
Bis-Cyclic Guanidine Heterocyclic Peptidomimetics as Opioid Ligands with Mixed μ-, κ- and δ-Opioid Receptor Interactions: A Potential Approach to Novel Analgesics

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
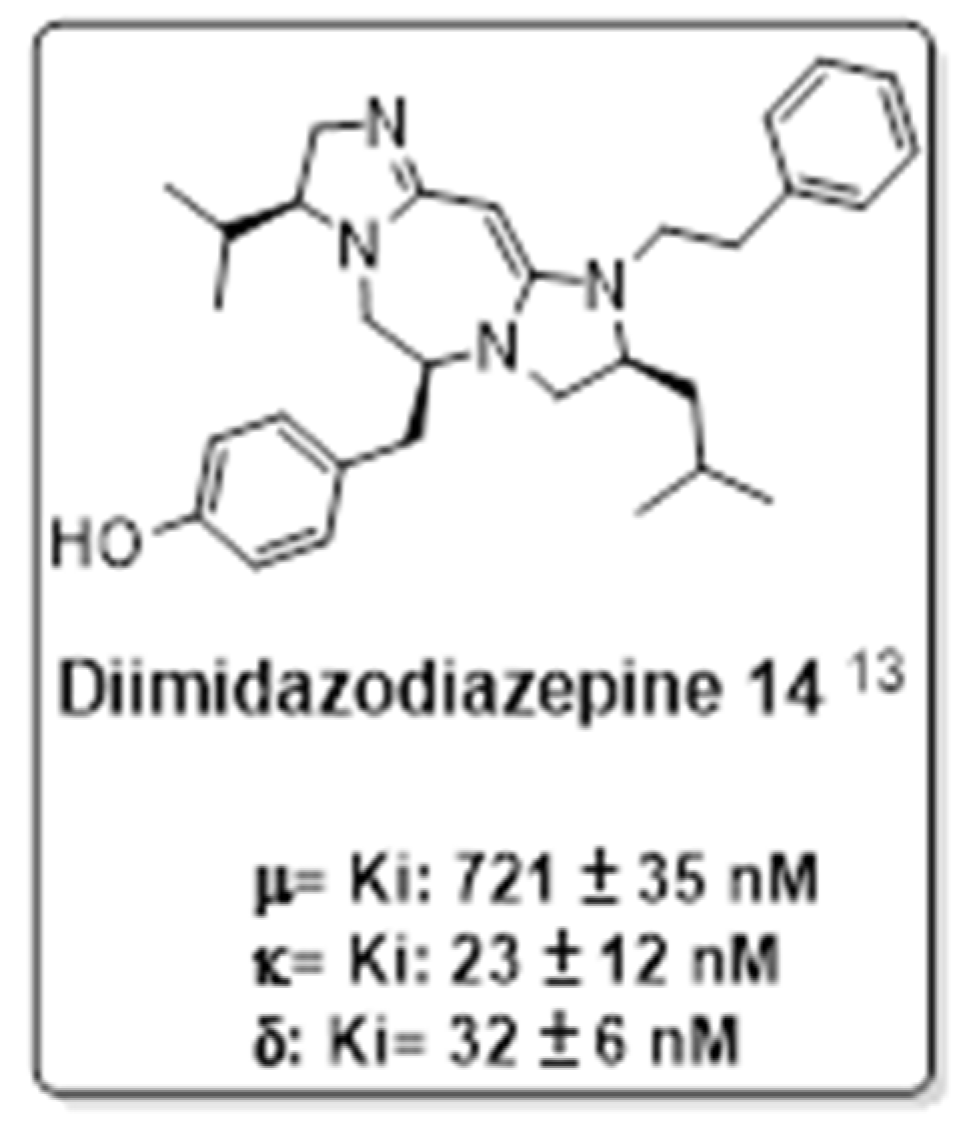
2.1. In Vivo Pharmacological Evaluation
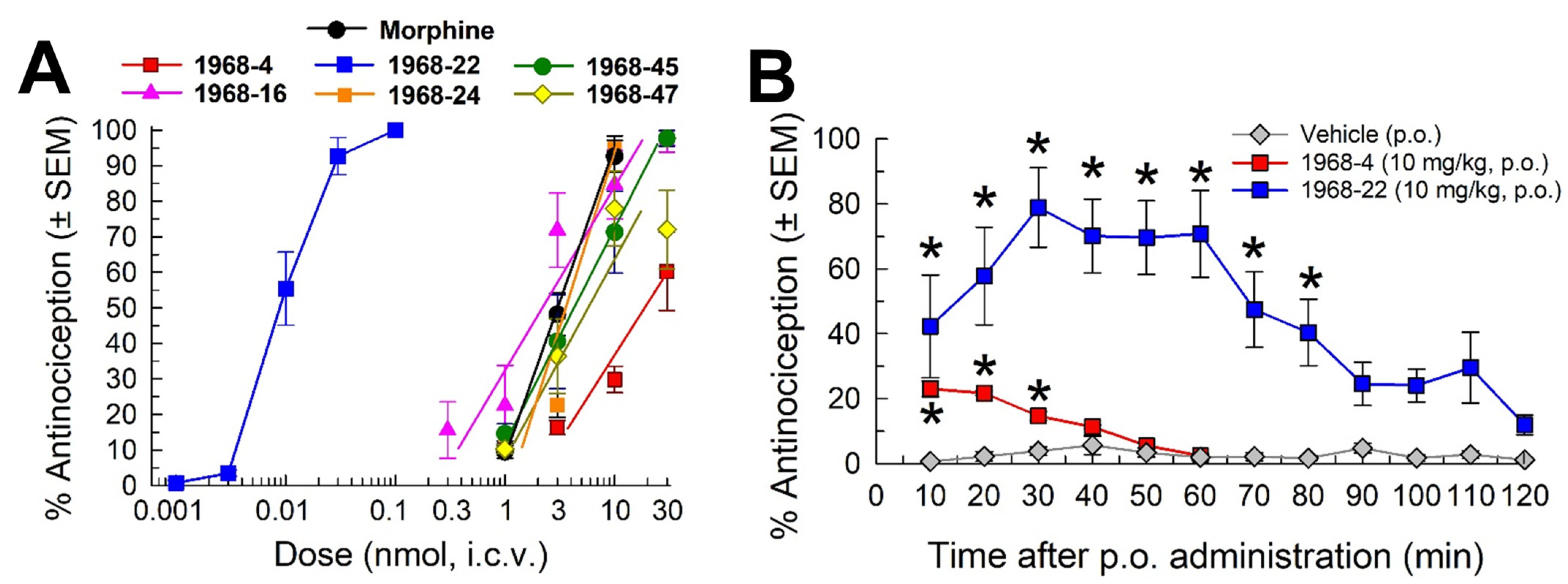
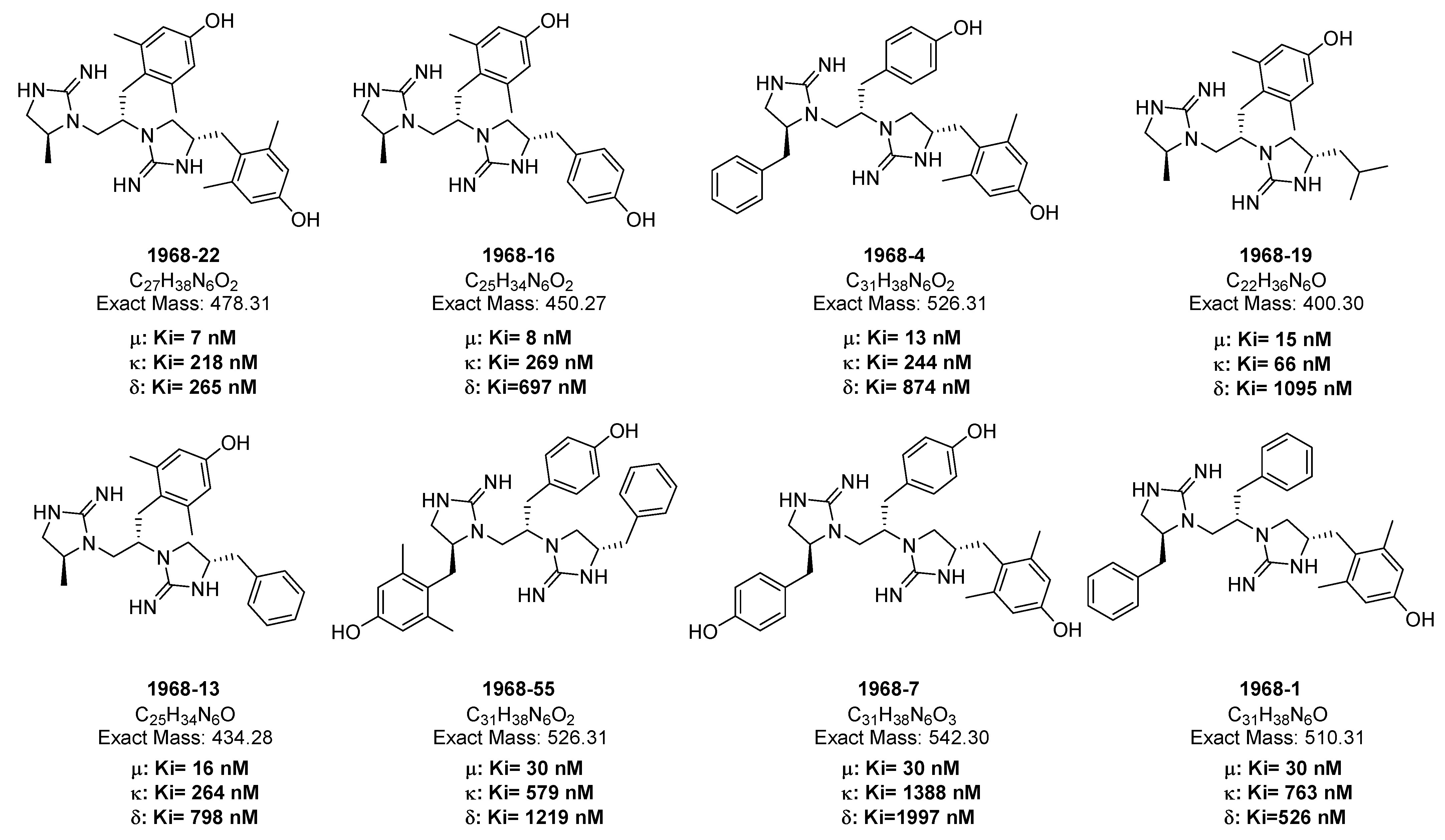
2.2. Opioid Receptor Selectivity of Antinociception Induced by 1968-Series Compounds
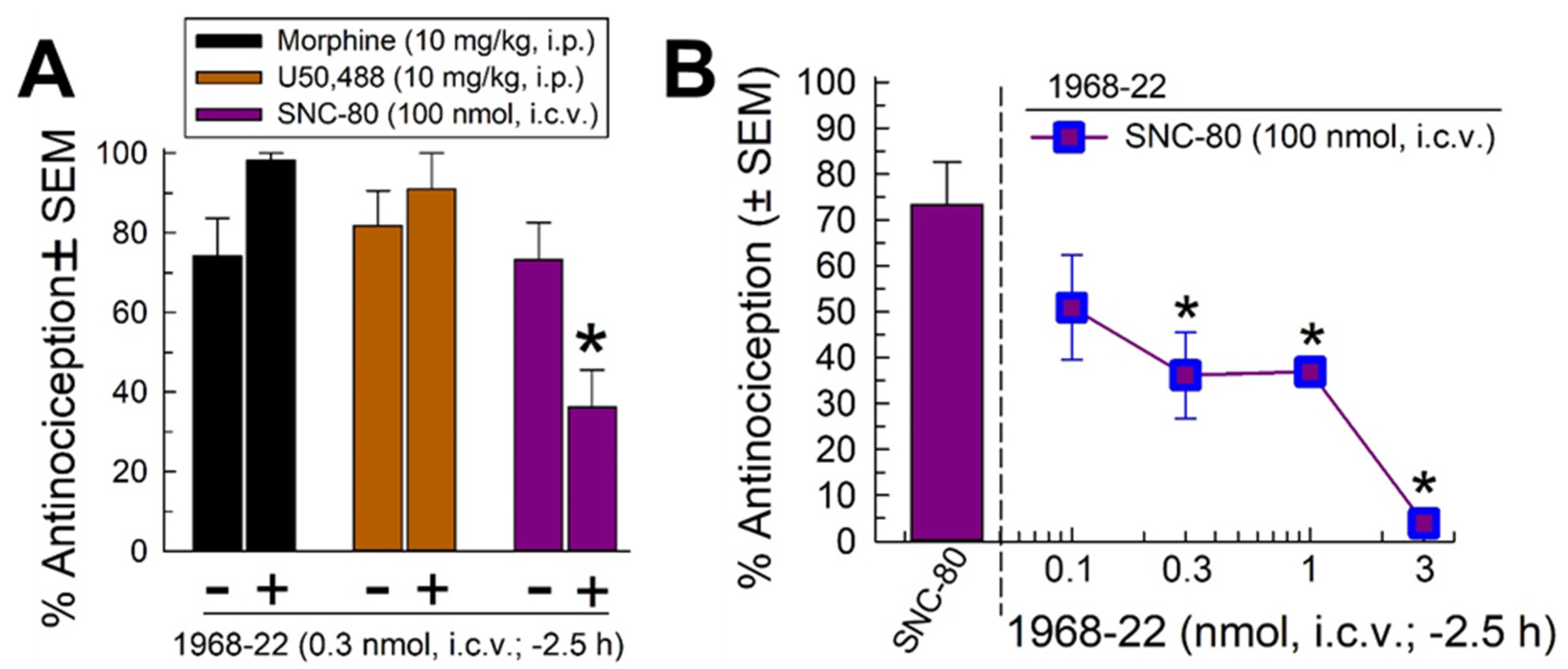
2.3. Determination of Opioid Receptor-Mediated Selective Antagonist Activity of 1968-22
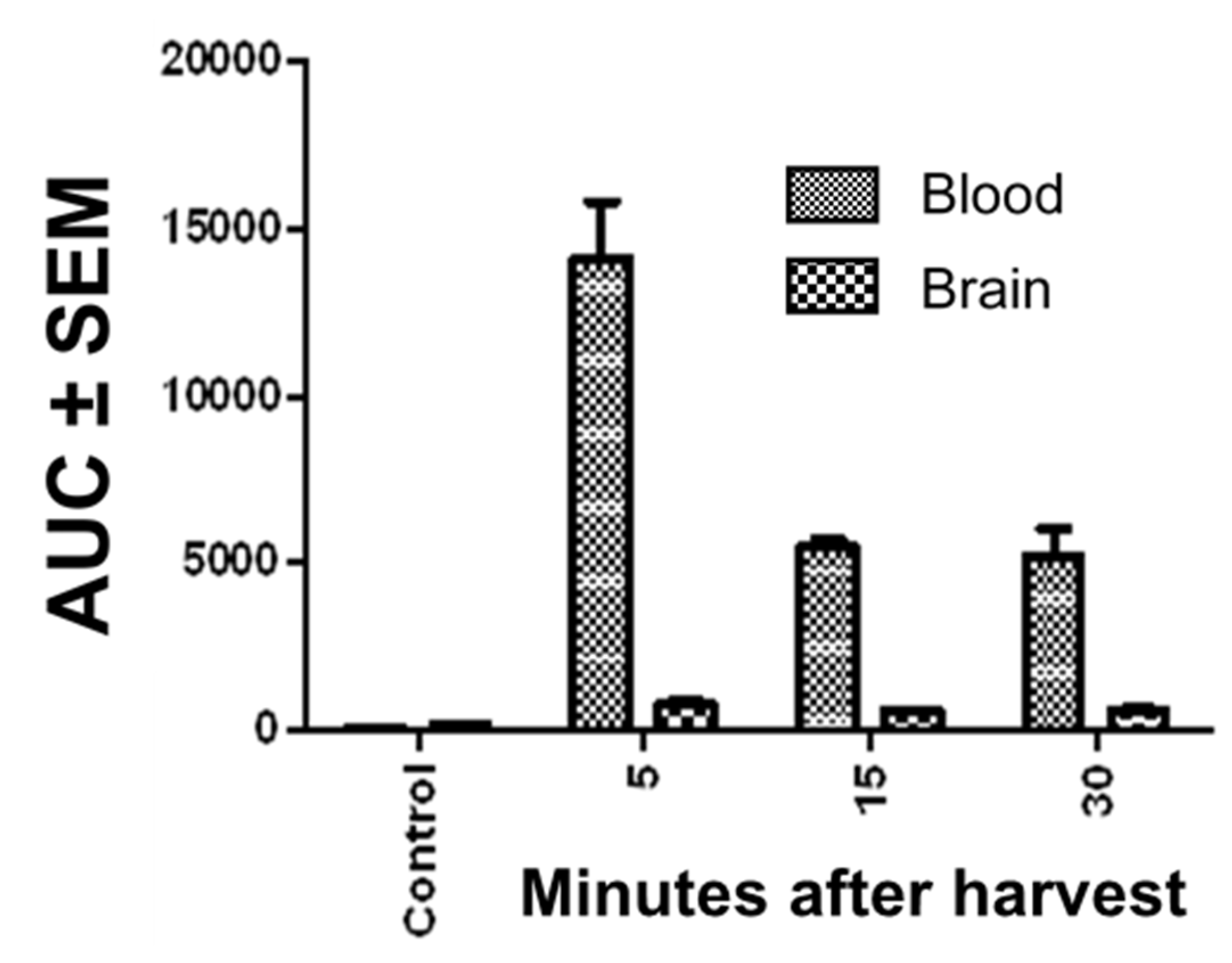
2.4. Assessment of Pharmacokinetic Stability and CNS Penetration of Bis-Cyclic Guanidine 1968-22
2.5. In Vivo Assessment of Opioid-Related Liabilities of 1968-22
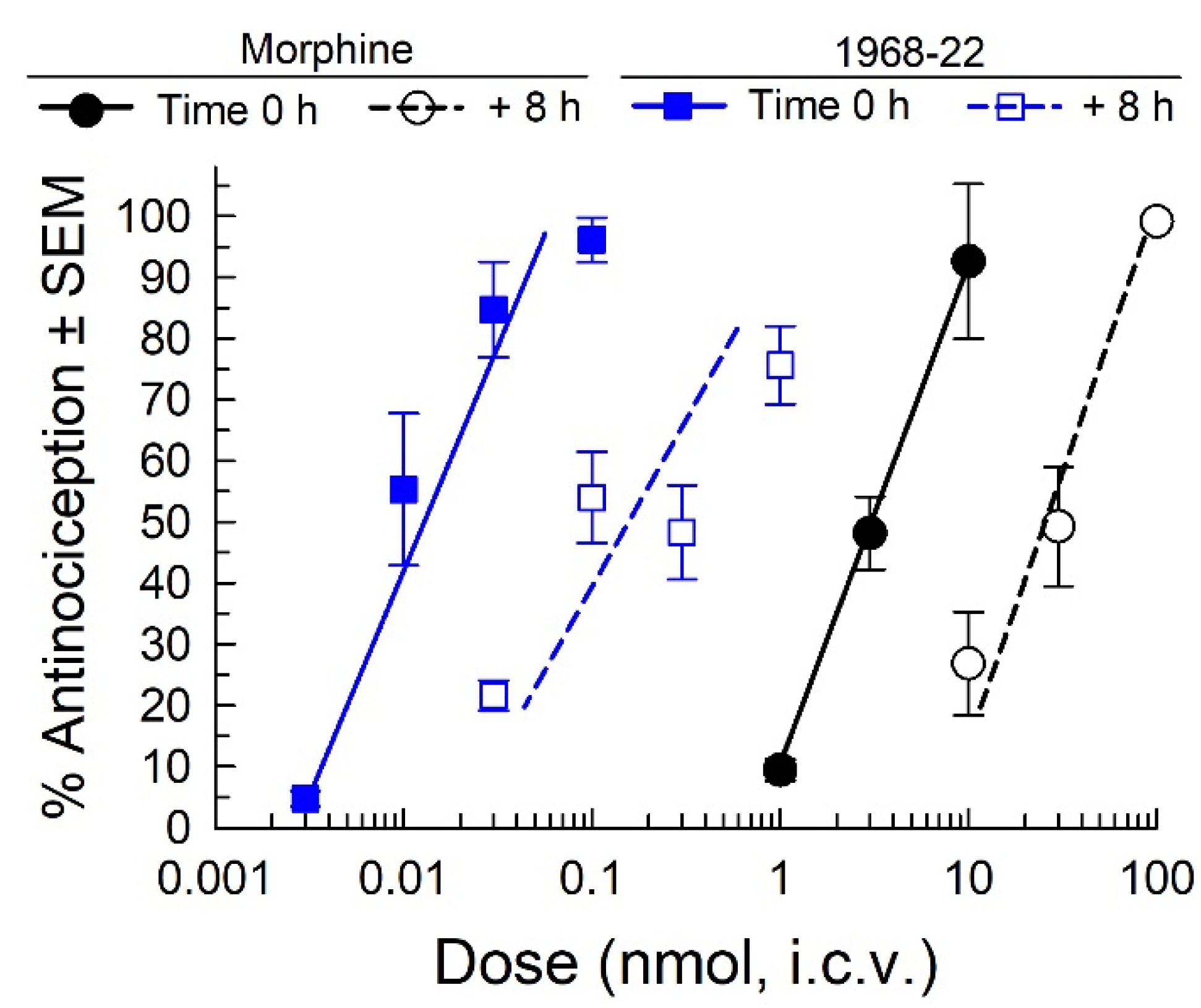
2.6. Assessment of Acute Antinociceptive Tolerance
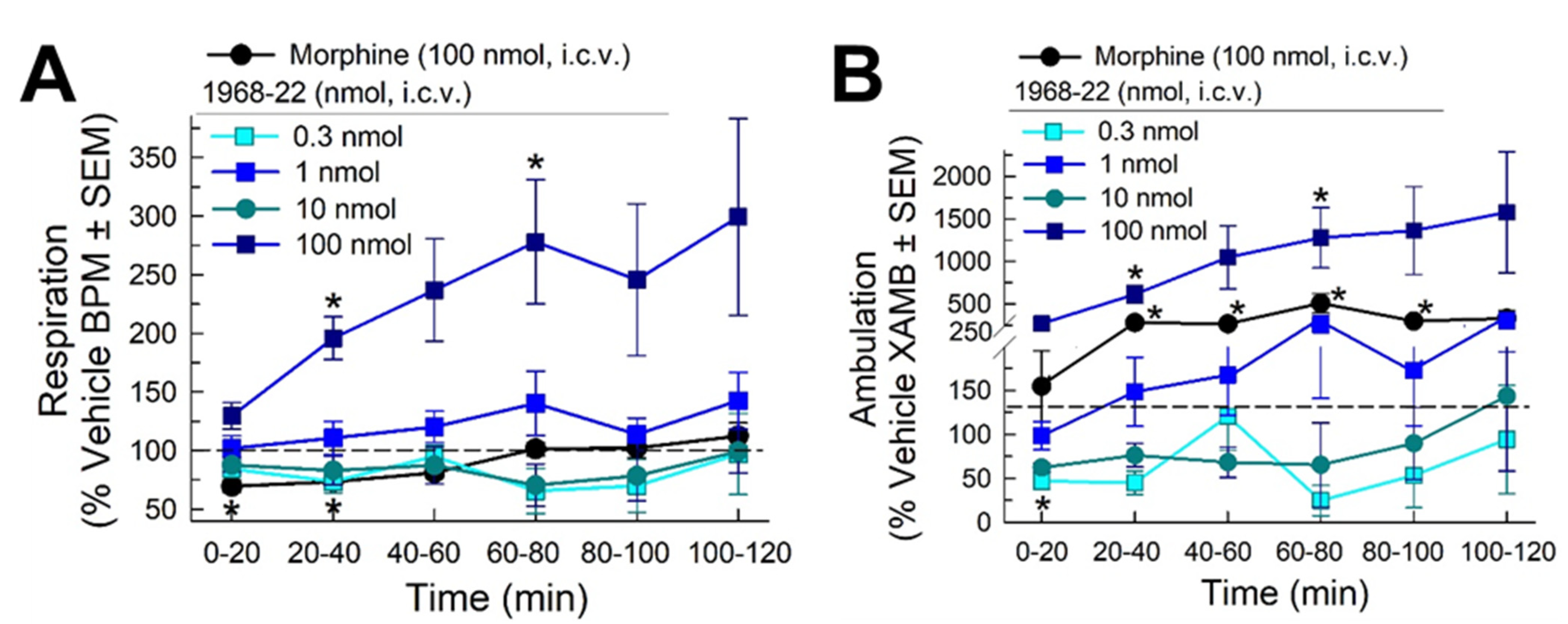
2.7. Evaluation of Respiratory and Spontaneous Locomotor Effects of 1968-22
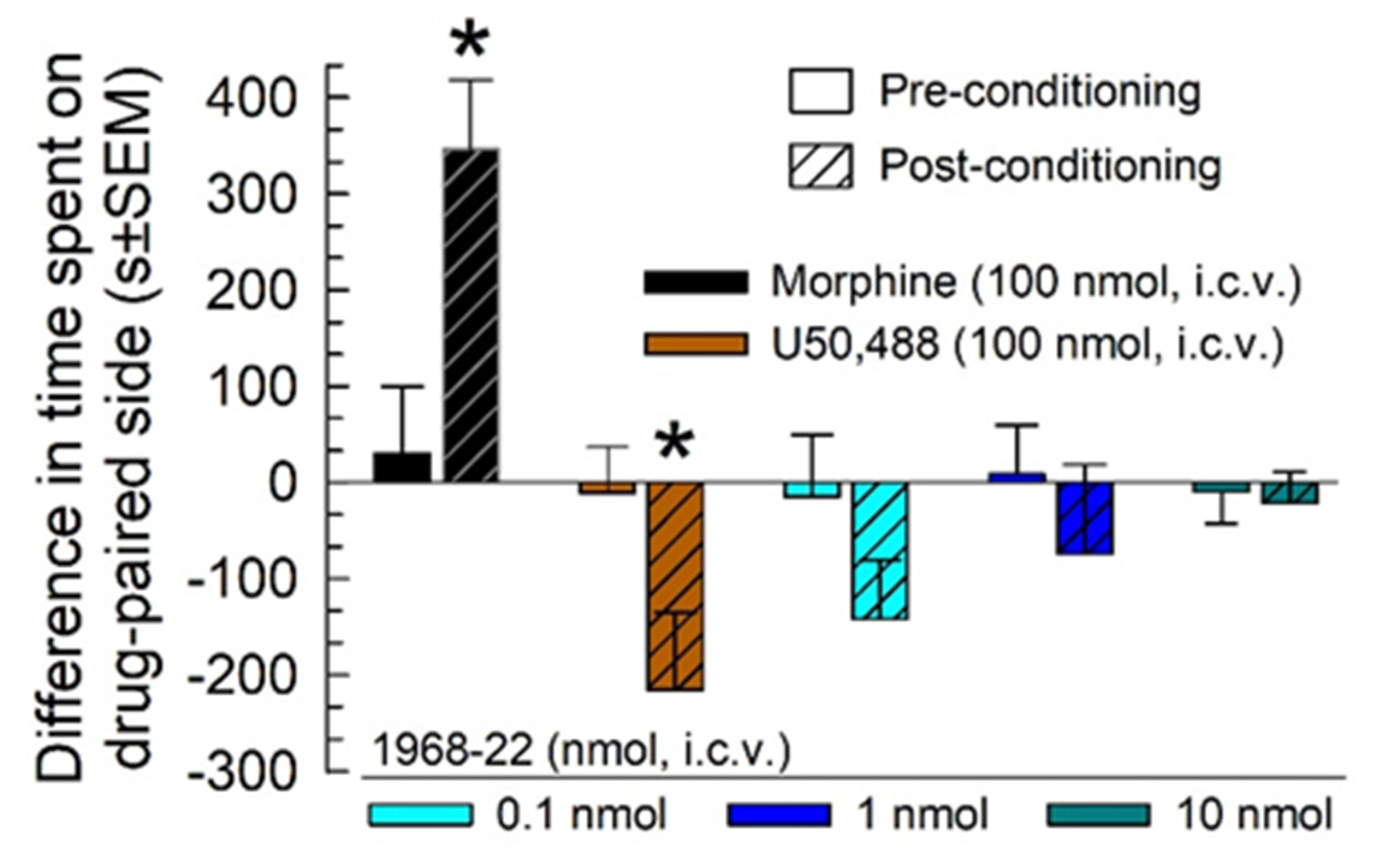
2.8. Evaluation of Potential Reinforcing or Aversive Properties of 1968-22
3. Discussion
4. Materials and Methods
4.1. Chemistry Synthesis
4.2. Typical Procedure for the Individual Synthesis of Bis-Cyclic Guanidine
4.3. In Vitro Radioligand Competition Binding Assays
4.4. In Vivo Testing
4.4.1. Animals and Drug Administration
4.4.2. Antinociceptive Testing
4.4.3. Acute Antinociceptive Tolerance Determination
4.4.4. Respiration and Ambulation
4.4.5. Evaluation of Potential Conditioned Place Preference and Conditioned Place Aversion
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BBB | blood–brain barrier |
| 13C NMR | C-nuclear magnetic resonance |
| CHO | Chinese hamster ovary |
| CI | confidential interval |
| CPP | conditioned-place preference |
| DAMGO | H-Tyr-D-Ala-Gly-NMe-Phe-Gly-ol |
| DMSO | dimethyl sulfoxide |
| Dmt | 2’,6’-dimethyltyrosine |
| DOR | delta opioid receptor |
| DPDPE | c[D-Pen2,D-Pen5]encephalin |
| EC50 | concentration for 50% of maximal effect |
| ED50 | dose for 50% of maximal effect |
| ESI-MS | electron spray ionization mass spectrometry |
| HBSS | Hank’s balanced salt solution |
| HCV | hepatitis C virus |
| 1H NMR | H-nuclear magnetic resonance |
| i.c.v. | intracerebroventricular |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| KOR | kappa opioid receptor |
| KOR KO | kappa opioid receptor gene-disrupted mice |
| LC-MS | liquid chromatography coupled to mass spectrometry |
| LP | 1,3-[(2R,6R,11R)-8-hydroxy-6,11-dimethyl-1,4,5,6-tetrahydro-2,6-methano-3-benzazocin-3(2H)-yl]-N-phenylpropanamide |
| MMP | matrix metalloproteinase |
| MOR | mu opioid receptor |
| MOR KO | mu opioid receptor gene-disrupted mice |
| MRM | multiple reaction monitoring |
| MS | mass spectrometry |
| nor-BNI | nor-binaltorphimine |
| NTPase | Nile virus nucleoside triphosphatases |
| PBS | phosphate buffered saline |
| PMSF | phenylmethanesulfonyl fluoride |
| RP-HPLC | reverse phase high-performance liquid chromatography |
| s.c. | subcutaneous |
| TACE | TNF-alpha converting enzyme |
| TFA | trifluoroacetic acid |
| UV | ultra violet |
References
- Ballantyne, J.C. The brain on opioids. Pain 2018, 159 (Suppl. S1), S24–S30. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2021, 26, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Inturrisi, C.E. Clinical pharmacology of opioids for pain. Clin. J. Pain 2002, 18, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. New concepts in opioid analgesia. Expert Opin. Investig. Drugs 2018, 27, 765–775. [Google Scholar] [CrossRef]
- Nuckols, T.K.; Anderson, L.; Popescu, I.; Diamant, A.L.; Doyle, B.; Di Capua, P.; Chou, R. Opioid prescribing: A systematic review and critical appraisal of guidelines for chronic pain. Ann. Intern. Med. 2014, 160, 38–47. [Google Scholar] [CrossRef]
- Mercadante, S.; Arcuri, E.; Santoni, A. Opioid-Induced Tolerance and Hyperalgesia. CNS Drugs 2019, 33, 943–955. [Google Scholar] [CrossRef]
- Ehrlich, A.T.; Kieffer, B.L.; Darcq, E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23, 315–326. [Google Scholar] [CrossRef]
- Spetea, M.; Asim, M.F.; Wolber, G.; Schmidhammer, H. The µ opioid receptor and ligands acting at the µ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef]
- Busserolles, J.; Lolignier, S.; Kerckhove, N.; Bertin, C.; Authier, N.; Eschalier, A. Replacement of current opioid drugs focusing on MOR-related strategies. Pharmacol. Ther. 2020, 210, 107519. [Google Scholar] [CrossRef]
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331. [Google Scholar] [CrossRef]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittalà, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef] [PubMed]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef] [PubMed]
- Eans, S.O.; Ganno, M.L.; Mizrachi, E.; Houghten, R.A.; Dooley, C.T.; McLaughlin, J.P.; Nefzi, A. Parallel Synthesis of Hexahydrodiimidazodiazepines Heterocyclic Peptidomimetics and Their in Vitro and in Vivo Activities at μ (MOR), δ (DOR), and κ (KOR) Opioid Receptors. J. Med. Chem. 2015, 58, 4905–4917. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.F.; Lambert, D.G. Simultaneous targeting of multiple opioid receptor types. Curr. Opin. Support. Palliat. Care 2015, 9, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Guerrini, R.; Calo, G.; Salvadori, S.; Rowbotham, D.J.; Lambert, D.G. Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 2009, 103, 38–49. [Google Scholar] [CrossRef]
- Günther, T.; Dasgupta, P.; Mann, A.; Miess, E.; Kliewer, A.; Fritzwanker, S.; Steinborn, R.; Schulz, S. Targeting multiple opioid receptors-improved analgesics with reduced side effects? Br. J. Pharmacol. 2018, 175, 2857–2868. [Google Scholar] [CrossRef]
- Anand, J.P.; Montgomery, D. Multifunctional Opioid Ligands. Handb. Exp. Pharmacol. 2018, 247, 21–51. [Google Scholar] [CrossRef]
- Iwamoto, E.T.; Martin, W.R. Multiple opioid receptors. Med. Res. Rev. 1981, 1, 411–440. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef]
- Toll, L.; Khroyan, T.V.; Polgar, W.E.; Jiang, F.; Olsen, C.; Zaveri, N.T. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: Implications for therapeutic applications. J. Pharmacol. Exp. Ther. 2009, 331, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, S.; Hayashi, T.; Kawase, M.; Saito, S.; Sugibayashi, K.; Morimoto, Y. Transdermal delivery of the potent analgesic dihydroetorphine: Kinetic analysis of skin permeation and analgesic effect in the hairless rat. J. Pharm. Pharmacol. 2000, 52, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, S.; Minami, M.; Nakagawa, T.; Iwamura, T.; Satoh, M. Pharmacological study of dihydroetorphine in cloned mu-, delta- and kappa-opioid receptors. Eur. J. Pharmacol. 1995, 291, 367–373. [Google Scholar] [CrossRef]
- Parenti, C.; Turnaturi, R.; Aricò, G.; Marrazzo, A.; Prezzavento, O.; Ronsisvalle, S.; Scoto, G.M.; Ronsisvalle, G.; Pasquinucci, L. Antinociceptive profile of LP1, a non-peptide multitarget opioid ligand. Life Sci. 2012, 90, 957–961. [Google Scholar] [CrossRef]
- Pasquinucci, L.; Parenti, C.; Amata, E.; Georgoussi, Z.; Pallaki, P.; Camarda, V.; Calò, G.; Arena, E.; Montenegro, L.; Turnaturi, R. Synthesis and Structure-Activity Relationships of (-)-cis-N-Normetazocine-Based LP1 Derivatives. Pharmaceuticals 2018, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Pasternak, G.W.; Negus, S.S. Confronting the opioid crisis with basic research in neuropharmacology. Neuropharmacology 2020, 166, 107972. [Google Scholar] [CrossRef]
- Coussens, N.P.; Sittampalam, G.S.; Jonson, S.G.; Hall, M.D.; Gorby, H.E.; Tamiz, A.P.; McManus, O.B.; Felder, C.C.; Rasmussen, K. The Opioid Crisis and the Future of Addiction and Pain Therapeutics. J. Pharmacol. Exp. Ther. 2019, 371, 396–408. [Google Scholar] [CrossRef]
- Nefzi, A.; Dooley, C.; Ostresh, J.M.; Houghten, R.A. Combinatorial chemistry: From peptides and peptidomimetics to small organic and heterocyclic compounds. Bioorg. Med. Chem. Lett. 1998, 8, 2273–2278. [Google Scholar] [CrossRef]
- Nefzi, A.; Santos, R.T. Efficient approaches toward the solid-phase synthesis of new heterocyclic azoniaspiro ring systems: Synthesis of tri- and tetrasubstituted 10-oxo- 3,9-diaza-6-azoniaspiro[5.5]undecanes. J. Org. Chem. 2005, 70, 9622–9625. [Google Scholar] [CrossRef]
- Nefzi, A.; Ostresh, J.M.; Yu, Y.; Houghten, R.A. Combinatorial chemistry: Libraries from libraries, the art of the diversity-oriented transformation of resin-bound peptides and chiral polyamides to low molecular weight acyclic and heterocyclic compounds. J. Org. Chem. 2004, 69, 3603–3609. [Google Scholar] [CrossRef]
- Nefzi, A.; Ostresh, J.M.; Appel, J.R.; Bidlack, J.; Dooley, C.T.; Houghten, R.A. Identification of potent and highly selective chiral tri-amine and tetra-amine mu opioid receptors ligands: An example of lead optimization using mixture-based libraries. Bioorg. Med. Chem. Lett. 2006, 16, 4331–4338. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.N.; Nefzi, A.; Ostresh, J.M.; Houghten, R.A. Tethered libraries: Solid-phase synthesis of substituted urea-linked bicyclic guanidines. J. Comb. Chem. 2001, 3, 189–195. [Google Scholar] [CrossRef]
- Ortiz, M.A.; Michaels, H.; Molina, B.; Toenjes, S.; Davis, J.; Marconi, G.D.; Hecht, D.; Gustafson, J.L.; Piedrafita, F.J.; Nefzi, A. Discovery of cyclic guanidine-linked sulfonamides as inhibitors of LMTK3 kinase. Bioorg. Med. Chem. Lett. 2020, 30, 127108. [Google Scholar] [CrossRef] [PubMed]
- Hensler, M.E.; Bernstein, G.; Nizet, V.; Nefzi, A. Pyrrolidine bis-cyclic guanidines with antimicrobial activity against drug-resistant Gram-positive pathogens identified from a mixture-based combinatorial library. Bioorg. Med. Chem. Lett. 2006, 16, 5073–5079. [Google Scholar] [CrossRef] [PubMed]
- Ostresh, J.M.; Schoner, C.C.; Hamashin, V.T.; Nefzi, A.; Meyer, J.-P.; Houghten, R.A. Solid-Phase Synthesis of Trisubstituted Bicyclic Guanidines via Cyclization of Reduced N-Acylated Dipeptides. J. Org. Chem. 1998, 63, 8622–8623. [Google Scholar] [CrossRef]
- Arutyunyan, S.; Nefzi, A. Synthesis of chiral polyaminothiazoles. J. Comb. Chem. 2010, 12, 315–317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nefzi, A.; Giulianotti, M.A.; Houghten, R.A. Solid-phase synthesis of bis-heterocyclic compounds from resin-bound orthogonally protected lysine. J. Comb. Chem. 2001, 3, 68–70. [Google Scholar] [CrossRef]
- Rohde, K.H.; Michaels, H.A.; Nefzi, A. Synthesis and antitubercular activity of 1,2,4-trisubstitued piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 2206–2209. [Google Scholar] [CrossRef]
- Hruby, V.J.; Agnes, R.S. Conformation-activity relationships of opioid peptides with selective activities at opioid receptors. Biopolymers 1999, 51, 391–410. [Google Scholar] [CrossRef]
- Garg, S.; Nurgali, K.; Mishra, V.K. Food Proteins as Source of Opioid Peptides-A Review. Curr. Med. Chem. 2016, 23, 893–910. [Google Scholar] [CrossRef]
- Cesselin, F. Opioid and anti-opioid peptides. Fundam. Clin. Pharmacol. 1995, 9, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Fricker, L.D.; Margolis, E.B.; Gomes, I.; Devi, L.A. Five Decades of Research on Opioid Peptides: Current Knowledge and Unanswered Questions. Mol. Pharmacol. 2020, 98, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W. Opioid peptide-derived analgesics. AAPS J. 2005, 7, E560–E565. [Google Scholar] [CrossRef]
- Remesic, M.; Lee, Y.S.; Hruby, V.J. Cyclic Opioid Peptides. Curr. Med. Chem. 2016, 23, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.T.; Houghten, R.A. New opioid peptides, peptidomimetics, and heterocyclic compounds from combinatorial libraries. Biopolymers 1999, 51, 379–390. [Google Scholar] [CrossRef]
- Houghten, R.A.; Ganno, M.L.; McLaughlin, J.P.; Dooley, C.T.; Eans, S.O.; Santos, R.G.; LaVoi, T.; Nefzi, A.; Welmaker, G.; Giulianotti, M.A.; et al. Direct Phenotypic Screening in Mice: Identification of Individual, Novel Antinociceptive Compounds from a Library of 734,821 Pyrrolidine Bis-piperazines. ACS Comb. Sci. 2016, 18, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Rinnová, M.; Nefzi, A.; Houghten, R.A. Opioid activity of 4-imidazolidinone positional analogues of Leu-Enkephalin. Bioorg. Med. Chem. Lett. 2002, 12, 3175–3178. [Google Scholar] [CrossRef]
- Hammami, S.; Mighri, Z.; Dooley, C.T.; Nefzi, A. Synthesis and analgesic activity of alkylated, reduced and constrained oligoheterocyclic peptidomimetic analogs of Leu-enkephalin. Bioorg. Med. Chem. Lett. 2014, 24, 4482–4485. [Google Scholar] [CrossRef] [PubMed]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; McLaughlin, J.P.; Houghten, R.A. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef]
- Montero, A.; Goya, P.; Jagerovic, N.; Callado, L.F.; Meana, J.J.; Girón, R.; Goicoechea, C.; Martín, M.I. Guanidinium and aminoimidazolinium derivatives of N-(4-piperidyl)propanamides as potential ligands for mu opioid and I2-imidazoline receptors: Synthesis and pharmacological screening. Bioorg. Med. Chem. 2002, 10, 1009–1018. [Google Scholar] [CrossRef]
- Martínez-Mayorga, K.; Medina-Franco, J.L.; Giulianotti, M.A.; Pinilla, C.; Dooley, C.T.; Appel, J.R.; Houghten, R.A. Conformation-opioid activity relationships of bicyclic guanidines from 3D similarity analysis. Bioorg. Med. Chem. 2008, 16, 5932–5938. [Google Scholar] [CrossRef] [PubMed]
- Nefzi, A.; Appel, J.; Arutyunyan, S.; Houghten, R.A. Parallel synthesis of chiral pentaamines and pyrrolidine containing bis-heterocyclic libraries. Multiple scaffolds with multiple building blocks: A double diversity for the identification of new antitubercular compounds. Bioorg. Med. Chem. Lett. 2009, 19, 5169–5175. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.T.; Chung, N.N.; Wilkes, B.C.; Schiller, P.W.; Bidlack, J.M.; Pasternak, G.W.; Houghten, R.A. An all D-amino acid opioid peptide with central analgesic activity from a combinatorial library. Science 1994, 266, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.M.; Eans, S.O.; Ganno, M.L.; Davis, J.C.; Dooley, C.T.; McLaughlin, J.P.; Nefzi, A. Antinociceptive activity of thiazole-containing cyclized DAMGO and Leu-(Met) enkephalin analogs. Org. Biomol. Chem. 2019, 17, 5305–5315. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.T.; Ny, P.; Bidlack, J.M.; Houghten, R.A. Selective ligands for the mu, delta, and kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J. Biol. Chem. 1998, 273, 18848–18856. [Google Scholar] [CrossRef]
- Li, Y.; Cazares, M.; Wu, J.; Houghten, R.A.; Toll, L.; Dooley, C. Potent μ-Opioid Receptor Agonists from Cyclic Peptides Tyr-c[D-Lys-Xxx-Tyr-Gly]: Synthesis, Biological, and Structural Evaluation. J. Med. Chem. 2016, 59, 1239–1245. [Google Scholar] [CrossRef]
- Vanderah, T.W.; Largent-Milnes, T.; Lai, J.; Porreca, F.; Houghten, R.A.; Menzaghi, F.; Wisniewski, K.; Stalewski, J.; Sueiras-Diaz, J.; Galyean, R.; et al. Novel D-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur. J. Pharmacol. 2008, 583, 62–72. [Google Scholar] [CrossRef]
- Lisowski, M.; Olczak, J.; Zabrocki, J. Circular dichroic properties of the tyrosine residues in tetrazole analogues of opioid peptides. J. Pept. Sci. 2006, 12, 297–302. [Google Scholar] [CrossRef]
- Bryant, S.D.; Jinsmaa, Y.; Salvadori, S.; Okada, Y.; Lazarus, L.H. Dmt and opioid peptides: A potent alliance. Biopolymers 2003, 71, 86–102. [Google Scholar] [CrossRef]
- Balboni, G.; Marzola, E.; Sasaki, Y.; Ambo, A.; Marczak, E.D.; Lazarus, L.H.; Salvadori, S. Role of 2′,6′-dimethyl-l-tyrosine (Dmt) in some opioid lead compounds. Bioorg. Med. Chem. 2010, 18, 6024–6030. [Google Scholar] [CrossRef][Green Version]
- Dolle, R.E.; Michaut, M.; Martinez-Teipel, B.; Belanger, S.; Graczyk, T.M.; DeHaven, R.N. Further studies of tyrosine surrogates in opioid receptor peptide ligands. Bioorg. Med. Chem. Lett. 2007, 17, 2656–2660. [Google Scholar] [CrossRef] [PubMed]
- Brice-Tutt, A.C.; Senadheera, S.N.; Ganno, M.L.; Eans, S.O.; Khaliq, T.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Phenylalanine Stereoisomers of CJ-15,208 and [d-Trp]CJ-15,208 Exhibit Distinctly Different Opioid Activity Profiles. Molecules 2020, 25, 3999. [Google Scholar] [CrossRef] [PubMed]
- Hoot, M.R.; Sypek, E.I.; Reilley, K.J.; Carey, A.N.; Bidlack, J.M.; McLaughlin, J.P. Inhibition of Gβγ-subunit signaling potentiates morphine-induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behav. Pharmacol. 2013, 24, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Ferracane, M.J.; Brice-Tutt, A.C.; Coleman, J.S.; Simpson, G.G.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Murray, T.F.; McLaughlin, J.P.; Aldrich, J.V. Design, Synthesis, and Characterization of the Macrocyclic Tetrapeptide cyclo[Pro-Sar-Phe-d-Phe]: A Mixed Opioid Receptor Agonist–Antagonist Following Oral Administration. ACS Chem. Neurosci. 2020, 11, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine-seeking behaviour. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef]
- Ko, M.C.; Husbands, S.M. Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J. Pharmacol. Exp. Ther. 2009, 328, 193–200. [Google Scholar] [CrossRef]
- Negus, S.S.; Schrode, K.; Stevenson, G.W. Micro/kappa opioid interactions in rhesus monkeys: Implications for analgesia and abuse liability. Exp. Clin. Psychopharmacol. 2008, 16, 386–399. [Google Scholar] [CrossRef]
- Sutters, K.A.; Miaskowski, C.; Taiwo, Y.O.; Levine, J.D. Analgesic synergy and improved motor function produced by combinations of mu-delta- and mu-kappa-opioids. Brain Res. 1990, 530, 290–294. [Google Scholar] [CrossRef]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and Exogenous Opioids in Pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- O’Brien, J.J.; Benfield, P. Dezocine. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1989, 38, 226–248. [Google Scholar] [CrossRef]
- Preston, K.L.; Jasinski, D.R. Abuse liability studies of opioid agonist-antagonists in humans. Drug Alcohol Depend. 1991, 28, 49–82. [Google Scholar] [CrossRef]
- Strain, E.C.; Preston, K.L.; Liebson, I.A.; Bigelow, G.E. Opioid antagonist effects of dezocine in opioid-dependent humans. Clin. Pharmacol. Ther. 1996, 60, 206–217. [Google Scholar] [CrossRef]
- Strain, E.C.; Stitzer, M.L.; Liebson, I.A.; Bigelow, G.E. Buprenorphine versus methadone in the treatment of opioid dependence: Self-reports, urinalysis, and addiction severity index. J. Clin. Psychopharmacol. 1996, 16, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Eder, H.; Fischer, G.; Gombas, W.; Jagsch, R.; Stühlinger, G.; Kasper, S. Comparison of buprenorphine and methadone maintenance in opiate addicts. Eur. Addict. Res. 1998, 4 (Suppl. S1), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, M.K.; Johanson, C.E.; Schuster, C.R. Opioid reinforcement in heroin-dependent volunteers during outpatient buprenorphine maintenance. Drug Alcohol Depend. 1999, 56, 191–203. [Google Scholar] [CrossRef]
- Miller, R.R. Evaluation of nalbuphine hydrochloride. Am. J. Hosp. Pharm. 1980, 37, 942–949. [Google Scholar] [CrossRef]
- Schmidt, W.K.; Tam, S.W.; Shotzberger, G.S.; Smith, D.H., Jr.; Clark, R.; Vernier, V.G. Nalbuphine. Drug Alcohol Depend. 1985, 14, 339–362. [Google Scholar] [CrossRef]
- Dykstra, L.A.; Gmerek, D.E.; Winger, G.; Woods, J.H. Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Ther. 1987, 242, 413–420. [Google Scholar]
- Shippenberg, T.S.; Herz, A. Differential effects of mu and kappa opioid systems on motivational processes. NIDA Res. Monogr. 1986, 75, 563–566. [Google Scholar]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef]
- Dosaka-Akita, K.; Tortella, F.C.; Holaday, J.W.; Long, J.B. The kappa opioid agonist U-50,488H antagonizes respiratory effects of mu opioid receptor agonists in conscious rats. J. Pharmacol. Exp. Ther. 1993, 264, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Haji, A.; Takeda, R. Effects of a kappa-receptor agonist U-50488 on bulbar respiratory neurons and its antagonistic action against the mu receptor-induced respiratory depression in decerebrate cats. Jpn. J. Pharmacol. 2001, 87, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Nefzi, A.; Giulianotti, M.A.; Houghten, R.A. Solid-Phase Synthesis of Substituted 2,3-Diketopiperazines from Reduced Polyamides. Tetrahedron 2000, 56, 3319–3326. [Google Scholar] [CrossRef]
- Nefzi, A.; Giulianotti, M.A.; Ong, N.A.; Houghten, R.A. Solid-phase synthesis of bis-2-imidazolidinethiones from resin-bound tripeptides. Org. Lett. 2000, 2, 3349–3350. [Google Scholar] [CrossRef]
- Acharya, A.N.; Ostresh, J.M.; Houghten, R.A. Solid-phase synthesis of bis-cyclic guanidines from tripeptides. Tetrahedron 2001, 57, 9911–9914. [Google Scholar] [CrossRef]
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. USA 1985, 82, 5131–5135. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef]
- Aldrich, J.V.; Kulkarni, S.S.; Senadheera, S.N.; Ross, N.C.; Reilley, K.J.; Eans, S.O.; Ganno, M.L.; Murray, T.F.; McLaughlin, J.P. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15,208. ChemMedChem 2011, 6, 1739–1745. [Google Scholar] [CrossRef]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: In vivo and in vitro characterization of mu-selective agonist and antagonist activity. J. Pharmacol. Exp. Ther. 1999, 289, 304–311. [Google Scholar]
- Jiang, Q.; Seyed-Mozaffari, A.; Sebastian, A.; Archer, S.; Bidlack, J.M. Preventing morphine antinociceptive tolerance by irreversible mu opioid antagonists before the onset of their antagonism. J. Pharmacol. Exp. Ther. 1995, 273, 680–688. [Google Scholar] [CrossRef]
- Way, E.L.; Loh, H.H.; Shen, F.H. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J. Pharmacol. Exp. Ther. 1969, 167, 1–8. [Google Scholar] [PubMed]
- Eans, S.O.; Ganno, M.L.; Reilley, K.J.; Patkar, K.A.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short acting κ opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ED50 (and 95% Confidence Interval (C.I.)) Values | ||
|---|---|---|
| Compound | i.c.v. (nmol) | Two-Way RM ANOVA |
| 1968-4 * | 20.8 (5.52–127.1) | F(24,224) = 4.51, p < 0.0001 |
| 1968-16 | 2.00 (0.5–7.45) | F(40,350) = 9.98, p < 0.0001 |
| 1968-22 | 0.011 (0.005–0.026) | F(35,294) = 19.8, p < 0.0001 |
| 1968-24 | 3.76 (2.21–6.52) | F(16,168) = 34.1, p < 0.0001 |
| 1968-45 | 4.29 (1.33–13.1) | F(21,196) = 7.09, p < 0.0001 |
| 1968-47 * | 5.72 (1.13–29.6) | F(21,196) = 5.79, p < 0.0001 |
| Morphine | 3.09 (2.76–3.50) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLaughlin, J.P.; Rayala, R.; Bunnell, A.J.; Tantak, M.P.; Eans, S.O.; Nefzi, K.; Ganno, M.L.; Dooley, C.T.; Nefzi, A. Bis-Cyclic Guanidine Heterocyclic Peptidomimetics as Opioid Ligands with Mixed μ-, κ- and δ-Opioid Receptor Interactions: A Potential Approach to Novel Analgesics. Int. J. Mol. Sci. 2022, 23, 9623. https://doi.org/10.3390/ijms23179623
McLaughlin JP, Rayala R, Bunnell AJ, Tantak MP, Eans SO, Nefzi K, Ganno ML, Dooley CT, Nefzi A. Bis-Cyclic Guanidine Heterocyclic Peptidomimetics as Opioid Ligands with Mixed μ-, κ- and δ-Opioid Receptor Interactions: A Potential Approach to Novel Analgesics. International Journal of Molecular Sciences. 2022; 23(17):9623. https://doi.org/10.3390/ijms23179623
Chicago/Turabian StyleMcLaughlin, Jay P., Ramanjaneyulu Rayala, Ashley J. Bunnell, Mukund P. Tantak, Shainnel O. Eans, Khadija Nefzi, Michelle L. Ganno, Colette T. Dooley, and Adel Nefzi. 2022. "Bis-Cyclic Guanidine Heterocyclic Peptidomimetics as Opioid Ligands with Mixed μ-, κ- and δ-Opioid Receptor Interactions: A Potential Approach to Novel Analgesics" International Journal of Molecular Sciences 23, no. 17: 9623. https://doi.org/10.3390/ijms23179623
APA StyleMcLaughlin, J. P., Rayala, R., Bunnell, A. J., Tantak, M. P., Eans, S. O., Nefzi, K., Ganno, M. L., Dooley, C. T., & Nefzi, A. (2022). Bis-Cyclic Guanidine Heterocyclic Peptidomimetics as Opioid Ligands with Mixed μ-, κ- and δ-Opioid Receptor Interactions: A Potential Approach to Novel Analgesics. International Journal of Molecular Sciences, 23(17), 9623. https://doi.org/10.3390/ijms23179623