.M.; Hill, A.F.; Stinear, T.P.; Kaparakis-Liaskos, M. Staphylococcus aureus membrane vesicles contain immunostimulatory DNA, RNA and peptidoglycan that activate innate immune receptors and induce autophagy. J. Extracell. Vesicles 2021, 10, e12080. [Google Scholar] [CrossRef]
- Bitto, N.J.; Zavan, L.; Johnston, E.L.; Stinear, T.P.; Hill, A.F.; Kaparakis-Liaskos, M. Considerations for the Analysis of Bacterial Membrane Vesicles: Methods of Vesicle Production and Quantification Can Influence Biological and Experimental Outcomes. Microbiol. Spectr. 2021, 9, e0127321. [Google Scholar] [CrossRef]
- Murase, K.; Aikawa, C.; Nozawa, T.; Nakatake, A.; Sakamoto, K.; Kikuchi, T.; Nakagawa, I. Biological Effect of Streptococcus pyogenes-Released Extracellular Vesicles on Human Monocytic Cells, Induction of Cytotoxicity, and Inflammatory Response. Front. Cell. Infect. Microbiol. 2021, 11, 711144. [Google Scholar] [CrossRef]
- Mehanny, M.; Kroniger, T.; Koch, M.; Hoppstädter, J.; Becher, D.; Kiemer, A.K.; Lehr, C.M.; Fuhrmann, G. Yields and Immunomodulatory Effects of Pneumococcal Membrane Vesicles Differ with the Bacterial Growth Phase. Adv. Healthc. Mater. 2022, 11, e2101151. [Google Scholar] [CrossRef]
- de Rezende Rodovalho, V.; da Luz, B.S.R.; Nicolas, A.; do Carmo, F.L.R.; Jardin, J.; Briard-Bion, V.; Jan, G.; Le Loir, Y.; de Carvalho Azevedo, V.A.; Guedon, E. Environmental conditions modulate the protein content and immunomodulatory activity of extracellular vesicles produced by the probiotic Propionibacterium freudenreichii. Appl. Environ. Microbiol. 2020, 87, e02263-20. [Google Scholar] [CrossRef]
- Woo, J.H.; Kim, S.; Lee, T.; Lee, J.C.; Shin, J.H. Production of Membrane Vesicles in Listeria monocytogenes Cultured with or without Sub-Inhibitory Concentrations of Antibiotics and Their Innate Immune Responses In Vitro. Genes 2021, 12, 415. [Google Scholar] [CrossRef]
- Wang, X.; Eagen, W.J.; Lee, J.C. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 3174–3184. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lim, Y.; An, S.J.; Choi, B.K. Characterization and immunostimulatory activity of extracellular vesicles from Filifactor alocis. Mol. Oral Microbiol. 2020, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Simpson, M.E.; Petri, W.A., Jr. TLR2 as a Therapeutic Target in Bacterial Infection. Trends. Mol. Med. 2020, 26, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Prados-Rosales, R.; Baena, A.; Martinez, L.R.; Luque-Garcia, J.; Kalscheuer, R.; Veeraraghavan, U.; Camara, C.; Nosanchuk, J.D.; Besra, G.S.; Chen, B.; et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J. Clin. Investig. 2011, 121, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Song, M.K.; Gho, Y.S.; Kim, H.H.; Choi, B.K. Extracellular vesicles derived from the periodontal pathogen Filifactor alocis induce systemic bone loss through Toll-like receptor 2. J. Extracell. Vesicles 2021, 10, e12157. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.C.; Begg, A.P.; Anderson, G.A.; Takai, S.; Lämmler, C.; Browning, G.F. Epidemiology of Rhodococcus equi strains on Thoroughbred horse farms. Appl. Environ. Microbiol. 2001, 67, 2167–2175. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Hao, X.; Li, M.; Luo, H. Rhodococcus equi-Derived Extracellular Vesicles Promoting Inflammatory Response in Macrophage through TLR2-NF-κB/MAPK Pathways. Int. J. Mol. Sci. 2022, 23, 9742. https://doi.org/10.3390/ijms23179742
Xu Z, Hao X, Li M, Luo H. Rhodococcus equi-Derived Extracellular Vesicles Promoting Inflammatory Response in Macrophage through TLR2-NF-κB/MAPK Pathways. International Journal of Molecular Sciences. 2022; 23(17):9742. https://doi.org/10.3390/ijms23179742
Chicago/Turabian StyleXu, Zhaokun, Xiujing Hao, Min Li, and Haixia Luo. 2022. "Rhodococcus equi-Derived Extracellular Vesicles Promoting Inflammatory Response in Macrophage through TLR2-NF-κB/MAPK Pathways" International Journal of Molecular Sciences 23, no. 17: 9742. https://doi.org/10.3390/ijms23179742
APA StyleXu, Z., Hao, X., Li, M., & Luo, H. (2022). Rhodococcus equi-Derived Extracellular Vesicles Promoting Inflammatory Response in Macrophage through TLR2-NF-κB/MAPK Pathways. International Journal of Molecular Sciences, 23(17), 9742. https://doi.org/10.3390/ijms23179742