
Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
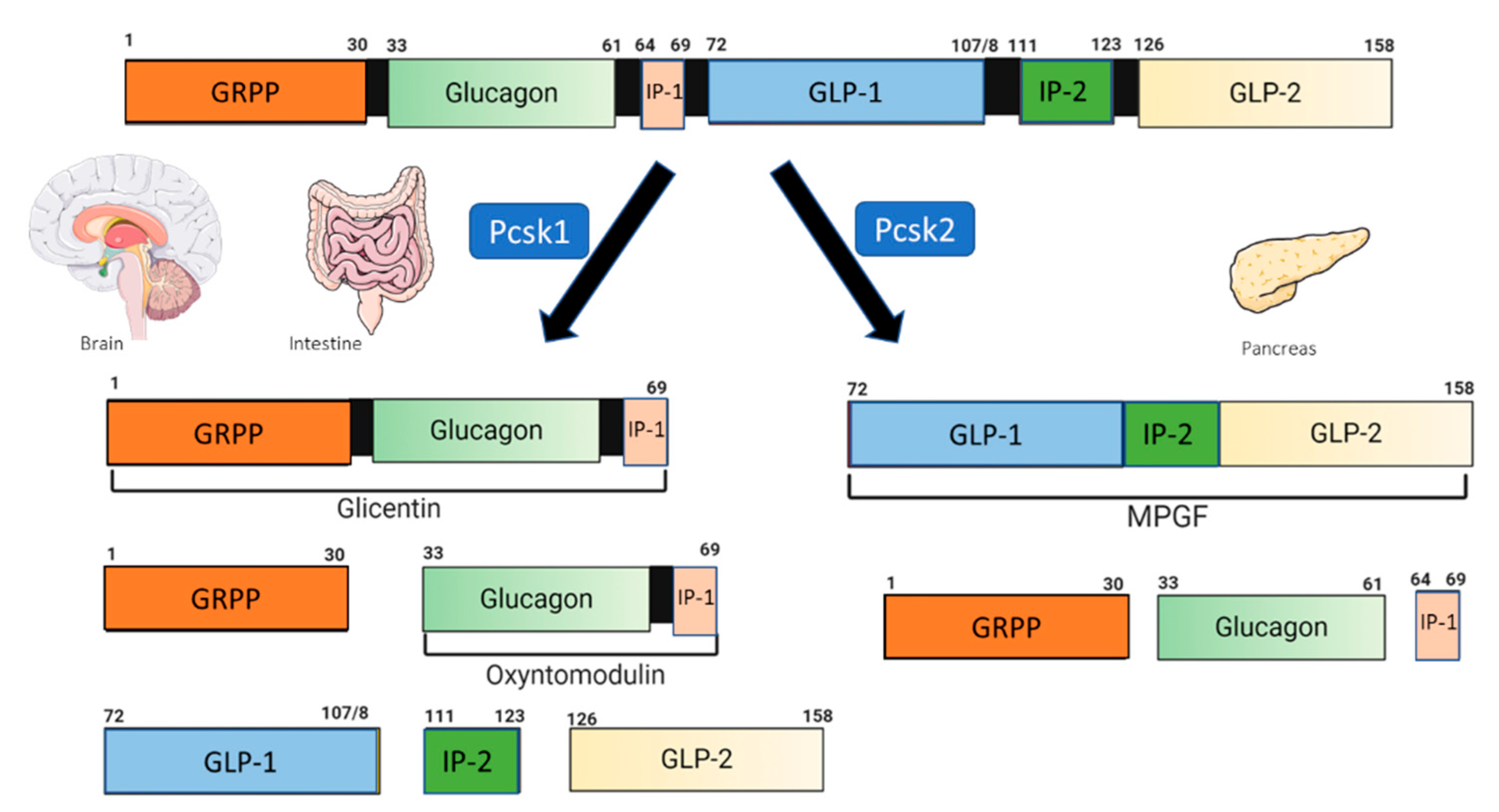
2. Glucagon-like Peptide 1 and the GLP-1 Receptor Agonist
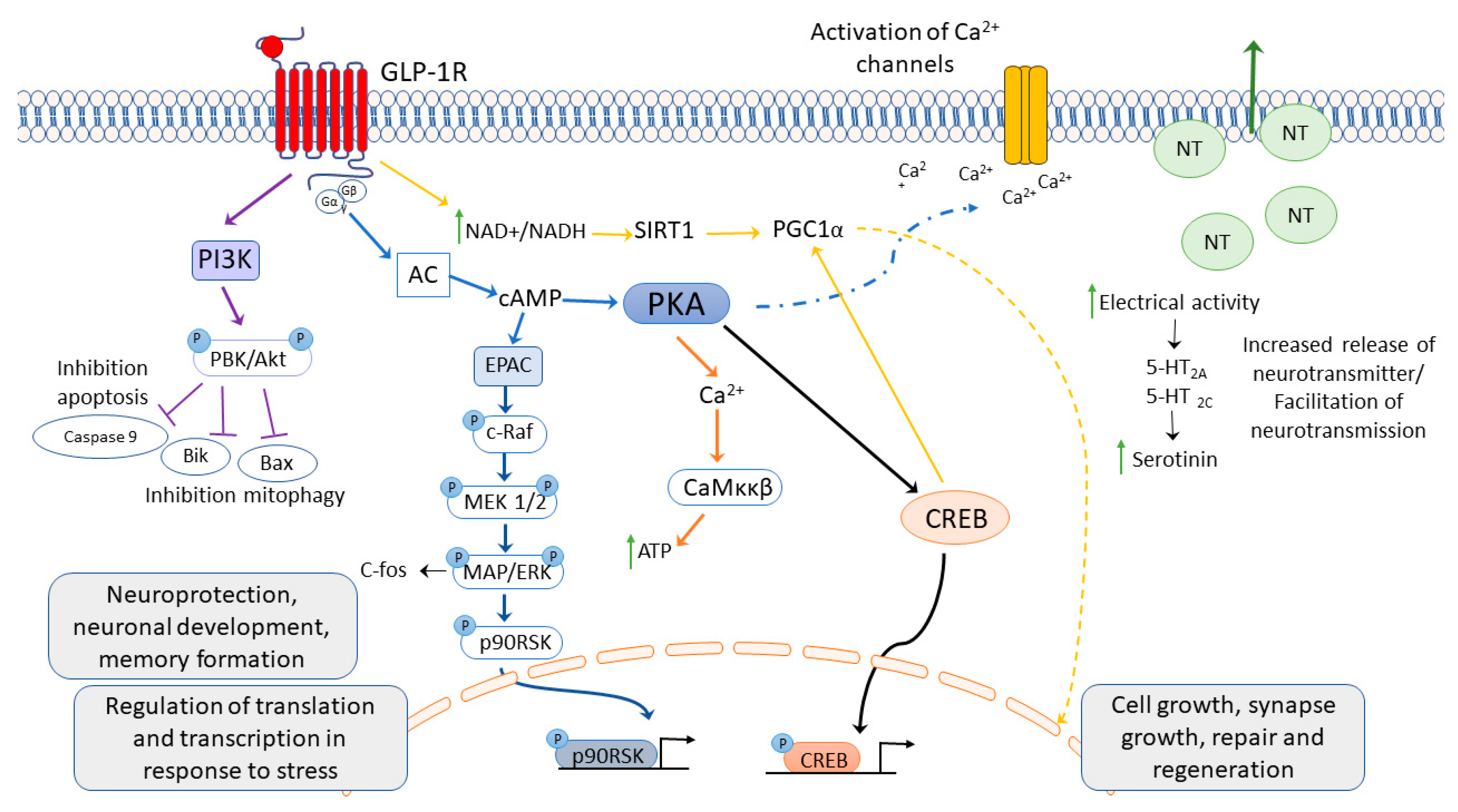
3. Glucagon-like Peptide 1 Receptor Activation
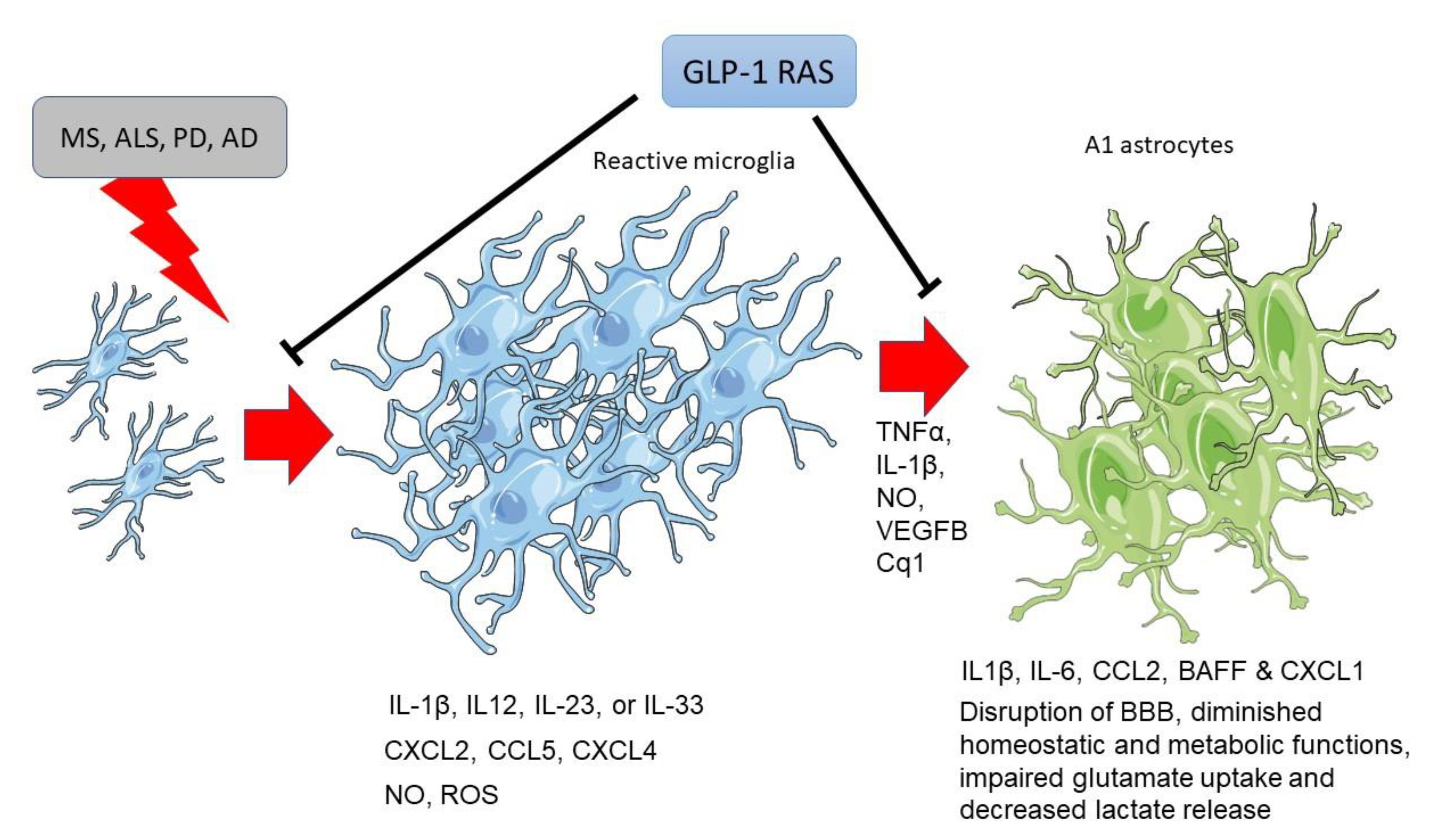
4. Anti-Inflammatory Effects of GLP-1R Activation in Neurodegenerative Diseases
4.1. Multiple Sclerosis
4.2. Amyotrophic Lateral Sclerosis (ALS)
4.3. Alzheimer’s Disease (AD)
4.4. Parkinson’s Disease (AD)
5. Discussion and Concluding Remarks
6. The Limitation of the Study
7. Conclusions
8. The Future Perspectives
Funding
Conflicts of Interest
References
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Concept of CNS Immune Privilege. Trends Immunol. 2015, 36, 569. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic Cells Permit Immune Invasion of the CNS in an Animal Model of Multiple Sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]
- Waisman, A.; Liblau, R.S.; Becher, B. Innate and Adaptive Immune Responses in the CNS. Lancet Neurol. 2015, 14, 945–955. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; el Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Plastini, M.J.; Desu, H.L.; Brambilla, R. Dynamic Responses of Microglia in Animal Models of Multiple Sclerosis. Front. Cell Neurosci. 2020, 14, 269. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of Innate Immune Responses in the Brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar]
- Astiz, M.; Pernía, O.; Barrios, V.; Garcia-Segura, L.M.; Diz-Chaves, Y. Short-Term High-Fat Diet Feeding Provides Hypothalamic but Not Hippocampal Protection against Acute Infection in Male Mice. Neuroendocrinology 2017, 104, 40–50. [Google Scholar] [CrossRef]
- Astiz, M.; Diz-Chaves, Y.; Garcia-Segura, L.M. Sub-Chronic Exposure to the Insecticide Dimethoate Induces a Proinflammatory Status and Enhances the Neuroinflammatory Response to Bacterial Lypopolysaccharide in the Hippocampus and Striatum of Male Mice. Toxicol. Appl. Pharmacol. 2013, 272, 263–271. [Google Scholar]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-Neuron Metabolic Cooperation Shapes Brain Activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Torres-Ceja, B.; Olsen, M.L. A Closer Look at Astrocyte Morphology: Development, Heterogeneity, and Plasticity at Astrocyte Leaflets. Curr. Opin. Neurobiol. 2022, 74, 102550. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A Common Thread in Neurological Disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar]
- Rossi, G.C.M.; Gandini Wheeler-Kingshott, C.A.M.; Toosy, A. Editorial: Neuroinflammation and the Visual System. Front. Neurol. 2021, 12, 1412. [Google Scholar] [CrossRef]
- Pape, K.; Tamouza, R.; Leboyer, M.; Zipp, F. Immunoneuropsychiatry—Novel Perspectives on Brain Disorders. Nat. Rev. Neurol. 2019, 15, 317–328. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Baylr, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Parkin, J.; Cohen, B. An Overview of the Immune System. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Insuela, D.B.R.; Carvalho, V.F. Glucagon and Glucagon-like Peptide-1 as Novel Anti-Inflammatory and Immunomodulatory Compounds. Eur. J. Pharmacol. 2017, 812, 64–72. [Google Scholar] [CrossRef]
- Hölscher, C. Protective Properties of GLP-1 and Associated Peptide Hormones in Neurodegenerative Disorders. Br. J. Pharmacol. 2022, 179, 695–714. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jun, H.S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediat. Inflamm. 2016, 2016, 3094642. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.N.; Stein, L.M.; Fortin, S.M.; Hayes, M.R. The Role of Glia in the Physiology and Pharmacology of Glucagon-like Peptide-1: Implications for Obesity, Diabetes, Neurodegeneration and Glaucoma. Br. J. Pharmacol. 2022, 179, 715–726. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like Peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Gil-Lozano, M.; González-Matías, L.C.; Mallo, F.; Vigo, E. Pulmonary GLP-1 Receptor Increases at Birth and Exogenous GLP-1 Receptor Agonists Augmented Surfactant-Protein Levels in Litters from Normal and Nitrofen-Treated Pregnant Rats. Endocrinology 2013, 154, 1144–1155. [Google Scholar] [CrossRef]
- Gault, V.A.; Hölscher, C. GLP-1 Receptor Agonists Show Neuroprotective Effects in Animal Models of Diabetes. Peptides 2018, 100, 101–107. [Google Scholar] [CrossRef]
- Diz-Chaves, Y.; Gil-Lozano, M.; Toba, L.; Fandiño, J.; Ogando, H.; González-Matías, L.C.; Mallo, F. Stressing Diabetes? The Hidden Links between Insulinotropic Peptides and the HPA Axis. J. Endocrinol. 2016, 230, R77–R94. [Google Scholar] [CrossRef]
- Gil-Lozano, M.; Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Vigo, E.; González-Matías, L.C.; Brubaker, P.L.; Mallo, F. Corticotropin-Releasing Hormone and the Sympathoadrenal System Are Major Mediators in the Effects of Peripherally Administered Exendin-4 on the Hypothalamic-Pituitary-Adrenal Axis of Male Rats. Endocrinology 2014, 155, 2511–2523. [Google Scholar] [CrossRef]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.-S.; Drucker, D.J.; Husain, M. Cardioprotective and Vasodilatory Actions of Glucagon-Like Peptide 1 Receptor Are Mediated through Both Glucagon-Like Peptide 1 Receptor-Dependent and -Independent Pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef]
- Fandiño, J.; Toba, L.; González-Matías, L.C.; Diz-Chaves, Y.; Mallo, F. GLP-1 Receptor Agonist Ameliorates Experimental Lung Fibrosis. Sci. Rep. 2020, 10, 18091. [Google Scholar] [CrossRef]
- Diz-Chaves, Y.; Herrera-Pérez, S.; González-Matías, L.C.; Lamas, J.A.; Mallo, F. Glucagon-like Peptide-1 (GLP-1) in the Integration of Neural and Endocrine Responses to Stress. Nutrients 2020, 12, 3304. [Google Scholar] [CrossRef]
- Shirazi, R.; Palsdottir, V.; Collander, J.; Anesten, F.; Vogel, H.; Langlet, F.; Jaschke, A.; Schurmann, A.; Prevot, V.; Shao, R.; et al. Glucagon-like Peptide 1 Receptor Induced Suppression of Food Intake, and Body Weight Is Mediated by Central IL-1 and IL-6. Proc. Natl. Acad. Sci. USA 2013, 110, 16199–16204. [Google Scholar] [CrossRef] [Green Version]
- Skibicka, K.P. The Central GLP-1: Implications for Food and Drug Reward. Front. Neurosci. 2013, 7, 181. [Google Scholar]
- Drucker, D.J.; Mojsov, S.; Habener, J.F. Cell-Specific Post-Translational Processing of Preproglucagon Expressed from a Metallothionein-Glucagon Fusion Gene. J. Biol. Chem. 1986, 261, 9637–9643. [Google Scholar]
- Diz-Chaves, Y.; Herrera-Pérez, S.; González-Matías, L.C.; Mallo, F. Effects of Glucagon-like Peptide 1 (GLP-1) Analogs in the Hippocampus. Vitam. Horm. 2022, 118, 457–478. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef]
- Tucker, J.D.; Dhanvantari, S.; Brubaker, P.L. Proglucagon Processing in Islet and Intestinal Cell Lines. Regul. Pept. 1996, 62, 29–35. [Google Scholar] [CrossRef]
- Chen, Y.C.; Taylor, A.J.; Verchere, C.B. Islet Prohormone Processing in Health and Disease. Diabetes Obes. Metab. 2018, 20, 64–76. [Google Scholar] [CrossRef]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like Peptide I Stimulates Insulin Gene Expression and Increases Cyclic AMP Levels in a Rat Islet Cell Line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar]
- Li, Y.; Glotfelty, E.J.; Karlsson, T.; Fortuno, L.V.; Harvey, B.K.; Greig, N.H. The Metabolite GLP-1 (9–36) Is Neuroprotective and Anti-Inflammatory in Cellular Models of Neurodegeneration. J. Neurochem. 2021, 159, 867–886. [Google Scholar] [CrossRef]
- Larsen, P.J.; Tang-Christensen, M.; Jessop, D.S. Central Administration of Glucagon-Like Peptide-1 Activates Hypothalamic Neuroendocrine Neurons in the Rat 1. Endocrinology 1997, 138, 4445–4455. [Google Scholar] [CrossRef]
- Ugleholdt, R.; Zhu, X.; Deacon, C.F.; Ørskov, C.; Steiner, D.F.; Holst, J.J. Impaired Intestinal Proglucagon Processing in Mice Lacking Prohormone Convertase 1. Endocrinology 2004, 145, 1349–1355. [Google Scholar] [CrossRef]
- Holt, M.K.; Pomeranz, L.E.; Beier, K.T.; Reimann, F.; Gribble, F.M.; Rinaman, L. Synaptic Inputs to the Mouse Dorsal Vagal Complex and Its Resident Preproglucagon Neurons. J. Neurosci. 2019, 39, 9767–9781. [Google Scholar] [CrossRef]
- Merchenthaler, I.; Lane, M.; Shughrue, P. Distribution of Pre-pro-Glucagon and Glucagon-like Peptide-1 Receptor Messenger RNAs in the Rat Central Nervous System. J. Comp. Neurol. 1999, 403, 261–280. [Google Scholar]
- Thiebaud, N.; Gribble, F.; Reimann, F.; Trapp, S.; Fadool, D.A. A Unique Olfactory Bulb Microcircuit Driven by Neurons Expressing the Precursor to Glucagon-like Peptide 1. Sci. Rep. 2019, 9, 15542. [Google Scholar] [CrossRef]
- Card, J.P.; Johnson, A.L.; Llewellyn-Smith, I.J.; Zheng, H.; Anand, R.; Brierley, D.I.; Trapp, S.; Rinaman, L. GLP-1 Neurons Form a Local Synaptic Circuit within the Rodent Nucleus of the Solitary Tract. J. Comp. Neurol. 2018, 526, 2149–2164. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Reimann, F.; Gribble, F.M.; Trapp, S. Preproglucagon Neurons Project Widely to Autonomic Control Areas in the Mouse Brain. Neuroscience 2011, 180, 111–121. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Marina, N.; Manton, R.N.; Reimann, F.; Gribble, F.M.; Trapp, S. Spinally Projecting Preproglucagon Axons Preferentially Innervate Sympathetic Preganglionic Neurons. Neuroscience 2015, 284, 872–887. [Google Scholar] [CrossRef]
- Llewellyn-Smith, I.J.; Gnanamanickam, G.J.E.; Reimann, F.; Gribble, F.M.; Trapp, S. Preproglucagon (PPG) Neurons Innervate Neurochemicallyidentified Autonomic Neurons in the Mouse Brainstem. Neuroscience 2013, 229, 130–143. [Google Scholar] [CrossRef]
- Alhadeff, A.L.; Baird, J.-P.; Swick, J.C.; Hayes, M.R.; Grill, H.J. Glucagon-Like Peptide-1 Receptor Signaling in the Lateral Parabrachial Nucleus Contributes to the Control of Food Intake and Motivation to Feed. Neuropsychopharmacology 2014, 39, 2233–2243. [Google Scholar]
- López-Ferreras, L.; Eerola, K.; Shevchouk, O.T.; Richard, J.E.; Nilsson, F.H.; Jansson, L.E.; Hayes, M.R.; Skibicka, K.P. The Supramammillary Nucleus Controls Anxiety-like Behavior; Key Role of GLP-1R. Psychoneuroendocrinology 2020, 119, 104720. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural Regulation of Endocrine and Autonomic Stress Responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The Short Half-Life of Glucagon-like Peptide-1 in Plasma Does Not Reflect Its Long-Lasting Beneficial Effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef]
- Müller, T.D.; Clemmensen, C.; Finan, B.; Dimarchi, R.D.; Tschöp, M.H. Anti-Obesity Therapy: From Rainbow Pills to Polyagonists. Pharm. Rev. 2018, 70, 712–746. [Google Scholar] [CrossRef]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef]
- Hölscher, C. Brain Insulin Resistance: Role in Neurodegenerative Disease and Potential for Targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar]
- Trujillo, J.M.; Nuffer, W.; Smith, B.A. GLP-1 Receptor Agonists: An Updated Review of Head-to-Head Clinical Studies. Adv. Endocrinol. Metab. 2021, 12, 2042018821997320. [Google Scholar] [CrossRef]
- Aroda, V.R. A Review of GLP-1 Receptor Agonists: Evolution and Advancement, through the Lens of Randomised Controlled Trials. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 22–33. [Google Scholar] [CrossRef] [Green Version]
- Neidigh, J.W.; Fesinmeyer, R.M.; Prickett, K.S.; Andersen, N.H. Exendin-4 and Glucagon-like-Peptide-1: NMR Structural Comparisons in the Solution and Micelle-Associated States. Biochemistry 2001, 40, 13188–13200. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Müller, T.D. Emerging Opportunities for the Treatment of Metabolic Diseases: Glucagon-like Peptide-1 Based Multi-Agonists. Mol. Cell Endocrinol. 2015, 418 Pt 1, 42–54. [Google Scholar] [CrossRef]
- Tan, Q.; Akindehin, S.E.; Orsso, C.E.; Waldner, R.C.; DiMarchi, R.D.; Müller, T.D.; Haqq, A.M. Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 2022, 13, 838410. [Google Scholar] [CrossRef]
- Glaesner, W.; Vick, A.M.; Millican, R.; Ellis, B.; Tschang, S.H.; Tian, Y.; Bokvist, K.; Brenner, M.; Koester, A.; Porksen, N.; et al. Engineering and Characterization of the Long-Acting Glucagon-like Peptide-1 Analogue LY2189265, an Fc Fusion Protein. Diabetes Metab. Res. Rev. 2010, 26, 287–296. [Google Scholar] [CrossRef]
- Willard, F.S.; Wainscott, D.B.; Showalter, A.D.; Stutsman, C.; Ma, W.; Cardona, G.R.; Zink, R.W.; Corkins, C.M.; Chen, Q.; Yumibe, N.; et al. Discovery of an Orally Efficacious Positive Allosteric Modulator of the Glucagon-like Peptide-1 Receptor. J. Med. Chem. 2021, 64, 3439–3448. [Google Scholar] [CrossRef]
- Nakane, A.; Gotoh, Y.; Ichihara, J.; Nagata, H. New Screening Strategy and Analysis for Identification of Allosteric Modulators for Glucagon-like Peptide-1 Receptor Using GLP-1 (9-36) Amide. Anal. Biochem. 2015, 491, 23–30. [Google Scholar] [CrossRef]
- Méndez, M.; Matter, H.; Defossa, E.; Kurz, M.; Lebreton, S.; Li, Z.; Lohmann, M.; Löhn, M.; Mors, H.; Podeschwa, M.; et al. Design, Synthesis, and Pharmacological Evaluation of Potent Positive Allosteric Modulators of the Glucagon-like Peptide-1 Receptor (GLP-1R). J. Med. Chem. 2020, 63, 2292–2307. [Google Scholar] [CrossRef]
- Thorens, B. Expression Cloning of the Pancreatic Beta Cell Receptor for the Gluco-Incretin Hormone Glucagon-like Peptide 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8641–8645. [Google Scholar] [CrossRef]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef]
- McLean, B.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef]
- Outeiriño-Iglesias, V.; Romani-Perez, M.; Gonzalez-Matias, L.C.; Vigo, E.; Mallo, F. GLP-1 Increases Preovulatory LH Source and the Number of Mature Follicles, As Well As Synchronizing the Onset of Puberty in Female Rats. Endocrinology 2015, 156, 4226–4237. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Moya, C.M.; Santisteban, P.; González-Matías, L.C.; Vigo, E.; Mallo, F. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology 2015, 156, 3559–3569. [Google Scholar] [CrossRef]
- Bullock, B.P.; Heller, R.S.; Habener, J.F. Tissue Distribution of Messenger Ribonucleic Acid Encoding the Rat Glucagon-like Peptide-1 Receptor. Endocrinology 1996, 137, 2968–2978. [Google Scholar] [CrossRef]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Ørskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 Receptor Localization in Monkey and Human Tissue: Novel Distribution Revealed With Extensively Validated Monoclonal Antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Baggio, L.L.; Yusta, B.; Mulvihill, E.E.; Cao, X.; Streutker, C.J.; Butany, J.; Cappola, T.P.; Margulies, K.B.; Drucker, D.J. GLP-1 Receptor Expression Within the Human Heart. Endocrinology 2018, 159, 1570–1584. [Google Scholar] [CrossRef]
- Ast, J.; Arvaniti, A.; Fine, N.H.F.; Nasteska, D.; Ashford, F.B.; Stamataki, Z.; Koszegi, Z.; Bacon, A.; Jones, B.J.; Lucey, M.A.; et al. Super-Resolution Microscopy Compatible Fluorescent Probes Reveal Endogenous Glucagon-like Peptide-1 Receptor Distribution and Dynamics. Nat. Commun. 2020, 11, 467. [Google Scholar] [CrossRef]
- Zhang, Y.; Parajuli, K.R.; Fava, G.E.; Gupta, R.; Xu, W.; Nguyen, L.U.; Zakaria, A.F.; Fonseca, V.A.; Wang, H.; Mauvais-Jarvis, F.; et al. GLP-1 Receptor in Pancreatic α-Cells Regulates Glucagon Secretion in a Glucose-Dependent Bidirectional Manner. Diabetes 2019, 68, 34–44. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andréasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef]
- Vendrell, J.; el Bekay, R.; Peral, B.; García-Fuentes, E.; Megia, A.; Macias-Gonzalez, M.; Real, J.F.; Jimenez-Gomez, Y.; Escoté, X.; Pachón, G.; et al. Study of the Potential Association of Adipose Tissue GLP-1 Receptor with Obesity and Insulin Resistance. Endocrinology 2011, 152, 4072–4079. [Google Scholar] [CrossRef]
- Ejarque, M.; Guerrero-Pérez, F.; de la Morena, N.; Casajoana, A.; Virgili, N.; López-Urdiales, R.; Maymó-Masip, E.; Pujol Gebelli, J.; Garcia Ruiz de Gordejuela, A.; Perez-Maraver, M.; et al. Role of Adipose Tissue GLP-1R Expression in Metabolic Improvement after Bariatric Surgery in Patients with Type 2 Diabetes. Sci. Rep. 2019, 9, 6274. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Camarena, V.; Sant, D.W.; Wang, G. Human Epicardial Fat Expresses Glucagon-like Peptide 1 and 2 Receptors Genes. Horm. Metab. Res. 2017, 49, 625–630. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, H.; Ma, X.; Zhang, Y.; Lu, S.; Wang, Y.; Zong, C.; Qin, D.; Wang, Y.; Yang, Y.Y.; et al. GLP-1/GLP-1R Signaling in Regulation of Adipocyte Differentiation and Lipogenesis. Cell. Physiol. Biochem. 2017, 42, 1165–1176. [Google Scholar] [CrossRef]
- Körner, M.; Stöckli, M.; Waser, B.; Reubi, J.C. GLP-1 Receptor Expression in Human Tumors and Human Normal Tissues: Potential for in Vivo Targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef]
- Andersen, D.B.; Grunddal, K.V.; Pedersen, J.; Kuhre, R.E.; Lund, M.L.; Holst, J.J.; Ørskov, C. Using a Reporter Mouse to Map Known and Novel Sites of GLP-1 Receptor Expression in Peripheral Tissues of Male Mice. Endocrinology 2021, 162, bqaa246. [Google Scholar] [CrossRef]
- Gray, S.M.; Xin, Y.; Ross, E.C.; Chazotte, B.M.; Capozzi, M.E.; El, K.; Svendsen, B.; Ravn, P.; Sloop, K.W.; Tong, J.; et al. Discordance between GLP-1R Gene and Protein Expression in Mouse Pancreatic Islet Cells. J. Biol. Chem. 2020, 295, 11529–11541. [Google Scholar] [CrossRef]
- Campos, R.V.; Lee, Y.C.; Drucker, D.J. Divergent Tissue-Specific and Developmental Expression of Receptors for Glucagon and Glucagon-like Peptide-1 in the Mouse. Endocrinology 1994, 134, 2156–2164. [Google Scholar] [CrossRef]
- Sandhu, H.; Wiesenthal, S.R.; MacDonald, P.E.; McCall, R.H.; Tchipashvili, V.; Rashid, S.; Satkunarajah, M.; Irwin, D.M.; Shi, Z.Q.; Brubaker, P.L.; et al. Glucagon-like Peptide 1 Increases Insulin Sensitivity in Depancreatized Dogs. Diabetes 1999, 48, 1045–1053. [Google Scholar] [CrossRef]
- Richards, P.; Parker, H.E.; Adriaenssens, A.E.; Hodgson, J.M.; Cork, S.C.; Trapp, S.; Gribble, F.M.; Reimann, F. Identification and Characterization of GLP-1 Receptor-Expressing Cells Using a New Transgenic Mouse Model. Diabetes 2014, 63, 1224–1233. [Google Scholar] [CrossRef]
- Heppner, K.M.; Kirigiti, M.; Secher, A.; Paulsen, S.J.; Buckingham, R.; Pyke, C.; Knudsen, L.B.; Vrang, N.; Grove, K.L. Expression and Distribution of Glucagon-like Peptide-1 Receptor MRNA, Protein and Binding in the Male Nonhuman Primate (Macaca Mulatta) Brain. Endocrinology 2015, 156, 255–267. [Google Scholar] [CrossRef]
- Alvarez, E.; Martínez, M.D.; Roncero, I.; Chowen, J.A.; García-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vázquez, P.; Maldonado, A.; de Cáceres, J.; et al. The Expression of GLP-1 Receptor MRNA and Protein Allows the Effect of GLP-1 on Glucose Metabolism in the Human Hypothalamus and Brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef]
- Reiner, D.J.; Mietlicki-Baase, E.G.; McGrath, L.E.; Zimmer, D.J.; Bence, K.K.; Sousa, G.L.; Konanur, V.R.; Krawczyk, J.; Burk, D.H.; Kanoski, S.E.; et al. Astrocytes Regulate GLP-1 Receptor-Mediated Effects on Energy Balance. J. Neurosci. 2016, 36, 3531–3540. [Google Scholar] [CrossRef]
- Chowen, J.A.; de Fonseca, F.R.; Alvarez, E.; Navarro, M.; García-Segura, L.M.; Blázquez, E. Increased Glucagon-like Peptide-1 Receptor Expression in Glia after Mechanical Lesion of the Rat Brain. Neuropeptides 1999, 33, 212–215. [Google Scholar] [CrossRef]
- Qian, Z.; Chen, H.; Xia, M.; Chang, J.; Li, X.; Ye, S.; Wu, S.; Jiang, S.; Bao, J.; Wang, B.; et al. Activation of Glucagon-like Peptide-1 Receptor in Microglia Attenuates Neuroinflammation-Induced Glial Scarring via Rescuing Arf and Rho GAP Adapter Protein 3 Expressions after Nerve Injury. Ivyspring Int. Publ. Int. J. Biol. Sci. 2022, 2022, 1328–1346. [Google Scholar] [CrossRef]
- Smith, C.; Patterson-Cross, R.; Woodward, O.; Lewis, J.; Chiarugi, D.; Merkle, F.; Gribble, F.; Reimann, F.; Adriaenssens, A. A Comparative Transcriptomic Analysis of Glucagon-like Peptide-1 Receptor- and Glucose-Dependent Insulinotropic Polypeptide-Expressing Cells in the Hypothalamus. Appetite 2022, 174, 106022. [Google Scholar] [CrossRef]
- Heiss, C.N.; Mannerås-Holm, L.; Lee, Y.S.; Serrano-Lobo, J.; Håkansson Gladh, A.; Seeley, R.J.; Drucker, D.J.; Bäckhed, F.; Olofsson, L.E. The Gut Microbiota Regulates Hypothalamic Inflammation and Leptin Sensitivity in Western Diet-Fed Mice via a GLP-1R-Dependent Mechanism. Cell Rep. 2021, 35, 109163. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide Lowers Body Weight in Rodents via Distributed Neural Pathways. JCI Insight 2020, 5, e133429. [Google Scholar] [CrossRef]
- Salameh, T.S.; Rhea, E.M.; Talbot, K.; Banks, W.A. Brain Uptake Pharmacokinetics of Incretin Receptor Agonists Showing Promise as Alzheimer’s and Parkinson’s Disease Therapeutics. Biochem. Pharm. 2020, 180, 114187. [Google Scholar] [CrossRef]
- Burcelin, R.; Gourdy, P.; Dalle, S. GLP-1-Based Strategies: A Physiological Analysis of Differential Mode of Action. Physiology 2014, 29, 108–121. [Google Scholar] [CrossRef]
- Bendotti, G.; Montefusco, L.; Lunati, M.E.; Usuelli, V.; Pastore, I.; Lazzaroni, E.; Assi, E.; Seelam, A.J.; el Essawy, B.; Jang, Y.; et al. The Anti-Inflammatory and Immunological Properties of GLP-1 Receptor Agonists. Pharm. Res. 2022, 182, 106320. [Google Scholar] [CrossRef]
- Richard, J.E.; Anderberg, R.H.; López-Ferreras, L.; Olandersson, K.; Skibicka, K.P. Sex and Estrogens Alter the Action of Glucagon-like Peptide-1 on Reward. Biol. Sex. Differ. 2016, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Trammell, T.S.; Henderson, N.L.; Madkour, H.S.; Stanwood, G.D.; Graham, D.L. GLP-1R Activation Alters Performance in Cognitive Tasks in a Sex-Dependent Manner. Neurol. Sci. 2021, 42, 2911–2919. [Google Scholar] [CrossRef]
- Diz-Chaves, Y.; Toba, L.; Fandiño, J.; González-Matías, L.C.; Garcia-Segura, L.M.; Mallo, F. The GLP-1 Analog, Liraglutide Prevents the Increase of Proinflammatory Mediators in the Hippocampus of Male Rat Pups Submitted to Maternal Perinatal Food Restriction. J. Neuroinflamm. 2018, 15, 337. [Google Scholar] [CrossRef]
- Cataldi, M.; Muscogiuri, G.; Savastano, S.; Barrea, L.; Guida, B.; Taglialatela, M.; Colao, A. Gender-Related Issues in the Pharmacology of New Anti-Obesity Drugs. Obes. Rev. 2019, 20, 375–384. [Google Scholar] [CrossRef]
- Overgaard, R.V.; Petri, K.C.; Jacobsen, L.V.; Jensen, C.B. Liraglutide 3.0 Mg for Weight Management: A Population Pharmacokinetic Analysis. Clin. Pharm. 2016, 55, 1413–1422. [Google Scholar] [CrossRef]
- Rentzeperi, E.; Pegiou, S.; Koufakis, T.; Grammatiki, M.; Kotsa, K. Sex Differences in Response to Treatment with Glucagon-like Peptide 1 Receptor Agonists: Opportunities for a Tailored Approach to Diabetes and Obesity Care. J. Pers. Med. 2022, 12, 454. [Google Scholar] [CrossRef]
- Cabrera-Vera, T.M.; Vanhauwe, J.; Thomas, T.O.; Medkova, M.; Preininger, A.; Mazzoni, M.R.; Hamm, H.E. Insights into G Protein Structure, Function, and Regulation. Endocr. Rev. 2003, 24, 765–781. [Google Scholar] [CrossRef]
- Hölscher, C. Central Effects of GLP-1: New Opportunities for Treatments of Neurodegenerative Diseases. J. Endocrinol. 2014, 221, T31–T41. [Google Scholar] [CrossRef]
- Kahles, F.; Meyer, C.; Möllmann, J.; Diebold, S.; Findeisen, H.M.; Lebherz, C.; Trautwein, C.; Koch, A.; Tacke, F.; Marx, N.; et al. GLP-1 Secretion Is Increased by Inflammatory Stimuli in an IL-6–Dependent Manner, Leading to Hyperinsulinemia and Blood Glucose Lowering. Diabetes 2014, 63, 3221–3229. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Mandard, S.; Dray, C.; Deckert, V.; Valet, P.; Besnard, P.; Drucker, D.J.; Lagrost, L.; Grober, J. Lipopolysaccharides-Mediated Increase in Glucose-Stimulated Insulin Secretion: Involvement of the GLP-1 Pathway. Diabetes 2014, 63, 471–482. [Google Scholar] [CrossRef]
- Cechin, S.R.; Pérez-Álvarez, I.; Fenjves, E.; Molano, R.D.; Pileggi, A.; Berggren, P.O.; Ricordi, C.; Pastori, R.L. Anti-Inflammatory Properties of Exenatide in Human Pancreatic Islets. Cell Transplant. 2012, 21, 633–648. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Park, M.S.; Choung, J.S.; Kim, S.S.; Oh, H.H.; Choi, C.S.; Ha, S.Y.; Kang, Y.; Kim, Y.; Jun, H.S. Glucagon-like Peptide-1 Inhibits Adipose Tissue Macrophage Infiltration and Inflammation in an Obese Mouse Model of Diabetes. Diabetologia 2012, 55, 2456–2468. [Google Scholar] [CrossRef]
- Wang, X.C.; Gusdon, A.M.; Liu, H.; Qu, S. Effects of Glucagon-like Peptide-1 Receptor Agonists on Non-Alcoholic Fatty Liver Disease and Inflammation. World J. Gastroenterol. 2014, 20, 14821–14830. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Bierhansl, L.; Hartung, H.-P.; Aktas, O.; Ruck, T.; Roden, M.; Meuth, S.G. Thinking Outside the Box: Non-Canonical Targets in Multiple Sclerosis. Nat. Rev. Drug Discov. 2022, 21, 578–600. [Google Scholar] [CrossRef]
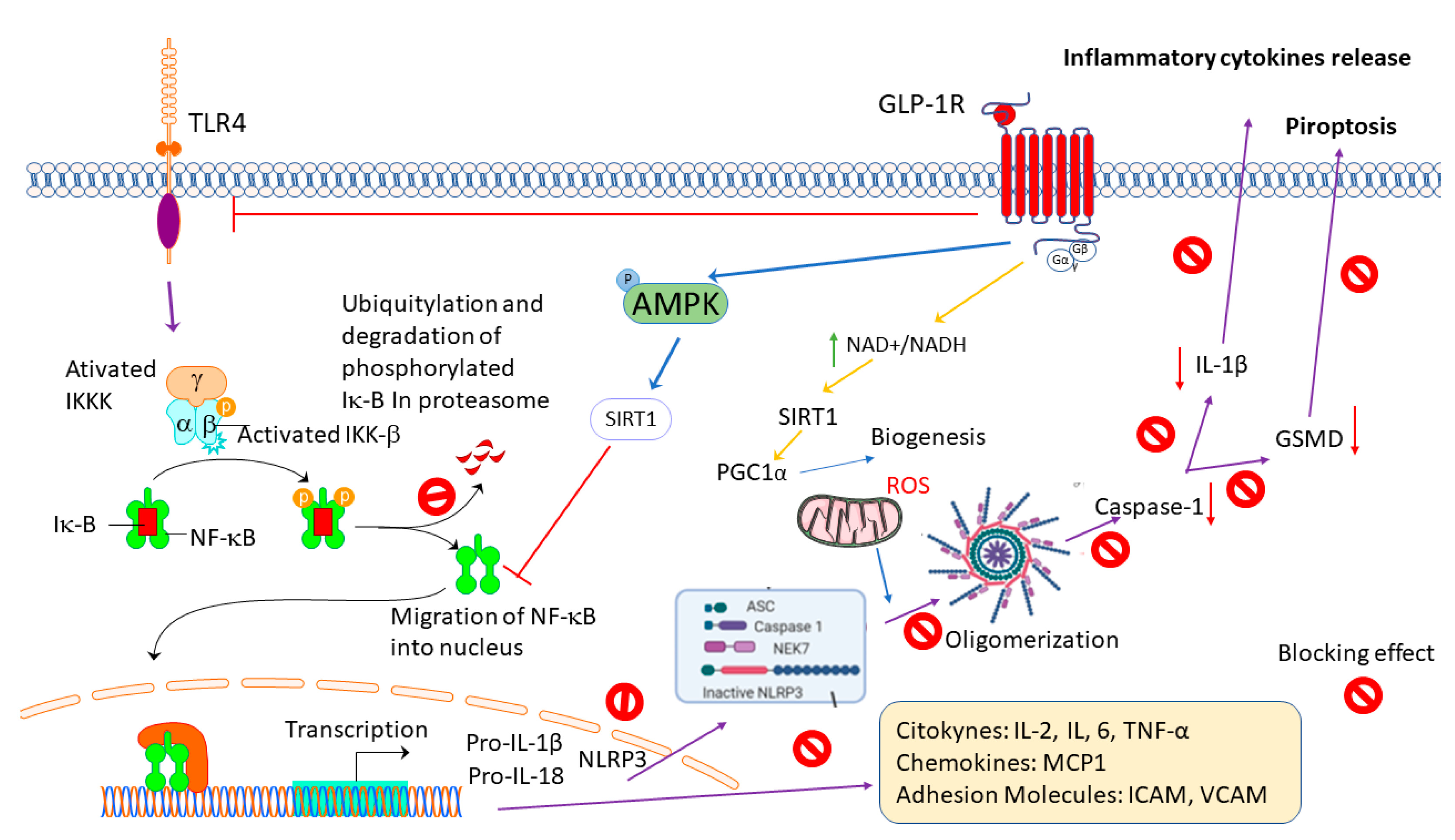
- Song, S.; Guo, R.; Mehmood, A.; Zhang, L.; Yin, B.; Yuan, C.; Zhang, H.; Guo, L.; Li, B. Liraglutide Attenuate Central Nervous Inflammation and Demyelination through AMPK and Pyroptosis-Related NLRP3 Pathway. CNS Neurosci. 2022, 28, 422–434. [Google Scholar] [CrossRef]
- Ammar, R.A.; Mohamed, A.F.; Kamal, M.M.; Safar, M.M.; Abdelkader, N.F. Neuroprotective Effect of Liraglutide in an Experimental Mouse Model of Multiple Sclerosis: Role of AMPK/SIRT1 Signaling and NLRP3 Inflammasome. Inflammopharmacology 2022, 30, 919–934. [Google Scholar] [CrossRef]
- Chiou, H.Y.C.; Lin, M.W.; Hsiao, P.J.; Chen, C.L.; Chiao, S.; Lin, T.Y.; Chen, Y.C.; Wu, D.C.; Lin, M.H. Dulaglutide Modulates the Development of Tissue-Infiltrating Th1/Th17 Cells and the Pathogenicity of Encephalitogenic Th1 Cells in the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 1584. [Google Scholar] [CrossRef]
- Elbaz, E.M.; Senousy, M.A.; El-Tanbouly, D.M.; Sayed, R.H. Neuroprotective Effect of Linagliptin against Cuprizone-Induced Demyelination and Behavioural Dysfunction in Mice: A Pivotal Role of AMPK/SIRT1 and JAK2/STAT3/NF-ΚB Signalling Pathway Modulation. Toxicol. Appl. Pharmacol. 2018, 352, 153–161. [Google Scholar] [CrossRef]
- Glatigny, S.; Bettelli, E. Experimental Autoimmune Encephalomyelitis (EAE) as Animal Models of Multiple Sclerosis (MS). Cold Spring Harb. Perspect. Med. 2018, 8, a028977. [Google Scholar] [CrossRef] [Green Version]
- Kipp, M.; Clarner, T.; Dang, J.; Copray, S.; Beyer, C. The Cuprizone Animal Model: New Insights into an Old Story. Acta Neuropathol. 2009, 118, 723–736. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Smith, M.D.; Sotirchos, E.S.; Jin, J.; Meyers, K.; Taylor, M.; Garton, T.; Bannon, R.; Lord, H.N.; Dawson, T.M.; et al. Therapeutic Potential of a Novel Glucagon-like Peptide-1 Receptor Agonist, NLY01, in Experimental Autoimmune Encephalomyelitis. Neurotherapeutics 2021, 18, 1834–1848. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of Murine Microglial Cells by a V-Raf/v-Myc Carrying Retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Lee, C.H.; Jeon, S.J.; Cho, K.S.; Moon, E.; Sapkota, A.; Jun, H.S.; Ryu, J.H.; Choi, J.W. Activation of Glucagon-Like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by Reducing Neuroinflammatory Responses. Mol. Neurobiol. 2018, 55, 3007–3020. [Google Scholar] [CrossRef]
- DellaValle, B.; Brix, G.S.; Brock, B.; Gejl, M.; Landau, A.M.; Møller, A.; Rungby, J.; Larsen, A. Glucagon-like Peptide-1 Analog, Liraglutide, Delays Onset of Experimental Autoimmune Encephalitis in Lewis Rats. Front. Pharmacol. 2016, 7, 433. [Google Scholar] [CrossRef]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-Related Factors as Biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-Microarray Analysis of Multiple Sclerosis Lesions Yields New Targets Validated in Autoimmune Encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Han, M.H.; Hwang, S.I.; Roy, D.B.; Lundgren, D.H.; Price, J.V.; Ousman, S.S.; Fernald, G.H.; Gerlitz, B.; Robinson, W.H.; Baranzini, S.E.; et al. Proteomic Analysis of Active Multiple Sclerosis Lesions Reveals Therapeutic Targets. Nature 2008, 451, 1076–1081. [Google Scholar] [CrossRef]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of Inflammasomes in Multiple Sclerosis and Their Potential as Therapeutic Targets. J. Neuroinflamm. 2020, 17, 260. [Google Scholar] [CrossRef]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome Activation in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Peixoto, C.A.; de Oliveira, W.H.; da Araújo, S.M.; Nunes, A.K.S. AMPK Activation: Role in the Signaling Pathways of Neuroinflammation and Neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef]
- Carling, D.; Thornton, C.; Woods, A.; Sanders, M.J. AMP-Activated Protein Kinase: New Regulation, New Roles? Biochem. J. 2012, 445, 11–27. [Google Scholar] [CrossRef]
- Muraleedharan, R.; Dasgupta, B. AMPK in the Brain: Its Roles in Glucose and Neural Metabolism. FEBS J. 2022, 289, 2247–2262. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Nelson, A.T.; Trotti, D. Altered Bioenergetics and Metabolic Homeostasis in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2022, 1–17. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients Are Toxic to Motor Neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef]
- Shandilya, A.; Mehan, S. Dysregulation of IGF-1/GLP-1 Signaling in the Progression of ALS: Potential Target Activators and Influences on Neurological Dysfunctions. Neurol. Sci. 2021, 42, 3145–3166. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammatory Processes in Amyotrophic Lateral Sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef]
- Sargsyan, S.A.; Blackburn, D.J.; Barber, S.C.; Grosskreutz, J.; de Vos, K.J.; Monk, P.N.; Shaw, P.J. A Comparison of in Vitro Properties of Resting SOD1 Transgenic Microglia Reveals Evidence of Reduced Neuroprotective Function. BMC Neurosci. 2011, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Chiu, I.M.; Morimoto, E.T.A.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef]
- Wen, D.; Cui, C.; Duan, W.; Wang, W.; Wang, Y.; Liu, Y.; Li, Z.; Li, C. The Role of Insulin-like Growth Factor 1 in ALS Cell and Mouse Models: A Mitochondrial Protector. Brain Res. Bull. 2019, 144, 1–13. [Google Scholar] [CrossRef]
- Laird, A.S.; van Hoecke, A.; de Muynck, L.; Timmers, M.; van den Bosch, L.; van Damme, P.; Robberecht, W. Progranulin Is Neurotrophic in Vivo and Protects against a Mutant TDP-43 Induced Axonopathy. PLoS ONE 2010, 5, e13368. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Boillée, S.; Cleveland, D.W. Toxicity from Different SOD1 Mutants Dysregulates the Complement System and the Neuronal Regenerative Response in ALS Motor Neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7319–7326. [Google Scholar] [CrossRef]
- Li, J.; Cui, L.; Sun, X.; Shen, D.; Yang, X.; Liu, Q.; Liu, M. Alterations in Metabolic Biomarkers and Their Potential Role in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2022, 9, 1027–1038. [Google Scholar] [CrossRef]
- Ferrer-donato, A.; Contreras, A.; Frago, L.M.; Chowen, J.A.; Fernandez-martos, C.M. Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2021, 22, 10305. [Google Scholar] [CrossRef]
- Sun, H.; Knippenberg, S.; Thau, N.; Ragancokova, D.; Körner, S.; Huang, D.; Dengler, R.; Döhler, K.; Petri, S. Therapeutic Potential of N-Acetyl-Glucagon-like Peptide-1 in Primary Motor Neuron Cultures Derived from Non-Transgenic and SOD1-G93A ALS Mice. Cell Mol. Neurobiol. 2013, 33, 347–357. [Google Scholar] [CrossRef]
- Li, Y.; Chigurupati, S.; Holloway, H.W.; Mughal, M.; Tweedie, D.; Bruestle, D.A.; Mattson, M.P.; Wang, Y.; Harvey, B.K.; Ray, B.; et al. Exendin-4 Ameliorates Motor Neuron Degeneration in Cellular and Animal Models of Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e32008. [Google Scholar] [CrossRef]
- Knippenberg, S.; Thau, N.; Schwabe, K.; Dengler, R.; Schambach, A.; Hass, R.; Petri, S. Intraspinal Injection of Human Umbilical Cord Blood-Derived Cells Is Neuroprotective in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2012, 9, 107–120. [Google Scholar] [CrossRef]
- Keerie, A.; Brown-Wright, H.; Kirkland, I.; Grierson, A.; Alix, J.J.P.; Holscher, C.; Mead, R.J. The GLP-1 Receptor Agonist, Liraglutide, Fails to Slow Disease Progression in SOD1 G93A and TDP-43 Q331K Transgenic Mouse Models of ALS. Sci. Rep. 2021, 11, 17027. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- de Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How Does Brain Insulin Resistance Develop in Alzheimer’s Disease? Alzheimers Dement 2014, 10, S26–S32. [Google Scholar] [CrossRef]
- de Felice, F.G. Alzheimer’s Disease and Insulin Resistance: Translating Basic Science into Clinical Applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid Beta-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383. [Google Scholar] [CrossRef]
- Ferreira, S.T. Brain Insulin, Insulin-like Growth Factor 1 and Glucagon-like Peptide 1 Signalling in Alzheimer’s Disease. J. Neuroendocr. 2021, 33, e12959. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and Central Immune System Crosstalk in Alzheimer Disease—A Research Prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Hölscher, C. Potential Role of Glucagon-like Peptide-1 (GLP-1) in Neuroprotection. CNS Drugs 2012, 26, 871–882. [Google Scholar] [CrossRef]
- Park, J.S.; Kam, T.I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.H.; Song, J.J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking Microglial Activation of Reactive Astrocytes Is Neuroprotective in Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Gao, R.; Chen, X.; Chen, R.; Chen, Z. Glucagon-like Peptide-1 Analogs Mitigate Neuroinflammation in Alzheimer’s Disease by Suppressing NLRP2 Activation in Astrocytes. Mol. Cell Endocrinol. 2022, 542, 111529. [Google Scholar] [CrossRef]
- Mcclean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The Diabetes Drug Liraglutide Prevents Degenerative Processes in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- Holubová, M.; Hrubá, L.; Popelová, A.; Bencze, M.; Pražienková, V.; Gengler, S.; Kratochvílová, H.; Haluzík, M.; Železná, B.; Kuneš, J.; et al. Liraglutide and a Lipidized Analog of Prolactin-Releasing Peptide Show Neuroprotective Effects in a Mouse Model of β-Amyloid Pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef]
- Carranza-Naval, M.J.; del Marco, A.; Hierro-Bujalance, C.; Alves-Martinez, P.; Infante-Garcia, C.; Vargas-Soria, M.; Herrera, M.; Barba-Cordoba, B.; Atienza-Navarro, I.; Lubian-Lopez, S.; et al. Liraglutide Reduces Vascular Damage, Neuronal Loss, and Cognitive Impairment in a Mixed Murine Model of Alzheimer’s Disease and Type 2 Diabetes. Front. Aging Neurosci. 2021, 13, 741923. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The Diabetes Drug Liraglutide Ameliorates Aberrant Insulin Receptor Localisation and Signalling in Parallel with Decreasing Both Amyloid-β Plaque and Glial Pathology in a Mouse Model of Alzheimer’s Disease. NeuroMol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic Liraglutide Treatment Prevents Amyloid Plaque Deposition, Chronic Inflammation and Memory Impairment in APP/PS1 Mice. Behav. Brain Res. 2015, 293, 96–106. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra e Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The Diabetes Drug Liraglutide Reverses Cognitive Impairment in Mice and Attenuates Insulin Receptor and Synaptic Pathology in a Non-Human Primate Model of Alzheimer’s Disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Maskery, M.; Goulding, E.M.; Gengler, S.; Melchiorsen, J.U.; Rosenkilde, M.M.; Hölscher, C. The Dual GLP-1/GIP Receptor Agonist DA4-JC Shows Superior Protective Properties Compared to the GLP-1 Analogue Liraglutide in the APP/PS1 Mouse Model of Alzheimer’s Disease. Am. J. Alzheimers Dis. Other Demen. 2020, 35, 1533317520953041. [Google Scholar] [CrossRef]
- Salles, G.N.; Calió, M.L.; Hölscher, C.; Pacheco-Soares, C.; Porcionatto, M.; Lobo, A.O. Neuroprotective and Restorative Properties of the GLP-1/GIP Dual Agonist DA-JC1 Compared with a GLP-1 Single Agonist in Alzheimer’s Disease. Neuropharmacology 2020, 162, 107813. [Google Scholar] [CrossRef]
- Cai, H.Y.; Yang, D.; Qiao, J.; Yang, J.T.; Wang, Z.J.; Wu, M.N.; Qi, J.S.; Hölscher, C. A GLP-1/GIP Dual Receptor Agonist DA4-JC Effectively Attenuates Cognitive Impairment and Pathology in the APP/PS1/Tau Model of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 83, 799–818. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Li, L.; Hölscher, C. A Novel Dual GLP-1/GIP Receptor Agonist Alleviates Cognitive Decline by Re-Sensitizing Insulin Signaling in the Alzheimer Icv. STZ Rat Model. Behav. Brain Res. 2017, 327, 65–74. [Google Scholar] [CrossRef]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Hölscher, C. Neuroprotective Effects of a Triple GLP-1/GIP/Glucagon Receptor Agonist in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the Effects of the Novel GLP-1 Analogue Liraglutide in Alzheimer’s Disease: Study Protocol for a Randomised Controlled Trial (ELAD Study). Trials 2019, 20, 191. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial Phenotypes in Parkinson’s Disease and Animal Models of the Disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s Disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Labandeira, C.; Fraga-Bau, A.; Arias Ron, D.; Alvarez-Rodriguez, E.; Vicente-Alba, P.; Lago-Garma, J.; Rodriguez-Perez, A. Parkinson’s Disease and Diabetes Mellitus: Common Mechanisms and Treatment Repurposing. Neural Regen. Res. 2022, 17, 1652–1658. [Google Scholar] [CrossRef]
- de Iuliis, A.; Montinaro, E.; Fatati, G.; Plebani, M.; Colosimo, C. Diabetes Mellitus and Parkinson’s Disease: Dangerous Liaisons between Insulin and Dopamine. Neural Regen. Res. 2022, 17, 523–533. [Google Scholar] [CrossRef]
- Houser, M.C.; Tansey, M.G. The Gut-Brain Axis: Is Intestinal Inflammation a Silent Driver of Parkinson’s Disease Pathogenesis? NPJ Parkinsons Dis. 2017, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s Disease: Its Role in Neuronal Death and Implications for Therapeutic Intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. The Glucagon-like Peptide 1 (GLP) Receptor as a Therapeutic Target in Parkinson’s Disease: Mechanisms of Action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef]
- de Jesús Esparza-Salazar, F.; Lezama-Toledo, A.; Rivera-Monroy, G.; Borlongan, C. Exendin-4 for Parkinson’s Disease. Brain Circ. 2021, 7, 41. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Jin, Q.Q.; Hölscher, C.; Li, L. Glucagon-like Peptide-1/Glucose-Dependent Insulinotropic Polypeptide Dual Receptor Agonist DA-CH5 Is Superior to Exendin-4 in Protecting Neurons in the 6-Hydroxydopamine Rat Parkinson Model. Neural Regen. Res. 2021, 16, 1660–1670. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, D.; Feng, P.; Xue, G.; Ji, C.; Li, G.; Hölscher, C. A Novel GLP-1/GIP Dual Agonist Is More Effective than Liraglutide in Reducing Inflammation and Enhancing GDNF Release in the MPTP Mouse Model of Parkinson’s Disease. Eur. J. Pharm. 2017, 812, 82–90. [Google Scholar] [CrossRef]
- Cao, B.; Zhang, Y.; Chen, J.; Wu, P.; Dong, Y.; Wang, Y. Neuroprotective Effects of Liraglutide against Inflammation through the AMPK/NF-ΚB Pathway in a Mouse Model of Parkinson’s Disease. Metab. Brain Dis. 2022, 37, 451–462. [Google Scholar] [CrossRef]
- Cao, L.; Li, D.; Feng, P.; Li, L.; Xue, G.F.; Li, G.; Hölscher, C. A Novel Dual GLP-1 and GIP Incretin Receptor Agonist Is Neuroprotective in a Mouse Model of Parkinson’s Disease by Reducing Chronic Inflammation in the Brain. Neuroreport 2016, 27, 384–391. [Google Scholar] [CrossRef]
- Lv, M.J.; Xue, G.F.; Cheng, H.F.; Meng, P.F.; Lian, X.; Hölscher, C.; Li, D.F. The GLP-1/GIP Dual-Receptor Agonist DA5-CH Inhibits the NF-ΚB Inflammatory Pathway in the MPTP Mouse Model of Parkinson’s Disease More Effectively than the GLP-1 Single-Receptor Agonist NLY01. Brain Behav. 2021, 11, e2231. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Semaglutide Is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Neuroprotective Effects of the Novel GLP-1 Long Acting Analogue Semaglutide in the MPTP Parkinson’s Disease Mouse Model. Neuropeptides 2018, 71, 70–80. [Google Scholar] [CrossRef]
- Feng, P.; Zhang, X.; Li, D.; Ji, C.; Yuan, Z.; Wang, R.; Xue, G.; Li, G.; Hölscher, C. Two Novel Dual GLP-1/GIP Receptor Agonists Are Neuroprotective in the MPTP Mouse Model of Parkinson’s Disease. Neuropharmacology 2018, 133, 385–394. [Google Scholar] [CrossRef]
- Kim, S.; Moon, M.; Park, S. Exendin-4 Protects Dopaminergic Neurons by Inhibition of Microglial Activation and Matrix Metalloproteinase-3 Expression in an Animal Model of Parkinson’s Disease. J. Endocrinol. 2009, 202, 431–439. [Google Scholar] [CrossRef]
- Fang, X.; Tian, P.; Zhao, X.; Jiang, C.; Chen, T. Neuroprotective Effects of an Engineered Commensal Bacterium in the 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine Parkinson Disease Mouse Model via Producing Glucagon-like Peptide-1. J. Neurochem. 2019, 150, 441–452. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the Treatment of Patients with Parkinson’s Disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. Evolution of Inflammatory Diseases. Curr. Biol. 2012, 22, R733–R740. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Izquierdo, P.; Attwell, D.; Madry, C. Ion Channels and Receptors as Determinants of Microglial Function. Trends Neurosci. 2019, 42, 278–292. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial Signatures and Their Role in Health and Disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Zhang, C.J.; Jiang, M.; Zhou, H.; Liu, W.; Wang, C.; Kang, Z.; Han, B.; Zhang, Q.; Chen, X.; Xiao, J.; et al. TLR-Stimulated IRAKM Activates Caspase-8 Inflammasome in Microglia and Promotes Neuroinflammation. J. Clin. Investig. 2018, 128, 5399–5412. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Brambilla, R. The Contribution of Astrocytes to the Neuroinflammatory Response in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef]
- Meoni, S.; Macerollo, A.; Moro, E. Sex Differences in Movement Disorders. Nat. Rev. Neurol. 2020, 16, 84–96. [Google Scholar] [CrossRef]
- Chowen, J.A.; Garcia-Segura, L.M. Role of Glial Cells in the Generation of Sex Differences in Neurodegenerative Diseases and Brain Aging. Mech. Ageing Dev. 2021, 196, 111473. [Google Scholar] [CrossRef]
- Dubal, D.B. Sex Difference in Alzheimer’s Disease: An Updated, Balanced and Emerging Perspective on Differing Vulnerabilities. Handb. Clin. Neurol. 2020, 175, 261–273. [Google Scholar] [CrossRef]
- Schwendimann, R.N.; Alekseeva, N. Gender Issues in Multiple Sclerosis. Int. Rev. Neurobiol. 2007, 79, 377–392. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diz-Chaves, Y.; Mastoor, Z.; Spuch, C.; González-Matías, L.C.; Mallo, F. Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 9583. https://doi.org/10.3390/ijms23179583
Diz-Chaves Y, Mastoor Z, Spuch C, González-Matías LC, Mallo F. Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(17):9583. https://doi.org/10.3390/ijms23179583
Chicago/Turabian StyleDiz-Chaves, Yolanda, Zainab Mastoor, Carlos Spuch, Lucas C. González-Matías, and Federico Mallo. 2022. "Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 17: 9583. https://doi.org/10.3390/ijms23179583
APA StyleDiz-Chaves, Y., Mastoor, Z., Spuch, C., González-Matías, L. C., & Mallo, F. (2022). Anti-Inflammatory Effects of GLP-1 Receptor Activation in the Brain in Neurodegenerative Diseases. International Journal of Molecular Sciences, 23(17), 9583. https://doi.org/10.3390/ijms23179583