
Transcriptome Dynamics Underlying Magnesium Deficiency Stress in Three Founding Saccharum Species

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Observation of Three Saccharum Species under Different Mg2+ Treatment
2.2. Transcriptome Sequencing and Gene Expression Analysis
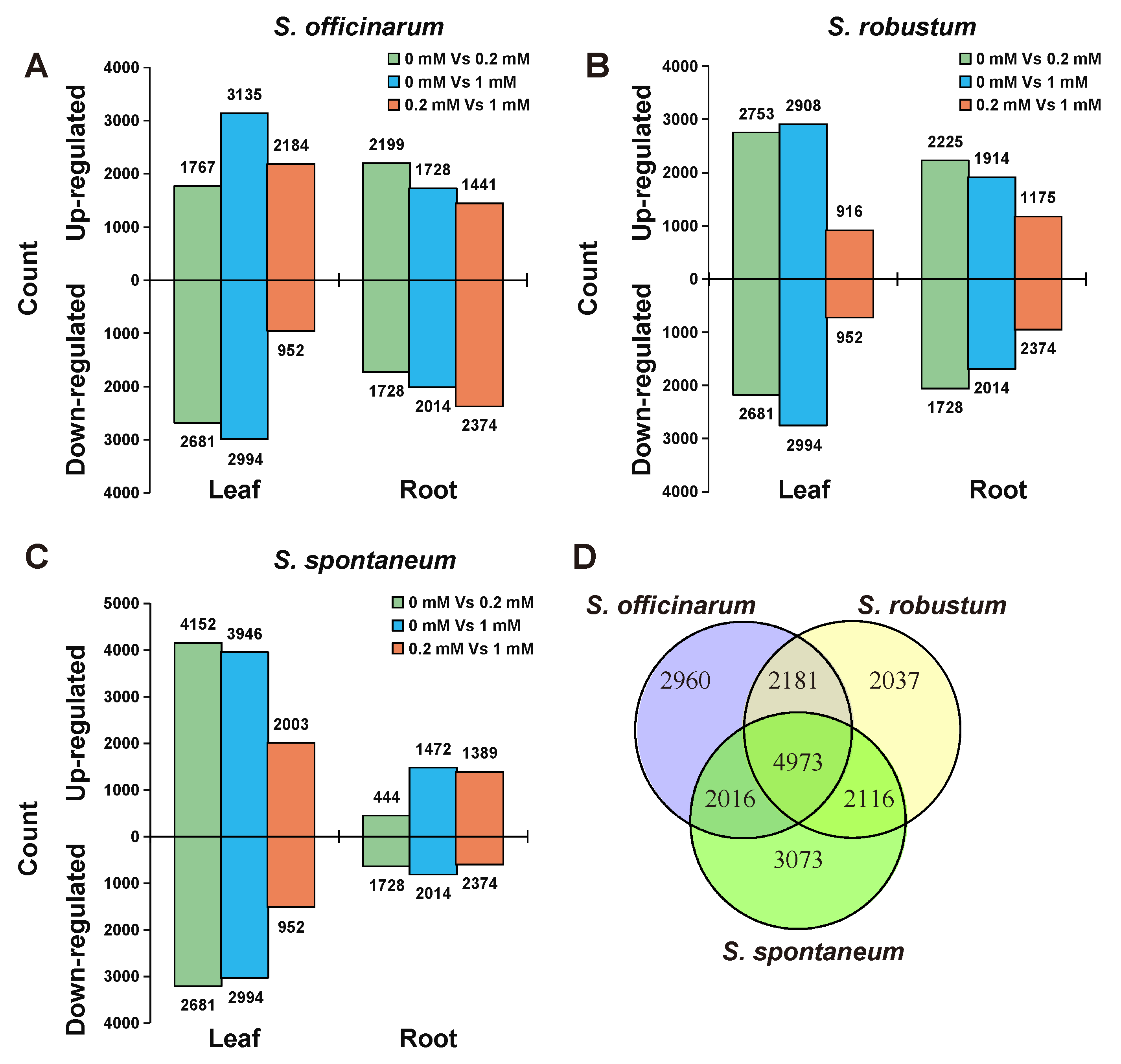
2.3. The Divergence of Transcriptome among the Three Species under Treatment of Mg2+
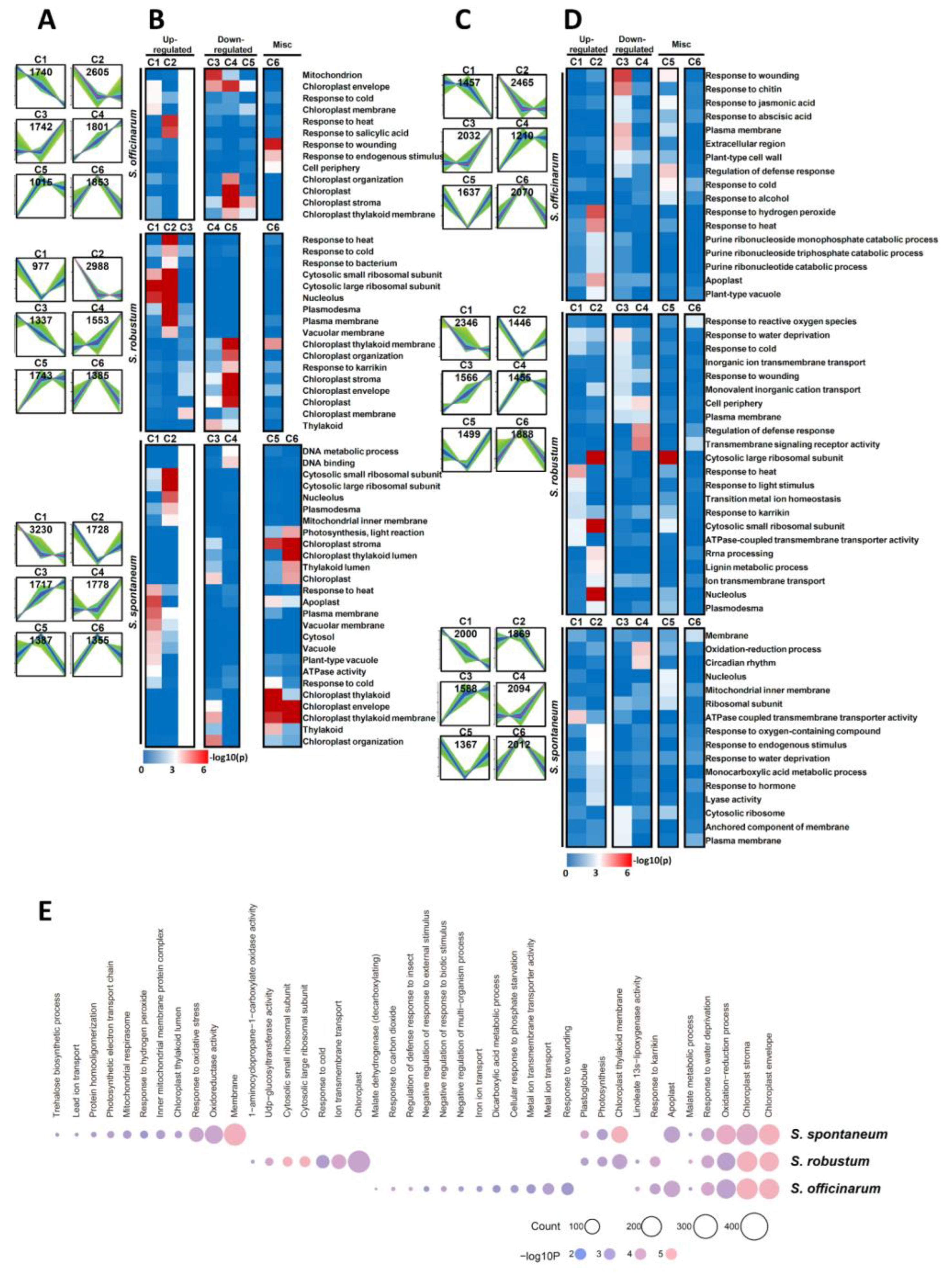
2.4. GO Category Enrichment of DEGs among Three Saccharum Species under MGD Stress
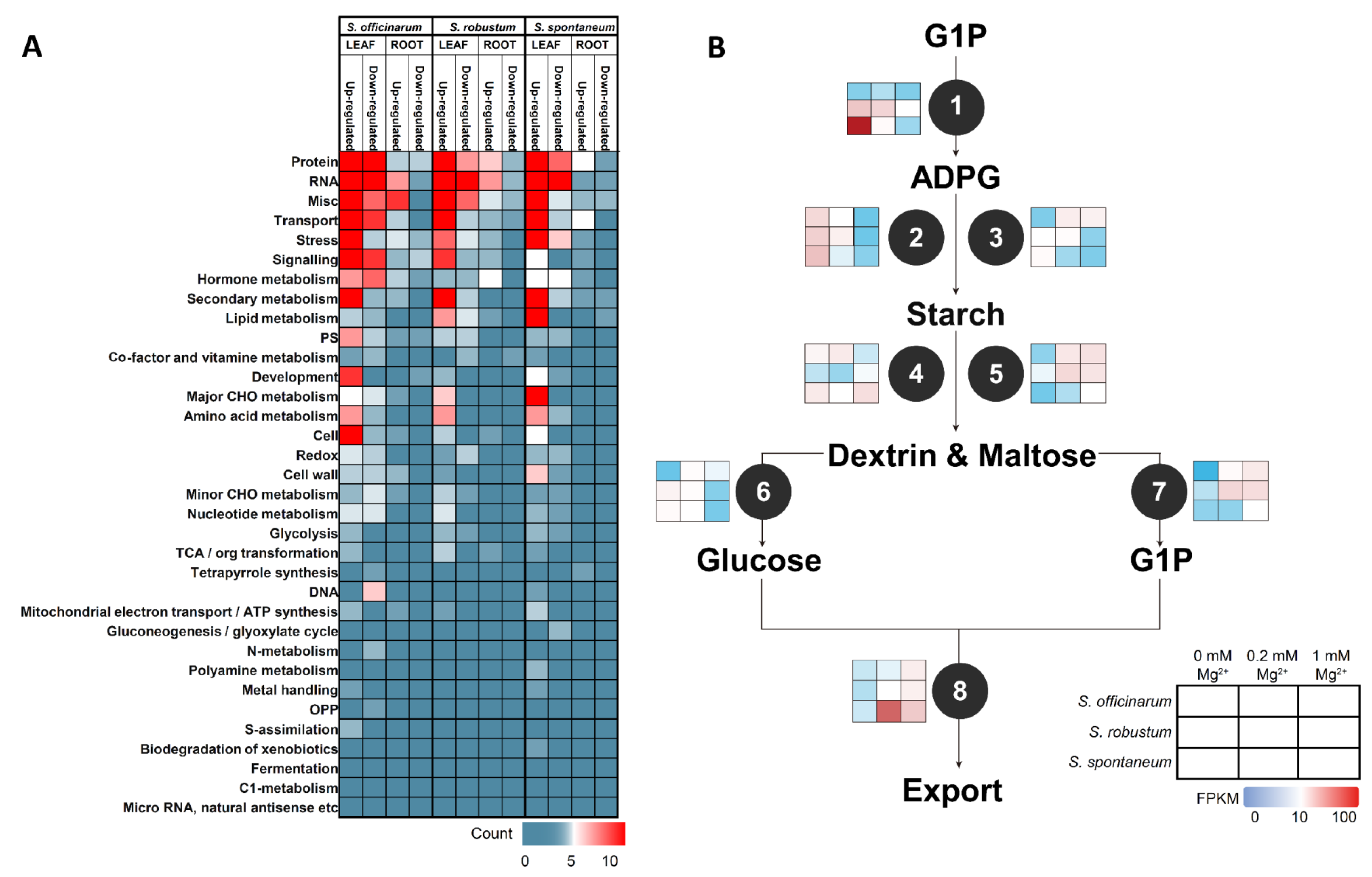
2.5. Mapman Annotation DEGs under MGD
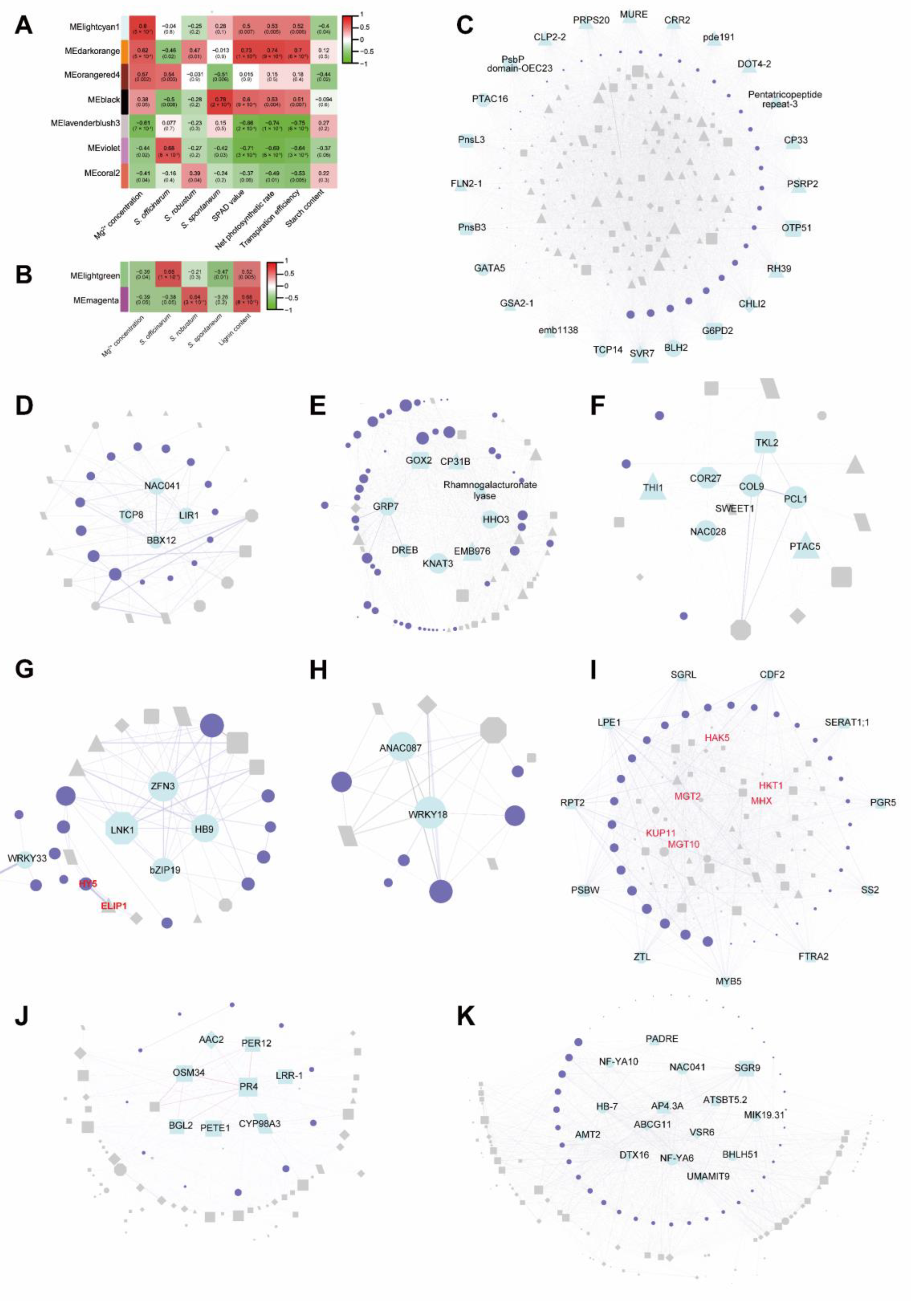
2.6. Co-Expression Networks Analysis by WGCNA
2.7. Hub Genes Investigating by WGCNA
2.8. Verification of Gene Expression Based on RNA-Seq by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Magnesium Stress Treatment
4.3. Determination of Lignin Content
4.4. Determination of Chlorophyll Content and Net Photosynthetic Efficiency
4.5. Detection of Starch Content
4.6. RNA Extraction and Library Construction
4.7. Gene Expression Analysis
4.8. Validation of FPKM Values for MGD Response Genes by qRT-PCR
4.9. Gene Co-Expression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silva, M.d.A.; Jifon, J.L.; Santos, C.M.d.; Jadoski, C.J.; Silva, J.A.G.D. Photosynthetic capacity and water use efficiency in sugarcane genotypes subject to water deficit during early growth phase. Braz. Arch. Biol. Technol. 2013, 56, 735–748. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Grivet, L.; Arruda, P. Sugarcane genomics: Depicting the complex genome of an important tropical crop. Curr. Opin. Plant Biol. 2002, 5, 122–127. [Google Scholar] [CrossRef]
- Talukdar, D.; Verma, D.K.; Malik, K.; Mohapatra, B.; Yulianto, R. Sugarcane as a potential biofuel crop. In Sugarcane Biotechnology: Challenges and Prospects; Springer: Cham, Switzerland, 2017; pp. 123–137. [Google Scholar]
- Roach, B.T. Nobilization of sugarcane. Proc. Int. Soc Sugar Cane Technol. 1972, 14, 206–216. [Google Scholar]
- Kaffka, S.R.; Grantz, D.A. Sugar Crops. In Encyclopedia of Agriculture and Food Systems; Van Alfen, N.K., Ed.; Academic Press: Oxford, UK, 2014; pp. 240–260. [Google Scholar]
- Irvine, J.E. Saccharum species as horticultural classes. Theor. Appl. Genet. 1999, 98, 186–194. [Google Scholar] [CrossRef]
- Endres, L.; Silva, J.V.; Ferreira, V.M.; Souza Barbosa, G.V.D. Photosynthesis and water relations in Brazilian sugarcane. Open Agric. J. 2010, 4, 31–37. [Google Scholar] [CrossRef]
- Harter, R.D. Acid Soils of the Tropics. ECHO Technical Note. 2007. Available online: http://courses.umass.edu/psoil370/Syllabus-files/Acid_Soils_of_the_Tropics.pdf (accessed on 10 July 2020).
- Sanchez, P.A.; Logan, T.J. Myths and science about the chemistry and fertility of soils in the tropics. SSSA Spec. Publ. 1992, 29, 35–46. [Google Scholar]
- Chen, Z.C.; Peng, W.T.; Li, J.; Liao, H. Functional Dissection and Transport Mechanism of Magnesium in Plants. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 142–152. [Google Scholar]
- Kurvits, A.; Kirkby, E. The uptake of nutrients by sunflower plants (Helianthus annum) growing in a continuous flowing culture system, supplied with nitrate or ammonium as nitrogen source. Z. Pflanz. Bodenkd. 1980, 143, 140–149. [Google Scholar] [CrossRef]
- Rhodes, R.; Miles, N.; Hughes, J.C. Interactions between potassium, calcium and magnesium in sugarcane grown on two contrasting soils in South Africa. Field Crops Res. 2018, 223, 1–11. [Google Scholar] [CrossRef]
- Heenan, D.; Campbell, L. Influence of potassium and manganese on growth and uptake of magnesium by soybeans (Glycine max (L.) Merr. cv. Bragg). Plant Soil 1981, 61, 447–456. [Google Scholar] [CrossRef]
- Broadley, M.; Brown, P.; Cakmak, I.; Rengel, Z.; Zhao, F. Chapter 7—Function of Nutrients: Micronutrients. In Marschners Mineral Nutrition of Higher Plants, 3rd ed.; Marschner, P., Ed.; Academic Press: San Diego, CA, USA, 2012; pp. 191–248. [Google Scholar]
- Marschner, H. Marschner’s Mineral Nutrition of Higher Plants; Academic Press: San Diego, CA, USA, 2012; Volume 89. [Google Scholar]
- Xie, K.; Cakmak, I.; Wang, S.; Zhang, F.; Guo, S. Synergistic and antagonistic interactions between potassium and magnesium in higher plants. Crop J. 2021, 9, 249–256. [Google Scholar] [CrossRef]
- Ogura, T.; Kobayashi, N.I.; Hermans, C.; Ichihashi, Y.; Shibata, A.; Shirasu, K.; Aoki, N.; Sugita, R.; Ogawa, T.; Suzuki, H. Short-term magnesium deficiency triggers nutrient retranslocation in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Cakmak, I.; Feng, J.; Yu, C.; Chen, X.; Xie, D.; Wu, L.; Song, Z.; Cao, J.; He, Y. Magnesium deficiency reduced the yield and seed germination in wax gourd by affecting the carbohydrate translocation. Front. Plant Sci. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Hauer-Jákli, M.; Tränkner, M. Critical leaf magnesium thresholds and the impact of magnesium on plant growth and photo-oxidative defense: A systematic review and meta-analysis from 70 years of research. Front. Plant Sci. 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Crusciol, C.A.C.; McCray, J.M.; Nascimento, C.A.C.; Martello, J.M.; de Siqueira, G.F.; Tarumoto, M.B. Magnesium as a Promoter of Technological Quality in Sugarcane. J. Soil Sci. Plant Nutr. 2020, 20, 19–30. [Google Scholar] [CrossRef]
- Weise, S.E.; van Wijk, K.J.; Sharkey, T.D. The role of transitory starch in C3, CAM, and C4 metabolism and opportunities for engineering leaf starch accumulation. J. Exp. Bot. 2011, 62, 3109–3118. [Google Scholar] [CrossRef]
- Cakmak, I.; Hengeler, C.; Marschner, H. Changes in phloem export of sucrose in leaves in response to phosphorus, potassium and magnesium deficiency in bean plants. J. Exp. Bot. 1994, 45, 1251–1257. [Google Scholar] [CrossRef]
- Farhat, N.; Smaoui, A.; Maurousset, L.; Porcheron, B.; Lemoine, R.; Abdelly, C.; Rabhi, M. Sulla carnosa modulates root invertase activity in response to the inhibition of long−distance sucrose transport under magnesium deficiency. Plant Biol. 2016, 18, 1031–1037. [Google Scholar] [CrossRef]
- Shabala, S.; Hariadi, Y. Effects of magnesium availability on the activity of plasma membrane ion transporters and light-induced responses from broad bean leaf mesophyll. Planta 2005, 221, 56–65. [Google Scholar] [CrossRef]
- Liang, S.; Qi, Y.; Zhao, J.; Li, Y.; Wang, R.; Shao, J.; Liu, X.; An, L.; Yu, F. Mutations in the Arabidopsis AtMRS2-11/AtMGT10/VAR5 gene cause leaf reticulation. Front. Plant Sci. 2017, 8, 2007. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and biological functions in plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- van de Mortel, J.E.; Almar Villanueva, L.; Schat, H.; Kwekkeboom, J.; Coughlan, S.; Moerland, P.D.; Ver Loren van Themaat, E.; Koornneef, M.; Aarts, M.G.M. Large expression differences in genes for iron and zinc homeostasis, stress response, and lignin biosynthesis distinguish roots of Arabidopsis thaliana and the related metal hyperaccumulator Thlaspi caerulescens. Plant Physiol. 2006, 142, 1127–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissenberg, K.; Claeijs, N.; Balcerowicz, D.; Schoenaers, S. Hormonal regulation of root hair growth and responses to the environment in Arabidopsis. J. Exp. Bot. 2020, 71, 2412–2427. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, Q.; Wu, J.; An, L.; Zhou, Z.; Wong, C.; Wu, M.; Yu, H.; Gan, Y. Zinc finger protein 5 (ZFP5) associates with ethylene signaling to regulate the phosphate and potassium deficiency-induced root hair development in Arabidopsis. Plant Mol. Biol. 2020, 102, 143–158. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Wang, Z.; Zhang, Z.; Wu, Z. Identifying key regulatory genes of maize root growth and development by RNA sequencing. Genomics 2020, 112, 5157–5169. [Google Scholar] [CrossRef]
- Mélida, H.; Largo−Gosens, A.; Novo−Uzal, E.; Santiago, R.; Pomar, F.; García, P.; García−Angulo, P.; Acebes, J.L.; Álvarez, J.; Encina, A. Ectopic lignification in primary cellulose−deficient cell walls of maize cell suspension cultures. J. Integr. Plant Biol. 2015, 57, 357–372. [Google Scholar] [CrossRef]
- Yan, J.; Aznar, A.; Chalvin, C.; Birdseye, D.S.; Baidoo, E.E.K.; Eudes, A.; Shih, P.M.; Loqué, D.; Zhang, A.; Scheller, H.V. Increased drought tolerance in plants engineered for low lignin and low xylan content. Biotechnol. Biofuels 2018, 11, 195. [Google Scholar] [CrossRef]
- Zanetti, M.E.; Rípodas, C.; Niebel, A. Plant NF-Y transcription factors: Key players in plant-microbe interactions, root development and adaptation to stress. Biochim. Biophys. Acta 2017, 1860, 645–654. [Google Scholar] [CrossRef]
- Yu, Y.; Bai, Y.; Wang, P.; Wang, Y.; Wan, H.; Liu, C.; Ni, Z. Soybean nuclear factor YA10 positively regulates drought resistance in transgenic Arabidopsis thaliana. Environ. Exp. Bot. 2020, 180, 104249. [Google Scholar] [CrossRef]
- Ma, X.; Zhu, X.; Li, C.; Song, Y.; Zhang, W.; Xia, G.; Wang, M. Overexpression of wheat NF-YA10 gene regulates the salinity stress response in Arabidopsis thaliana. Plant Physiol. Biochem. 2015, 86, 34–43. [Google Scholar] [CrossRef]
- Ye, X.; Huang, H.-Y.; Wu, F.-L.; Cai, L.-Y.; Lai, N.-W.; Deng, C.-L.; Guo, J.-X.; Yang, L.-T.; Chen, L.-S. Molecular mechanisms for magnesium-deficiency-induced leaf vein lignification, enlargement and cracking in Citrus sinensis revealed by RNA-Seq. Tree Physiol. 2021, 41, 280–301. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Mochizuki, N.; Yoshimura, N.; Motohashi, K.; Hisabori, T.; Masuda, T. Functional analysis of Arabidopsis thaliana isoforms of the Mg-chelatase CHLI subunit. Photochem. Photobiol. Sci. 2008, 7, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.Y.; Liao, L.L.; Liu, S.; Nie, M.M.; Li, J.; Zhang, L.D.; Ma, J.F.; Chen, Z.C. Magnesium deficiency triggers SGR–mediated chlorophyll degradation for magnesium remobilization. Plant Physiol. 2019, 181, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Arro, J.; Chen, Y.; Ming, R. Haplotype analysis of sucrose synthase gene family in three Saccharumspecies. BMC Genom. 2013, 14, 314. [Google Scholar] [CrossRef]
- Epstein, E. Mineral Nutrition of Plants: Principles and Perspectives; John Wiley: New York, NY, USA, 1972. [Google Scholar]
- Liu, Y.; Yan, J.; Wang, K.; Li, D.; Yang, R.; Luo, H.; Zhang, W. MiR396−GRF module associates with switchgrass biomass yield and feedstock quality. Plant Biotechnol. J. 2021, 19, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Sunkar, R.; Zhou, C.; Shen, H.; Zhang, J.Y.; Matts, J.; Wolf, J.; Mann, D.G.; Stewart, C.N., Jr.; Tang, Y. Overexpression of miR156 in switchgrass (Panicum virgatum L.) results in various morphological alterations and leads to improved biomass production. Plant Biotechnol. J. 2012, 10, 443–452. [Google Scholar] [CrossRef]
- Xiao, L.; Jiang, S.; Huang, P.; Chen, F.; Wang, X.; Cheng, Z.; Miao, Y.; Liu, L.; Searle, I.; Liu, C. Two Nucleoporin98 homologous genes jointly participate in the regulation of starch degradation to repress senescence in Arabidopsis. BMC Plant Biol. 2020, 20, 292. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Giesen, P.; Berry, C.M. Reconstruction and growth of the early tree Calamophyton (Pseudosporochnales, Cladoxylopsida) based on exceptionally complete specimens from Lindlar, Germany (Mid-Devonian): Organic connection of Calamophyton branches and Duisbergia trunks. Int. J. Plant Sci. 2013, 174, 665–686. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | GO | Biological Process | Log10 (P) |
|---|---|---|---|
| lightcyan1 (R = 0.8, p value = 5 × 10−7) | GO:0070413 | trehalose metabolism in response to stress | −5.7 |
| GO:0001666 | response to hypoxia | −4.2 | |
| GO:1905039 | carboxylic acid transmembrane transport | −3.32 | |
| darkorange (R = 0.62, p value = 5 × 10−4) | GO:0014070 | response to organic cyclic compound | −2.88 |
| GO:0007623 | circadian rhythm | −2.46 | |
| GO:0009737 | response to abscisic acid | −2.37 | |
| orangered4 (R = 0.57, p value = 0.002) | GO:0009657 | plastid organization | −21.44 |
| GO:0015979 | photosynthesis | −16.04 | |
| GO:0015994 | chlorophyll metabolic process | −8.9 | |
| black (R = 0.38, p value = 0.05) | GO:0001666 | response to hypoxia | −4.67 |
| GO:0000967 | rRNA 5’ −end processing | −4.46 | |
| GO:0009640 | photomorphogenesis | −4.44 | |
| lavenderblush3 (R = −0.61, p value = 7 × 10−4) | GO:0010035 | response to inorganic substance | −4.47 |
| GO:0044282 | small molecule catabolic process | −4.14 | |
| GO:0043648 | dicarboxylic acid metabolic process | −3.78 | |
| violet (R = −0.44, p value = 0.02) | GO:0015849 | organic acid transport | −5.64 |
| GO:0015979 | photosynthesis | −5.45 | |
| GO:0009639 | response to red or far red light | −4.95 | |
| coral2 (R = −0.41, p value = 0.04) | GO:0042273 | ribosomal large subunit biogenesis | −11.17 |
| GO:0002181 | cytoplasmic translation | −8.91 | |
| GO:0043648 | dicarboxylic acid metabolic process | −4.9 |
| Module | GO | Biological Process | Log10 (P) |
|---|---|---|---|
| lightgreen (R = −0.39, p value = 0.04) | GO:0009808 | lignin metabolic process | −4.83 |
| GO:0045492 | xylan biosynthetic process | −4.09 | |
| GO:0044282 | small molecule catabolic process | −3.98 | |
| magenta (R = −0.39, p value = 0.05) | GO:0009699 | phenylpropanoid biosynthetic process | −6.28 |
| GO:0010035 | response to inorganic substance | −5.95 | |
| GO:0009308 | amine metabolic process | −4.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, Y.; Hua, X.; Zhang, Z.; Fan, T.; Yao, W.; Zhang, M.; Zhang, J. Transcriptome Dynamics Underlying Magnesium Deficiency Stress in Three Founding Saccharum Species. Int. J. Mol. Sci. 2022, 23, 9681. https://doi.org/10.3390/ijms23179681
Wang Y, Li Y, Hua X, Zhang Z, Fan T, Yao W, Zhang M, Zhang J. Transcriptome Dynamics Underlying Magnesium Deficiency Stress in Three Founding Saccharum Species. International Journal of Molecular Sciences. 2022; 23(17):9681. https://doi.org/10.3390/ijms23179681
Chicago/Turabian StyleWang, Yongjun, Yihan Li, Xiuting Hua, Zhe Zhang, Tianqu Fan, Wei Yao, Muqing Zhang, and Jisen Zhang. 2022. "Transcriptome Dynamics Underlying Magnesium Deficiency Stress in Three Founding Saccharum Species" International Journal of Molecular Sciences 23, no. 17: 9681. https://doi.org/10.3390/ijms23179681
APA StyleWang, Y., Li, Y., Hua, X., Zhang, Z., Fan, T., Yao, W., Zhang, M., & Zhang, J. (2022). Transcriptome Dynamics Underlying Magnesium Deficiency Stress in Three Founding Saccharum Species. International Journal of Molecular Sciences, 23(17), 9681. https://doi.org/10.3390/ijms23179681