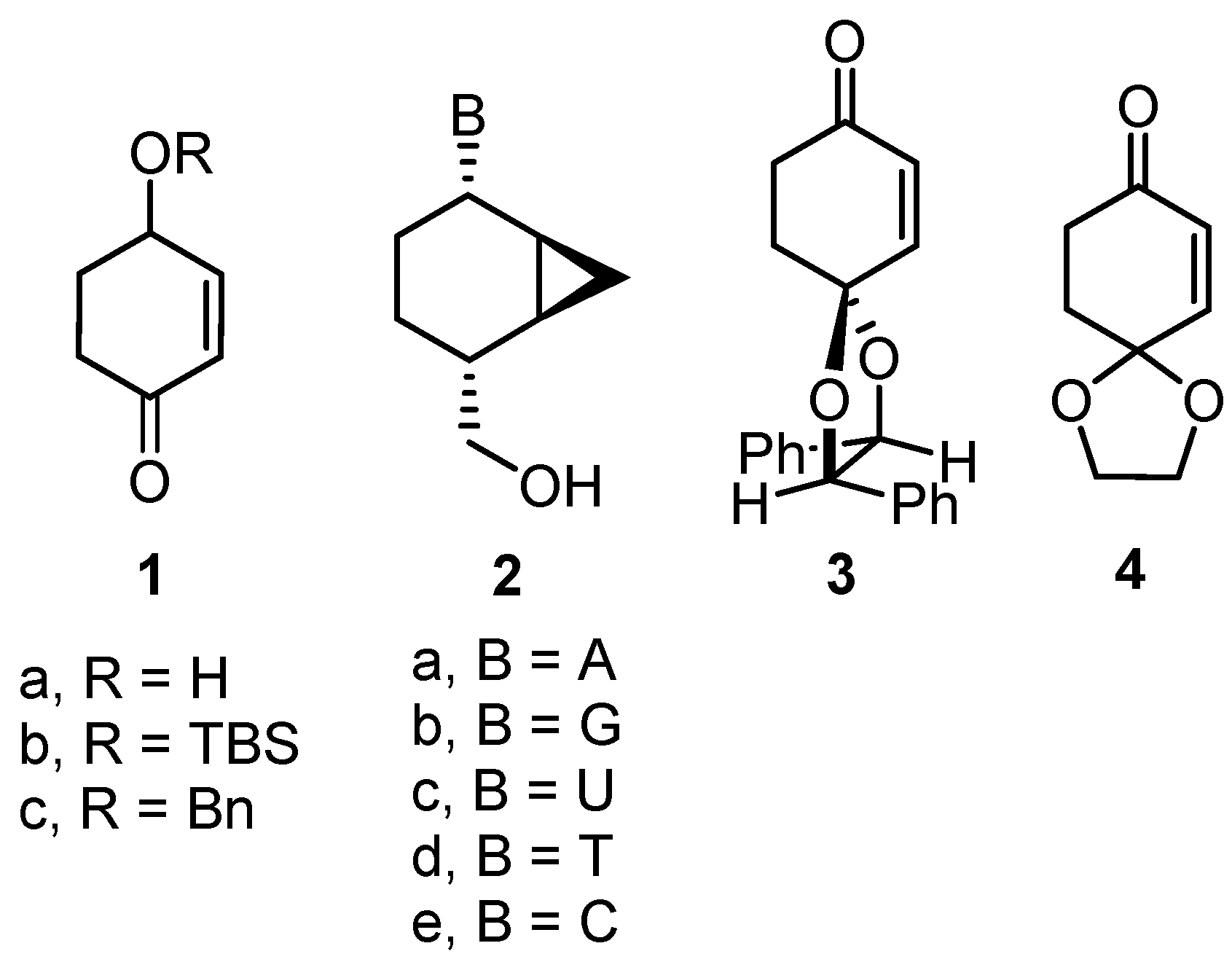
Synthetic Procedures
(8R)-1,4-Dioxaspiro [4.5]dec-6-en-8-ol [(R)-6]. To a stirred solution of enone
4 (104 mg, 0.68 mmol) in a biphasic medium of CH
2Cl
2 (2.3 mL) and water (2.3 mL), tetrabutylammonium chloride (57 mg, 0.20 mmol), sodium formate (139 mg, 2.04 mmol), and (
R,R)-Noyori-I [(
R,R)-
5] catalyst (13 mg, 0.020 mmol) were sequentially added at room temperature. The reaction mixture was allowed to stir for 3 h. Then, the phases were separated, and the aqueous layer was extracted with additional CH
2Cl
2 (2 × 2 mL) and the organic layers were concentrated under reduced pressure, affording a yellow oil that was purified by column chromatography (diethyl ether) to furnish allylic alcohol (
R)-
6 (83 mg, 0.53 mmol, 79%): [α]
D20 = +39.0 (
c 1.2, CHCl
3) [lit: [
37] [α]
D20 = +39.8 (
c 1.21, CHCl
3), ee = 93.1%] as a yellowish oil:
1H NMR (250 MHz, CDCl
3) δ 5.94 (ddd,
J7,6 = 10.1 Hz,
J7,8 = 2.8 Hz,
J7,9 = 1.1 Hz, 1H, H-7), 5.61 (dt,
J6,7 = 10.1 Hz,
J6,8 =
J6,10 =1.5 Hz, 1H, H-6), 4.29–4.12 (m, 1H, H-8), 4.05–3.85 (m, 4H, H-2, H-3), 2.17–2.02 (m, 1H, H-9), 2.00–1.89 (m, 1H, H-9), 1.85–1.65 (m, 2H, H-10);
13C NMR (90 MHz, CDCl
3) δ 135.3 (C-7), 129.0 (C-6), 105.2 (C-5), 66.0 (C-8), 64.8/64.6 (C-2, C-3), 31.0/30.6 (C-9, C-10).
(8S)-1,4-Dioxaspiro [4.5]dec-6-en-8-ol [(S)-6]. To a stirred solution of enone
4 (76 mg, 0.49 mmol) in a biphasic medium of CH
2Cl
2 (1.3 mL) and water (1.3 mL), tetrabutylammonium chloride (45 mg, 0.16 mmol), sodium formate (140 mg, 2.06 mmol), and (
S,S)-Noyori-I catalyst, [(
S,S)-
5], (9.1 mg, 0.015 mmol) were sequentially added at room temperature. The reaction mixture was allowed to stir for 24 h. Then, the phases were separated, and the aqueous layer was extracted with CH
2Cl
2 (2 × 10 mL). The organic layers were concentrated under reduced pressure, affording a yellow oil that was purified by column chromatography (diethyl ether) to obtain allylic alcohol (
S)-
6 (60 mg, 0.38 mmol, 78% yield) as a yellow oil: [α]
D20 = −38.7 (
c 1.3, CHCl
3) [lit: [
38] [α]
D20 = −40.5 (
c 1.24, CHCl
3) ee > 98.4%];
1H NMR (250 MHz, CDCl
3) δ: 5.93 (ddd,
J7,6 = 10.1 Hz,
J7,8 = 2.8 Hz,
J7,9 = 1.0 Hz, 1H, H-7), 5.60 (dt,
J6,7 = 10.1 Hz,
J6,8 =
J6,10 = 1.4 Hz, 1H, H-6), 4.26–4.16 (m, 1H, H-8), 4.05–3.85 (m, 4H, H-2, H-3), 2.19–2.05 (m, 1H, H-9), 2.04–1.90 (m, 1H, H-9), 1.90–1.68 (m, 2H, H-10).
(4R)-4-Hydroxycyclohex-2-en-1-one [(R)-1a]. Montmorillonite K-10 (832 mg) was added to a solution of (
R)-
6 (77 mg, 0.49 mmol) in CH
2Cl
2 (7.6 mL) and the mixture was stirred at room temperature for 4 h. Then, it was filtered, and the solvent was removed under vacuum to furnish the enone (
R)-
1a (34 mg, 0.31 mmol, 64% yield) as a yellowish oil: CHPLC (Daicel Chiralpak IC): 92% ee; [α]
D20 = +90.0 (
c 0.2, CHCl
3) [lit: [
39] [α]
D20 = +92.3 (
c 0.7, CHCl
3) ee > 99%];
1H NMR (250 MHz, CDCl
3) δ 6.95 (ddd,
J3,2 = 10.2 Hz,
J3,4 = 2.1 Hz,
J3,5eq = 1.6 Hz, 1H, H-3), 5.98 (ddd,
J2,3 = 10.2 Hz,
J2,4 = 2.1 Hz,
J2,6 = 1.0 Hz, 1H, H-2), 4.58 (ddt,
J4,5ax = 9.2 Hz,
J4,5eq = 4.4 Hz,
J4,2 =
J4,3 =2.1 Hz, 1H, H-4), 2.60 (dt,
Jgem = 9.8 Hz,
J6eq,5 =
J6eq,4 = 4.5 Hz, 1H, H-6eq), 2.45–2.28 (m, 2H, H-6ax, H-5eq), 2.10–1.90 (m, 1H, H-5ax);
13C NMR (90 MHz, CDCl
3) δ 199.4 (C-1), 153.6 (C-3), 129.0 (C-2), 66.2 (C-4), 35.4 (C-6), 32.4 (C-5).
(4S)-4-Hydroxycyclohex-2-en-1-one [(S)-1a]. Montmorillonite K-10 (852 mg) was added to a solution of (
S)-
6 (79 mg, 0.50 mmol) in CH
2Cl
2 (7.8 mL) and the mixture was stirred at rt for 4 h. Then, it was filtered and the solvent was removed under vacuum to furnish the known enone (
S)-
1a (35 mg, 0.31 mmol, 62% yield) as a yellowish oil: CHPLC (Daicel Chiralpak IC): 92% ee; [α]
D 20 =−92.3 (c 0.50, CHCl
3) [lit: [
9] [α]
D20= −92.0 (c 0.50, CHCl
3) ee > 99%];
1H NMR (250 MHz, CDCl
3) δ 6.93 (ddd,
J3,2 = 10.2 Hz,
J3,4 = 2.4 Hz,
J3,5eq = 1.6 Hz, 1H, H-3), 5.95 (ddd,
J2,3 = 10.2 Hz,
J2,4 = 2.0 Hz,
J2,6 = 1.0 Hz, 1H, H-2), 4.68–4.46 (m, 1H, H-4), 2.80–2.68 (m, 1H, OH), 2.57 (ddd,
Jgem = 9.8 Hz,
J6eq,4 = 5.0Hz,
J6eq,5 = 4.0 Hz, 1H, H-6eq), 2.47–2.24 (m, 2H, H-6ax, H-5eq), 2.00 (ddd,
Jgem = 12.8 Hz,
J5ax,6ax = 9.5 Hz,
J5ax,4 = 5.3 Hz, 1H, H-5ax).
(4R)-4-((tert-Butyldimethylsilyl)oxy)cyclohex-2-en-1-one [(R)-1b]. To a stirred solution of (
R)-
6 (102 mg, 0.65 mmol) in CH
2Cl
2 (0.5 mL), imidazole (56 mg, 0.82 mmol) and a solution of TBSCl (124 mg, 0.82 mmol) in CH
2Cl
2 (0.4 mL) were added at room temperature. The mixture was allowed to stir overnight. Then, a saturated aqueous NaHCO
3 solution (1 mL) was added, the aqueous layer was extracted with CH
2Cl
2 (3 × 1 mL), and the organic layers were dried and concentrated under vacuum. The crude was dissolved in CH
2Cl
2 (4 mL) and montmorillonite K-10 (513 mg) was added. The reaction mixture was allowed to stir at room temperature for 1 h. Then, it was filtered, and the solvent was removed under vacuum to furnish enone (
R)-
1b (128 mg, 0.56 mmol, 87% yield) as a yellowish oil: [α]
D20 = +92.3 (
c 1.02, CHCl
3) [lit: [
39] [α]
D20 = +97.9 (
c 1.2, CHCl
3) ee > 99%];
1H NMR (400 MHz, CDCl
3) δ 6.82 (ddd,
J3,2 = 10.2 Hz,
J3,4 = 2.1 Hz,
J3,5eq = 1.6 Hz, 1H, H-3), 5.91 (ddd,
J2,3 = 10.2 Hz,
J2,4 = 2.1 Hz,
J2,6eq = 1.0 Hz, 1H, H-2), 4.51 (ddt,
J4,5ax = 9.1 Hz,
J4,5eq = 4.6 Hz,
J4,2 =
J4,3 = 2.1 Hz, 1H, H-4), 2.56 (dt,
Jgem = 16.8 Hz,
J6eq,5ax =
J6eq,5eq = 4.5 Hz, 1H, H-6eq), 2.33 (ddd,
Jgem = 16.8 Hz,
J6ax,5ax = 12.8 Hz,
J6ax,5eq = 4.7 Hz, 1H, H-6ax), 2.20 (dqd,
Jgem = 12.8 Hz,
J5eq,4 =
J5eq,6eq =
J5eq,6ax = 4.6 Hz,
J5eq,3 = 1.6 Hz, 1H, H-5eq), 1.99 (tdd,
Jgem =
J5ax,6ax = 12.8 Hz,
J5ax,4 = 9.1 Hz,
J5ax,6eq = 4.5 Hz, 1H, H-5ax), 0.90 (s, 9H, H-
tBu), 0.12 (s, 3H, SiCH
3), (s, 3H, SiCH
3).
(4S)-4-((tert-Butyldimethylsilyl)oxy)cyclohex-2-en-1-one [(S)-1b]. To a stirred solution of (
S)-
6 (49 mg, 0.32 mmol) in CH
2Cl
2 (0.4 mL), imidazole (34 mg, 0.49 mmol) and a solution of TBSCl (74 mg, 0.49 mmol) in CH
2Cl
2 (0.2 mL) were added at room temperature. The mixture was allowed to stir overnight. Then, a saturated aqueous NaHCO
3 solution (1 mL) was added, the aqueous layer was extracted with more CH
2Cl
2 (3 × 1 mL), and the organic layers were dried and concentrated under vacuum. The crude was dissolved in CH
2Cl
2 (4 mL) and montmorillonite K-10 (294 mg) was added. The reaction mixture was allowed to stir at room temperature for 1 h. Then, it was filtered, and the solvent was removed under reduced pressure to afford enone (
S)-
1b (64 mg, 0.28 mmol, 88% yield) as a yellowish oil: [α]
D20= −93.7 (
c 0.70, CHCl
3) [lit: [
6] [α]
D20 = −97.0 (
c 1.26, CHCl
3), ee >99%];
1H NMR (250 MHz, CDCl
3) δ 6.81 (ddd,
J3,2 = 10.2 Hz,
J3,4 = 2.1 Hz,
J3,5eq = 1.6 Hz, 1H, H-3), 5.90 (ddd,
J2,3 = 10.2 Hz,
J2,4 = 2.1 Hz,
J2,6eq = 1.0 Hz, 1H, H-2), 4.50 (ddt,
J4,5ax = 9.0 Hz,
J4,5eq = 4.8 Hz,
J4,3 =
J4,2 = 2.1 Hz, 1H, H-4), 2.55 (dt,
Jgem = 16.7 Hz,
J6eq.5ax =
J6eq,5eq = 4.8 Hz, 1H, H-6eq), 2.32 (ddd,
Jgem = 16.7 Hz,
J6ax,5ax = 12.8 Hz,
J6ax,5eq = 4.8 Hz, 1H, H-6ax), 2.19 (dqd,
Jgem = 12.8 Hz,
J5eq,6eq =
J5eq,4 =
J5eq,6ax = 4.8 Hz,
J5eq,3 = 1.6 Hz, 1H, H-5eq), 1.97 (tdd,
Jgem =
J5ax,6ax = 12.8 Hz,
J5ax,4 = 9.0 Hz,
J5ax,6eq = 4.8 Hz, 1H, H-5ax), 0.89 (s, 9H, H-
tBu), 0.11 (s, 3H, SiCH
3), 0.10 (s, 3H, SiCH
3).
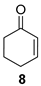
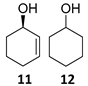
(1R)-2-Cyclohexen-1-ol [(R)-11]. To a stirred solution of enone 8 (250 µL, 2.59 mmol) in a biphasic medium of CH2Cl2 (8.6 mL) and water (8.6 mL) and under a nitrogen flux, tetrabutylammonium chloride (222 mg, 0.8 mmol), sodium formate (532 mg, 7.82 mmol), and (R,R)-Noyori-I catalyst, [(R,R)-5] (49.6 mg, 0.08 mmol) were sequentially added at room temperature. The reaction mixture was allowed to stir for 24 h. Then, CH2Cl2 (10 mL) and water (10 mL) were added, the two phases were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The volatiles of the organic phase were removed under reduced pressure and the resulting residue was purified by column chromatography (CHCl3), affording an inseparable mixture of allylic alcohol (R)-11 and cyclohexanol 12 (210 mg, 2.14 mmol, 82% yield) in a 2:1 ratio as a yellowish oil that was analyzed by NMR spectroscopy and CHPLC (Daicel Chiralpak IC): 92% ee; 1H NMR (250 MHz, CDCl3) ((R)-11) δ 5.82 (dtd, J3,2 = 10.0 Hz, J3,4eq = J3,4ax = 3.3 Hz, J3,1 = 1.1 Hz, 1H, H-3), 5.73 (ddt, J2,3 = 10.0, J2,1 = 3.4, J2,4 = J2,6 = 1.8 Hz, 1H, H-2), 4.42–4.13 (m, 1H, H-1), 2.07–1.89 (m, 1H, H-6), 1.92–1.81 (m, 1H, H-6), 1.79–1.64 (m, 2H, H-4, H-5), 1.64–1.48 (m, 2H, H-4, H-5); (12) δ 3.66–3.52 (m, 1H, H-1′), 1.64–1.48 (m, 4H, H-2′, H-6′), 1.38–0.96 (m, 6H, H-3′, H-4′, H-5′).
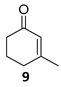
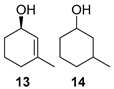
(1R)-3-Methyl-2-cyclohexen-1-ol [(R)-13]. To a stirred solution of enone
9 (510 µL, 495 mg, 4.49 mmol) in a biphasic medium of CH
2Cl
2 (14.7 mL) and water (14.7mL) and under a nitrogen flux, tetrabutylammonium chloride (367 mg, 1.32 mmol), sodium formate (899 mg, 13.2 mmol), and (
R,R)-Noyori-I catalyst [(
R,R)-
5] (84 mg, 0.13 mmol) were sequentially added at room temperature. The reaction mixture was allowed to stir for 72 h. Then, CH
2Cl
2 (10 mL) and water (10 mL) were added, the two phases were separated, and the aqueous layer was extracted with CH
2Cl
2 (3 × 5 mL). The volatiles of the organic phase were removed under reduced pressure and the resulting residue was purified by column chromatography (CHCl
3), yielding allylic alcohol (
R)-
13 (327 mg, 2.92 mmol, 65% yield) as a yellowish oil: CHPLC (Daicel Chiralpak IC): 88% ee; [α]
D20 = +89.8 (
c 0.1, CHCl
3) [lit: [
40] [α]
D20 = +62.4 (
c 1.0, CHCl
3), ee = 96%];
1H NMR (250 MHz, CDCl
3) δ 5.47 (dq,
J2,1 = 3.3 Hz,
J2,1′ = 1.6 Hz, 1H, H-2), 4.22–4.02 (m, 1H, H-1), 2.06–1.81 (m, 2H, H-6), 1.86–1.57 (m, 2H, H-5), 1.67 (s, 3H, H-1′), 1.64–1.34 (m, 2H, H-4).
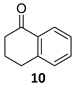
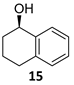
(1R)-1,2,3,4-Tetrahydro-1-naphthalenol [(R)-15]. To a stirred solution of α-tetralone
10 (100 µL, 110 mg, 0.73 mmol) in a biphasic medium of CH
2Cl
2 (2.4 mL) and water (2.4 mL) and under a nitrogen flux, tetrabutylammonium chloride (61 mg, 0.22 mmol), sodium formate (149 mg, 2.19 mmol), and (
R,R)-Noyori-I catalyst [(
R,R)-
5] (13.9 mg, 0.022 mmol) were sequentially added at room temperature. The reaction mixture was allowed to stir for 24 h. Then, CH
2Cl
2 (2 mL) and water (2 mL) were added, the two phases were separated, and the aqueous layer was extracted with CH
2Cl
2 (3 × 1 mL). The volatiles of the organic phase were removed under reduced pressure and the resulting residue was purified by column chromatography (CHCl
3), yielding alcohol (
R)-
15 (97 mg, 0.65 mmol, 87% yield) as a yellowish oil: CHPLC (Daicel Chiralpack OD-H): 94% ee; [α]
D20 = −36.5 (
c 0.98, CHCl
3) [lit: [
40] [α]
D20 = −33.2 (
c 1.0, CHCl
3), ee = 99%];
1H NMR (250 MHz, CDCl
3) δ 7.43 (dd,
J8,7 = 5.5 Hz,
J8,6 = 3.5 Hz, 1H, H-8), 7.25–7.17 (m, 2H, H-6, H-7), 7.11 (dd,
J5,6 = 5.5 Hz,
J5,7 = 3.5 Hz 1H, H-5), 4.76 (t,
J1,2ax =
J1,2eq = 4.5 Hz, 1H, H-1), 2.94–2.62 (m, 2H, H-4), 2.05–1.84 (m, 4H, H-2, H-3);
13C NMR (100 MHz, CDCl
3) δ 138.9 (C-8a), 137.3 (C-4a), 129.2/128.8 (C-5, C-8), 127.7/126.3 (C-6, C-7), 68.3 (C-1), 32.4 (C-2), 29.4 (C-4), 18.9 (C-3).
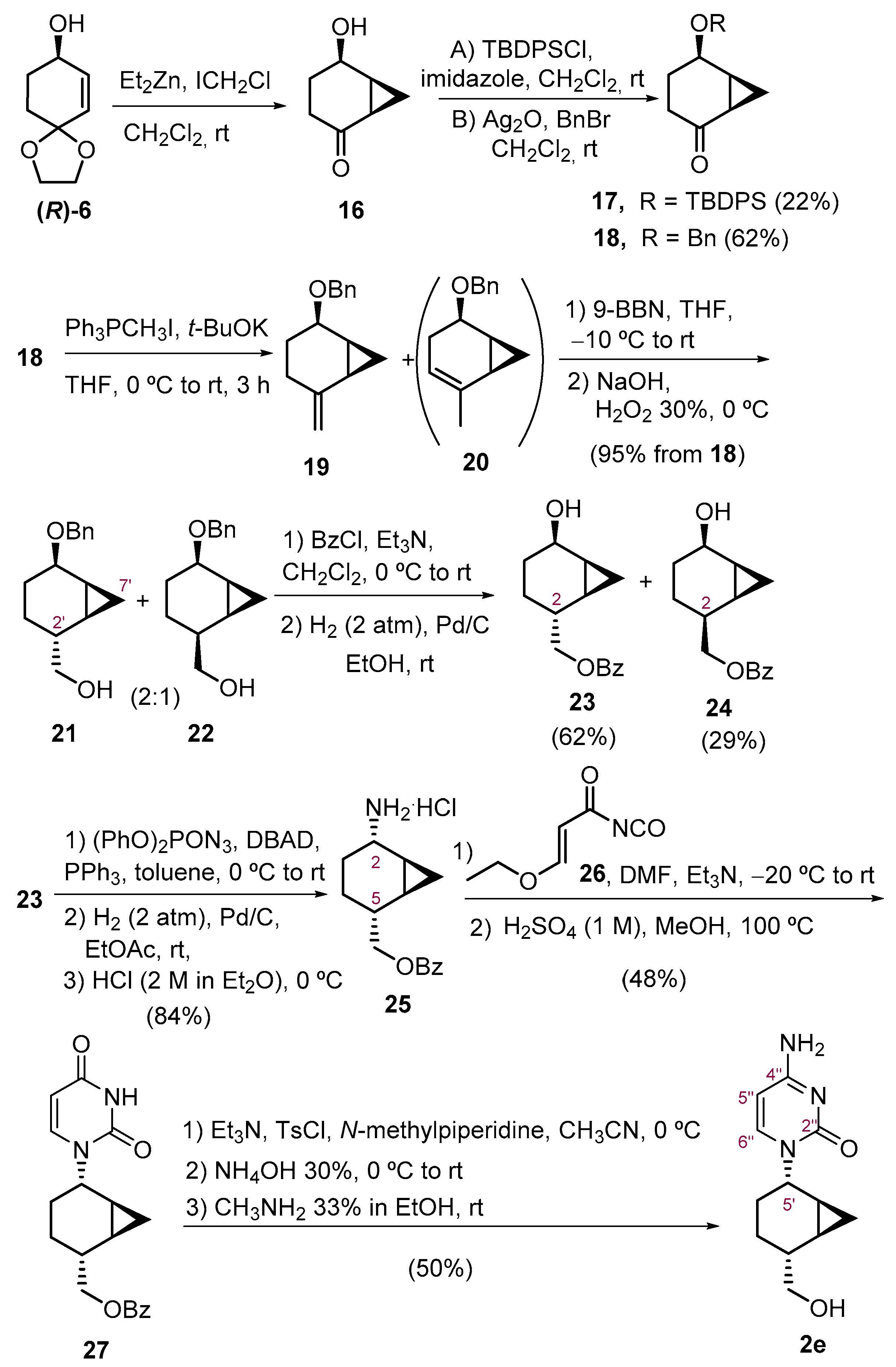
(1R,5R,6S)-5-(Benzyloxy)bicyclo [4.1.0]heptan-2-one(18). To a stirred solution of allylic alcohol (R)-6 (314 mg, 2.01 mmol) in anhydrous CH2Cl2 (10 mL), Et2Zn (4 mL, 4.02 mmol, 1 M in hexane) at 0 ᵒC was added and the mixture was stirred for 5 min at this temperature. Then, a solution of ICH2Cl (600 µL, 8.30 mmol) in CH2Cl2 (3 mL) was added dropwise via syringe at 0 ᵒC, and the mixture was stirred overnight, allowing it to warm to room temperature. Then, a saturated aqueous NaHCO3 solution (10 mL) was added, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The organic layer was dried (Na2SO4), concentrated under reduced pressure (with vacuum controller) and purified by column chromatography (hexane-EtO2, 4:1) furnishing ketone 16 (198 mg, 1.57 mmol, 78% yield) as a colorless oil. Due to the volatility of compound 16, the crude was used without further purification prior to the next step.
To a solution of crude of the hydroxyacetone 16 in CH2Cl2 (4 mL), Ag2O (560 mg, 2.41 mmol) and benzyl bromide (340 µL, 2.81 mmol) were added. The mixture was stirred at room temperature for 24 h, then it was filtered through a Celite® pad and concentrated in vacuo. The crude was purified by column chromatography (hexanes-EtOAc, 6:1) to furnish 18 (270 mg, 1,24 mmol, 62% yield from (R)-6) as a pale oil.
16: [α]
D20 = +78.5 (
c 0.65, CHCl
3) [lit: [
41] [α]
D20 = −80.7 (
c 1.37, CHCl
3), for its enantiomer, ee = 100%];
1H NMR (400 MHz, CDCl
3) δ 4.42 (dddd,
J5,4ax = 10.1 Hz,
J5,6 = 5.2 Hz,
J5,4eq = 4.1 Hz,
J5,3ax = 0.8 Hz, 1H, H-5), 2.37 (ddd,
Jgem = 18.4 Hz,
J3eq,4ax = 5.6 Hz,
J3eq,4eq = 3.6 Hz, 1H, H-3eq), 2.15 (dddd,
Jgem = 18.4 Hz,
J3ax,4ax = 12.1 Hz,
J3ax,4eq = 6.6 Hz,
J3ax,5 = 0.8 Hz, 1H, H-3ax), 1.98–1.83 (m, 3H, H-1, H-6, H-4eq), 1.63 (dddd,
Jgem = 13.8 Hz,
J4ax,3ax = 12.1 Hz,
J4ax,5 = 10.3 Hz,
J4ax,3eq = 5.6 Hz, 1H, H-4ax), 1.44 (q,
Jgem =
J7endo,1 =
J7endo,6 = 5.4 Hz, 1H, H-7endo), 1.14 (ddd,
J7exo,1/6 = 9.8 Hz,
J7exo,1/6 = 7.7 Hz,
Jgem = 5.4 Hz, 1H, H-7exo);
13C NMR (100 MHz, CDCl
3) δ 207.5 (C-2), 65.3 (C-5), 34.8 (C-3), 26.8 (C-1), 26.7 (C-4), 23.3 (C-6), 8.0 (C-7); IR (ATR) ν 3359, 2919, 2850, 1659, 1345, 1066, 1041 (cm
−1). HRMS (ESI+) Calcd. for [C
7H
10O
2+Na]
+ 149.0573, found: 149.0576.
18: [α]D20 = +62.6 (c 1.45, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.34 (m, 4H, H-Ar), 7.32–7.28 (m, 1H, H-Ar), 4.73 (d, Jgem = 11.9 Hz, 1H, CH2-Ph), 4.64 (d, Jgem = 11.9 Hz, 1H, CH2-Ph), 4.15 (dt, J5,4ax = 9.3 Hz, J5,4eq = J5,6 = 4.9 Hz, 1H, H-5), 2.42 (dt, Jgem = 17.7 Hz, J3eq,4 = 5.5 Hz, 1H, H-3eq), 2.12 (ddd, Jgem = 17.7 Hz, J3ax,4x = 10.7 Hz, J3ax,4eq = 6.5 Hz, 1H, H-3ax), 2.05–1.93 (m, 2H, H-4), 1.88–1.71 (m, 2H, H-1, H-6), 1.50 (q, Jgem = J7endo,6 = J7endo,1 = 5.4 Hz, 1H, H-7endo), 1.24 (td, J7exo,6 = J7exo,1 = 9.1 Hz, Jgem = 5.4 Hz, 1H, H-7exo); 13C NMR (100 MHz, CDCl3) δ 208.2 (C-2), 138.5 (C-Ar), 128.6/128.2/127.9/127.8 (C-Ar), 71.3 (C-5), 70.6 (CH2-Ph), 34.4 (C-3), 26.5 (C-4), 25.5 (C-1), 21.3 (C-6), 9.5 (C-7); IR (ATR) ν 3028, 2857, 1692 (C=O), 1342, 1075, 1028, 881, 631 cm−1. HRMS (ESI+) Calcd. for [C14H16O2+H]+ 217.1229, Found: 217.1231.
[(1S’,2R’,5R’,6S’)-5′-(Benzyloxy)bicyclo [4.1.0]hept-2′-yl]methanol (21) and its (2′S)-diastereoisomer (22). To a stirring solution of Ph3PCH3I (1.452 g, 3.59 mmol) in anhydrous THF (4 mL) at 0 °C, t-BuOK (402 mg, 3.58 mmol) was added, under nitrogen atmosphere, and the resulting yellow mixture was allowed to react for 1 h. Then, a solution of ketone 18 (155 mg, 0.71 mmol) in dry THF (1 mL) was added and the mixture was allowed to warm to room temperature and stirred for 3 h. Then, diethyl ether (10 mL) was added and the crude was filtered through a short pad of silica and Celite®, using additional diethyl ether as eluent. The volatiles were removed under vacuum to obtain an orange oil of alkene 19 that was used for the next step without further purification, as it isomerizes to the more stable endocyclic regioisomer 20 at room temperature.
The crude of alkene 19 was rapidly dissolved in anhydrous THF (7 mL) and 9-BBN (4.30 mL, 2.15 mmol, 0.5 M in THF) was added at −10 °C. The mixture was allowed to warm to room temperature and stirred overnight. Then, water (1.2 mL), NaOH (1.5 mL, 3 M in water), and H2O2 (1.4 mL, 30% in water) were added at 0 °C. After stirring for 15 min, the mixture was diluted with brine (15 mL) and CH2Cl2 (15 mL) and the aqueous phase was extracted with additional CH2Cl2 (2 × 10 mL). The organic layers were dried (Na2SO4), concentrated under reduced pressure, and purified by column chromatography (gradient hexane-EtOAc, 5:1 → 2:1) to provide a chromatographically inseparable mixture of alcohols 21 and 22 (158 mg, 0.68 mmol, 95% overall yield from 18) in a ca. 2:1 diastereomeric ratio as a colorless oil. After repeated purification by column chromatography, enriched fractions were obtained and were analyzed by NMR.
19: 1H NMR (400 MHz, CDCl3) δ 7.46–7.25 (m, 5H, H-Ar), 4.90 (br s, 1H, H-1’), 4.79 (br s, 1H, H-1′), 4.74 (d, Jgem = 11.9 Hz, 1H, CH2-Ph), 4.56 (d, Jgem = 11.9 Hz, 1H, CH2-Ph), 4.05 (q, J2,3ax = J2,3eq = J2,1 = 5.9 Hz, 1H, H-2), 2.21 (dddt, Jgem = 14.9 Hz, J4ax,3ax = 8.5 Hz, J4ax,3eq = 4.4 Hz, J4ax,1′ = 1.4 Hz, 1H, H-4ax), 2.03 (dddt, Jgem = 14.9 Hz, J4eq,3ax = 7.2 Hz, J4eq,3eq = 4.3 Hz, J4eq,1′ = 1.4 Hz, 1H, H-4eq), 1.88–1.75 (m, 1H, H-6), 1.71–1.50 (m, 3H, H-1, H-3), 0.94–0.84 (m, 2H, H-7); 13C NMR (100 MHz, CDCl3) δ 146.3 (C-5), 139.1 (C-ipso), 128.4/127.7/127.5 (C-Ar), 108.1 (C-1′), 72.0 (C-2), 69.7 (CH2-Ph), 28.8 (C-4), 27.7 (C-3), 19.9 (C-1), 17.0 (C-6), 8.7 (C-7); IR (ATR) ν 3070, 2930, 1471, 1427, 1106, 806, 741 cm−1. HRMS (ESI+) Calcd. for [C15H18O+H]+ 215.1436, Found: 215.1465.
20: 1H NMR (400 MHz, CDCl3) δ 7.42–7.36 (m, 3H, H-Ar), 7.35–7.25 (m, 2H, H-Ar), 5.01 (dq, J3,4eq = 5.3 Hz, J3,4ax = J3,1′ = 1.6 Hz, 1H, H-3), 4.69 (d, Jgem = 12.0 Hz, 1H, CH2-Ph), 4.65 (d, Jgem = 12.0 Hz, 1H, CH2-Ph), 3.98 (ddd, J5,4ax = 9.1 Hz, J5,4eq = 6.8 Hz, J5,6 = 4.1 Hz, 1H, H-5), 2.32 (ddd, Jgem = 15.3 Hz, J4eq,5 = 6.8 Hz, J4eq,3 = 5.2 Hz, 1H, H-4eq), 1.89–1.75 (m, 4H, H-1′, H-4ax) 1.64–1.51 (m, 1H, H-6), 1.30–1.23 (m, 1H, H-1), 0.90–0.85 (m, 2H, H-7); 13C NMR (100 MHz, CDCl3): δ 139.6 (C-ipso), 136.7 (C-2), 128.8/128.2/127.9 (C-Ar), 114.3 (C-3), 73.4 (C-5), 70.6 (CH2-Ph), 28.0 (C-4), 23.5 (C-1′), 18.0/17.8 (C-1, C-6), 10.1 (C-7); IR (ATR) ν 3064, 2910, 2852, 1667, 1452, 1070, 734, 697 cm−1. HRMS (ESI+) Calcd. for [C15H18O+H]+ 215.1436, Found: 215.1434.
21 and 22 mixture: 1H NMR (400 MHz, CDCl3) (ca. 84% trans-isomer 21) δ 7.40–7.31 (m, 4H, H-Ar), 7.30–7.24 (m, 1H, H-Ar), 4.71 (d, Jgem = 12.0 Hz, 1H, CH2-Ph), 4.59 (d, Jgem = 12.0 Hz, 1H, CH2-Ph), 3.95 (dt, J5′,4′ax = 10.0 Hz, J5′,4′eq = J5′,6′ = 5.7 Hz, 1H, H-5′), 3.57 (d, J1,2′ = 6.6 Hz, 2H, H-1), 1.79 (dtd, Jgem = 13.3 Hz, J4′eq,5′ = J4′eq,3′ax = 5.7 Hz, J4′eq,3′eq = 2.4 Hz, 1H, H-4′eq), 1.70 (dddd, J2′,3′ax = 8.5 Hz, J2′,1 = 6.6 Hz, J2′,3′eq = 5.4 Hz, J2′,1′ = 1.9 Hz, 1H, H-2′), 1.55 (dtd, Jgem = 13.2 Hz, J3′eq,4′ax = J3′eq,2′ = 5.4 Hz, J3′eq,4′eq = 2.4 Hz, 1H, H-3′eq), 1.29–1.26 (m, 1H, H-6′), 1.04 (tdd, Jgem = J4′ax,3′ax = 13.3 Hz, J4′ax,5′ = 10.0 Hz, J4′ax,3′eq = 5.4 Hz, 1H, H-4′ax), 0.99–0.83 (m, 2H, H-1′, H-3′ax), 0.74 (td, J7′exo,6′ = J7′exo,1′ = 8.8 Hz, Jgem = 5.4 Hz, 1H, H-7′exo), 0.46 (q, Jgem = J7′endo,6′ = J7′endo,1′ = 5.4 Hz, 1H, H-7′endo); 1H NMR (400 MHz, CDCl3) (ca. 16% cis-isomer 22, observable signals) δ 7.40–7.31 (m, 4H, H-Ar), 7.30–7.24 (m, 1H, H-Ar), 4.72 (d, Jgem = 11.8 Hz, 1H, CH2-Ph), 4.47 (d, Jgem = 11.8 Hz, 1H, CH2-Ph), 4.01 (ddd, J5′,4′ax = 7.0 Hz, J5′,4′eq = 5.1 Hz, J5′,6′ = 3.4 Hz, 1H, H-5′), 3.57 (m, 1H, H-1a), 3.49 (dd, Jgem = 10.5 Hz, J1b,2′ = 6.6 Hz, 1H, H-1b), 2.05 (dddd, J2′,3′ax = 12.3 Hz, J2′,1a = 10.2 Hz, J2′,1b = 6.6 Hz, J2′,1′ = 5.4 Hz, 1H, H-2′), 1.66–1.57(m, 1H, H-4′a), 1.44–1.33 (m, 2H, H-3′a, H-4′b), 1.18 (tt, J1′,6′ = J1′,7′exo = 8.5 Hz, J1′,7′endo = J1′,2′ = 5.4 Hz, 1H, H-1′), 1.02–0.98 (m, 1H, H-3′b), 0.75 (m, 1H, H-6′), 0.60–0.51 (m, 2H, H-7′); 13C NMR (101 MHz, CDCl3) (ca. 84% trans-isomer 20): δ 139.25 (C-ipso), 128.43 (C-meta), 127.82 (C-para), 127.49 (C-orto), 74.44 (C-5′), 69.62 (CH2-Ph), 67.98 (C-1), 38.10 (C-2′), 25.31 (C-4′), 25.23 (C-3′), 15.90 (C-1′), 14.88 (C-6′), 8.21 (C-7′); 13C NMR (100 MHz, CDCl3) (ca. 16% cis-isomer 21, observable signals) δ 139.22 (C-ipso), 128.43 (C-meta), 127.80 (C-para), 127.44 (C-orto), 70.87 (C-5′), 69.57 (CH2-Ph) 67.78 (C-1), 35.64 (C-2′), 28.43 (C-4′), 18.93 (C-3′), 14.19 (C-1′), 13.37 (C-6′), 1.98 (C-7′). HRMS (ESI+) Calcd. for [C15H20O2+H]+ 233.1542, Found: 233.1537.
[(1′S,2′R,5′R,6′S)-5′-Hydroxybicyclo [4.1.0]hept-2′-yl]methyl benzoate (23) and its (2′S)-diastereoisomer (24). To a stirred solution of a 2:1 mixture of 21 and 22 (158 mg, 0.68 mmol) in dry CH2Cl2 (7 mL) at 0 °C, anhydrous Et3N (100 µL, 0.71 mmol) and benzoyl chloride (91 µL, 0.78 mmol) were sequentially added under argon atmosphere. The mixture was subsequently allowed to attain room temperature overnight. Then, the solution was treated with HCl 10% solution (7 mL) and CH2Cl2 (7 mL), the two phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 7 mL). The organic layers were washed with brine (30 mL), dried (Na2SO4), and concentrated under reduced pressure to obtain a colorless oil that was directly used for the next step without further purification. Accordingly, the crude was dissolved in EtOH (7 mL) and hydrogenated in the presence of Pd/C (23 mg, 10% wt.) at 2 atm overnight. The mixture was filtered through a short pad of Celite® and washed with additional EtOH. The solvent was evaporated under reduced pressure and purified by column chromatography (hexanes-EtOAc, 10:1 → 5:1) to afford alcohol 23 (104 mg, 0.42 mmol, 62% yield) as a colorless syrup and its isomer 24 (49 mg, 0.20 mmol, 29% yield) as a colorless syrup.
23: [α]D20 = +48.7 (c 1,02, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.05 (d, Jorto,meta = 7.6 Hz, 2H, H-orto), 7.61–7.51 (m, 1H, H-para), 7.44 (t, Jmeta,orto = Jmeta,para = 7.6 Hz, 2H, H-meta), 4.28 (dd, J1,2′ = 6.8 Hz, J1,1′ = 2.0 Hz, 2H, H-1), 4.20 (dt, J5′,4′ax = 9.5 Hz, J5′,4′eq = J5′,6′ = 5.7 Hz, 1H, H-5′), 2.0 (dddd, J2′,3′ax = 13.8 Hz, J2′,1 = 6.8 Hz, J2′,3′eq = 4.4 Hz, J2′,1′ = 1.9 Hz, 1H, H-2′), 1.86–1.72 (m, 1H, H-4′), 1.70–1.61 (m, 2H, H-3′), 1.33 (tt, J6′,7′endo = J6′,7′exo = 8.8 Hz, J6′,5′ = J6′,1′ = 5.7 Hz, 1H, H-6′), 1.13–0.87 (m, 2H, H-1′, H-4′), 0.71 (td, J7′exo,1′ = J7′exo,6′ = 8.8 Hz, Jgem = 4.9 Hz, 1H, H-7′exo), 0.40 (q, J7′endo,1′ = J7′endo,6′ = Jgem = 5.3 Hz, 1H, H-7′endo); 13C NMR (100 MHz, CDCl3) δ 166.8 (C=O), 133.1 (C-para), 130.4 (C-ipso), 129.7 (C-orto), 128.5 (C-meta), 69.4 (C-1), 68.0 (C-5′), 34.5 (C-2′), 28.0 (C-4′), 26.0 (C-3′), 18.1 (C-6′), 16.2 (C-1′), 6 (C-7′).
24: [α]D20 = −12.3 (c 1.16, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.05 (d, Jorto,meta = 7.2 Hz, 2H, H-orto), 7.56 (t, Jpara,meta = 7.4 Hz, 1H, H-para), 7.44 (t, Jmeta,orto = Jmeta,para = 7.6 Hz, 2H, H-meta), 4.34 (dt, J5′,4′ax = 7.3 Hz, J5′,4′eq = J5′,6′ = 4.7 Hz, 1H, H-5′), 4.29 (dd, Jgem = 10.7 Hz, J1a,2′ = 6.9 Hz, 1H, H-1a), 4.18 (dd, Jgem = 10.7 Hz, J1b,2′ = 7.3 Hz, 1H, H-1b), 2.36 (dq, J2′,3′ax = 12.6 Hz, J2′,1a = J2′,1b = J2′,3′eq = 6.7 Hz, 1H, H-2′), 1.51–1.37 (m, 4H, H-6′, 2H-4′, H-3′), 1.27 (tt, J1′,2′ = J1′,7′exo = 8.5 Hz, J1′,7′endo = J1′,6′ = 5.7 Hz, 1H, H-1′), 1.23–1.13 (m, 1H, H-3′), 0.57 (q, Jgem = J7′endo,6′ = J7′endo,1′ = 5.3 Hz, 1H, H-7′endo), 0.46 (td, J7′exo,1′ = J7′exo,6′ = 8.9 Hz, Jgem = 5.1 Hz, 1H, H-7′exo); 13C NMR (100 MHz, CDCl3) δ 166.8 (C=O), 133.0 (C-para), 130.6 (C-ipso), 129.7 (C-orto), 128.5 (C-meta), 68.9 (C-1), 64.7 (C-5′), 31.7 (C-2′), 29.9 (C-4′), 19.4 (C-3′), 17.1 (C-1′), 14.7 (C-6′), 1.7 (C-7′).
(1S,2S,5R,6R)-5-[(Benzoyloxy)methyl)]bicyclo [4.1.0]heptan-2-amonium chloride (25). To a stirred solution of Ph3P (170 mg, 0.65 mmol) in dry toluene (4.5 mL), DBAD (150 mg, 0.65 mmol) was slowly added under argon atmosphere and the mixture was stirred for 45 min at 0 °C. After 15 min, a white suspension appeared. Then, diphenylphosphoryl azide (DPPA, 100 µL, 0.45 mmol) and a solution of 23 (105 mg, 0.43 mmol) in dry toluene (1 mL) were sequentially added. The mixture was allowed to warm to room temperature and stirred overnight. Then, the solvent was removed, and the crude was purified by column chromatography (hexanes-EtOAc, 20:1 to 15:1) to obtain a crude that was dissolved in EtOAc (2 mL) and hydrogenated in the presence of Pd/C (15 mg, 10% wt.) at 2 atm for 24 h. Then, the mixture was filtered through a short pad of Celite® and washed with additional EtOAc. The solvent was evaporated under reduced pressure and the crude was treated with 2 M HCl in Et2O (2 mL, 4 mmol) at 0 °C, stirred for 2 h, and filtered to furnish the ammonium salt 25 (101 mg, 0.36 mmol, 84% yield) as a brown solid: Mp 130–132 °C (from EtO2); [α]D20 = +14.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 3H, NH3+), 8.08–7.97 (m, 2H, H-Ar), 7.56–7.50 (m, 1H, H-Ar), 7.44–7.36 (m, 2H, H-Ar), 4.45 (d, J1′,5 = 7.7 Hz, 2H, H-1′), 3.84 (br s, 1H, H-2), 2.15–1.89 (m, 2H, H-5, H-4), 1.60–1.46 (m, 3H, H-3, H-4), 1.31–1.22 (m, 1H, H-1), 1.12–1.02 (m, 1H, H-6), 0.91 (td, J7exo,1 = J7exo,6 = 9.1 Hz, Jgem = 5.1 Hz, 1H, H-7exo), 0.19 (q, Jgem = J7endo,1 = J7endo,6 = 5.5 Hz, 1H, H-7endo); 13C NMR (100 MHz, CDCl3) δ 166.6 (C=O), 133.1 (C-para), 130.4 (C-ipso), 129.7 (C-orto), 128.5 (C-meta), 69.0 (C-1′), 47.1 (C-2), 33.9 (C-5), 23.2 (C-4), 19.4 (C-3), 14.0 (C-6), 12.5 (C-1), 10.4 (C-7); IR (ATR) ν 3404, 2929, 1712, 1273, 1113, 713 cm−1. HRMS (ESI+) Calcd. for [C15H18NO2]+ 244.1338, Found: 244.1334.
(E)-3-Ethoxyacryloyl isocyanate (26). Vinyl ether (5.75 mL, 60 mmol) was added dropwise to oxalyl chloride (7.60 mL, 90 mmol) at 0 °C. The reaction mixture was maintained for 2 h at 0 °C and then warmed to room temperature overnight. Excess oxalyl chloride was distilled off and the black residue was heated at 120 °C for 30 min. Then, the residue was purified by vacuum distillation, using a short Vigreux column, to obtain (2E)-3-ethoxyacryloyl chloride (4.30 g, 31.97 mmol, 53% yield) as a colorless liquid.
Silver cyanate (90 mg, 0.60 mmol), previously dried over phosphorus pentoxide at 80 °C for 3 h, in dry benzene (2 mL), was heated to reflux for 30 min and a solution of (2E)-3-ethoxyacryloyl chloride (45 mg, 0.31 mmol) in dry benzene (0.8 mL) was then added dropwise. The mixture was stirred for 30 min before allowing the solid to settle. The supernatant, which is a solution of the isocyanate 25, was then decanted and used directly in the next reaction.
[(1′R,2′R,5′S,6′S)-5′-(2″,4″-dioxo-3″,4″-dihydropyrimidin-1″(2H)-yl] bicyclo [4.1.0]heptan-2′-yl)methyl benzoate (27). The ammonium chloride 25 (43 mg, 0.153 mmol) was dissolved in dry DMF (1.8 mL), and Et3N (22 µL, 0.157 mmol) was added. The mixture was cooled to −20 °C and a freshly prepared solution of the isocyanate 26 was added slowly to avoid an increase in the temperature. The reaction mixture was stirred overnight at room temperature. The solvent was evaporated in vacuo, and then water (2 mL) was added. The residue was extracted with EtOAc (2 × 2 mL), washed with brine (2 mL), dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue was dissolved in MeOH (0.40 mL), H2SO4 (1 M, 0.62 mL) was added, and the mixture was heated to reflux for 3 h. Then, the mixture was concentrated under reduced pressure and purified by column chromatography (CH2Cl2 100% to CH2Cl2-MeOH, 20:1) to provide 27 (25 mg, 0.073 mmol, 48% yield) as a yellowish oil: 1H NMR (400 MHz, MeOH-d4) δ 8.05 (d, Jorto,meta = 7.6 Hz, 2H, H-orto), 7.96 (d, J6″,5″ = 8.0 Hz, 1H, H-6″), 7.64 (d, Jpara,meta = 7.6 Hz, 1H, H-para), 7.52 (t, Jmeta,orto = Jmeta,para = 7.6 Hz, 2H, H-meta), 5.46 (d, J5″,6″ = 8.0 Hz, 1H, H-5″), 4.83 (dt, J5′,4′ax = 4.3 Hz, J5′,4eq = J5′,6′ = 2.3 Hz, 1H, H-5′), 4.45 (d, J1,2′ = 6.0 Hz, 2H, H-1), 2.21 (dq, J2′,3′ax = 11.9 Hz, J2′,3′eq = J2′1 = 6.0 Hz, 1H, H-2′), 1.78–1.68 (m, 1H, H-4′eq), 1.54 (tt, Jgem = J4′ax,3′ax = 14.7 Hz, J4′ax,5′ = J4′ax,3′eq = 4.3 Hz, 1H, H-4′ax), 1.49–1.40 (m, 1H, H-3′), 1.32–1.21 (m, 2H, H-3′, H-6′), 1.12–1.04 (m, 1H, H-1′), 0.97 (td, J7exo,1 = J7exo,6 = 9.4 Hz, Jgem = 5.0 Hz, 1H, H-7′exo), 0.39 (q, Jgem = J7endo,1 = J7endo,6 = 5.3 Hz, 1H, H-7′endo); 13C NMR (101 MHz, MeOH-d4): δ 168.1 (C=O), 166.3 (C-4″), 152.7 (C-2″), 145.1 (C-6″), 134.4 (C-para), 131.5 (C-ipso), 130.5 (C-orto), 129.7 (C-meta), 101.6 (C-5″), 69.9 (C-1), 52.5 (C-5′), 34.7 (C-2′), 24.5 (C-4′), 20.1 (C-3′), 15.3 (C-6′), 13.7 (C-1′), 10.4 (C-7′). HRMS (ESI+) Calcd. for [C19H20N2O4+Na]+ 363.1321, Found: 363.1312.
4-Amino-1-[(1′S,2′S,5′R,6′R)-5′-(hydroxymethyl)bicyclo [4.1.0]hept-2′-yl]pyrimidin-2(1H)-one (2e). A solution of TsCl (31 mg, 0.16 mmol) in dry CH3CN (104 µL) was added to a mixture of 27 (27 mg, 0.08 mmol), Et3N (22 µL, 0.16 mmol), and N-methylpiperidine (12 µL, 0.1 mmol) in dry CH3CN (135 µL) at 0 °C, and the reaction mixture was stirred for 3 h. Then, 30% NH4OH was added at 0 °C, and the reaction solution was stirred at room temperature overnight. The mixture was diluted with water (1 mL) and EtOAc (1 mL) and the aqueous phase was extracted with additional CH2Cl2 (2 × 1 mL). The organic layers were dried (Na2SO4), concentrated under reduced pressure, and purified by column chromatography (CH2Cl2-EtOAc, 10:2 → CH2Cl2-MeOH 15:1) to provide the protected cytosine analogue as a yellowish oil that was dissolved in a 33% solution of methylamine in EtOH (17 mL) and stirred for 24 h. Then, the mixture was concentrated under reduced pressure and purified by column chromatography (CH2Cl2-MeOH, 20:1) to provide cytosine nucleoside analogue 2e (9 mg, 38 μmol, 50% yield) as a yellowish solid: 2e: [α]D20 = +36.2 (c 0.4, MeOH-d4); 1H NMR (400 MHz, MeOH-d4) δ 8.04 (d, J6,5 = 7.4 Hz, 1H, H-6), 5.90 (d, J5,6 = 7.4 Hz, 1H, H-5), 4.91–4.87 (m, 1H, H-2′) 3.64 (dd, Jgem = 10.7 Hz, J1″a,5′ = 5.9 Hz, 1H, H-1″a), 3.59 (dd, Jgem = 10.7 Hz, J1″a,5′ = 5.9 Hz, 1H, H-1″b), 1.85 (dq, J5′,4′ax = 11.4 Hz, J5′4′eq = J5′,1″a = J5′,1″b = 5.9 Hz, 1H, H-5′), 1.68 (dt, Jgem = 13.7 Hz, J3′eq,4′ax = J3′eq,5′ = 4.4 Hz, 1H, H-3′eq), 1.48 (tt, Jgem = J3′ax,4′ax = 13.7 Hz, J3′ax, 4′eq = J3′ax,5′ = 3.6 Hz, 1H, H-3′ax), 1.34–1.26 (m, 2H, H-1′, H-4′a), 1.19–1.07 (m, 1H, H-4′b), 1.00 (td, J6′,7′exo = J6′,1′ = 9.4 Hz, J6′,7′endo = 5.2 Hz, 1H, H-6′), 0.89 (td, J7exo,1 = J7exo,6 = 9.4, Jgem = 5.2 Hz, 1H, H-7′exo), 0.30 (q, Jgem = J7endo,1 = J7endo,6 = 5.2 Hz, 1H, H-7′endo); 13C NMR (100 MHz, MeOH-d4): δ 167.3 (C-4), 159.0 (C-2), 145.6 (C-6), 95.0 (C-5), 67.6 (C-1″), 52.8 (C-2′), 37.5 (C-5′), 24.6 (C-3′), 19.6 (C-4′), 15.7 (C-1′), 13.81 (C-6′), 10.26 (C-7′). HRMS (ESI+) Calcd. for [C12H17N3O2+H]+ 236.1399, Found: 236.1394.


 ,
, 


{kind=link}
{kind=link}
{kind=link}