3.2. Preparation of Sulfuric Acid Adsorbed on Silica Gel (SiO2-H2SO4)
This was previously reported [
29]. To a suspension of silica gel (29.5 g, 230–400 mesh size) in EtOAc (60 mL), H
2SO
4 (1.5 g, 15.5 mmol, 0.8 mL of a 98% aq. solution of H
2SO
4) was added, and the mixture was stirred magnetically for 30 min at room temperature. EtOAc was removed under reduced pressure (rotary evaporator), and the residue was heated at 100 °C for 72 h under vacuum to afford SiO
2-H
2SO
4 as a free-flowing powder.
General Experimental Procedures
A mixture of benzoic acid (0.122 g, 1 mmol), benzaldehyde (0.10 mL, 1 mmol), and isocyanide (0.15 g 1 mmol) was vigorously stirred in 2 mL of water at room temperature for 10 min in the presence of 0.02 g of immobilized sulfuric acid on silica gel (SiO2-H2SO4). Upon completion, the organic layer was separated and collected by a separating funnel. The combined organic phases were concentrated under reduced pressure, and the residue was purified by column chromatography using DCM/hexane (3:1) as an eluent to afford the desired products. Typical yields ranged from 60 to 98%. All other products (A-X) were obtained by a similar approach.
- A.
(2-nitrophenylcarbamoyl)(phenyl)methyl benzoate
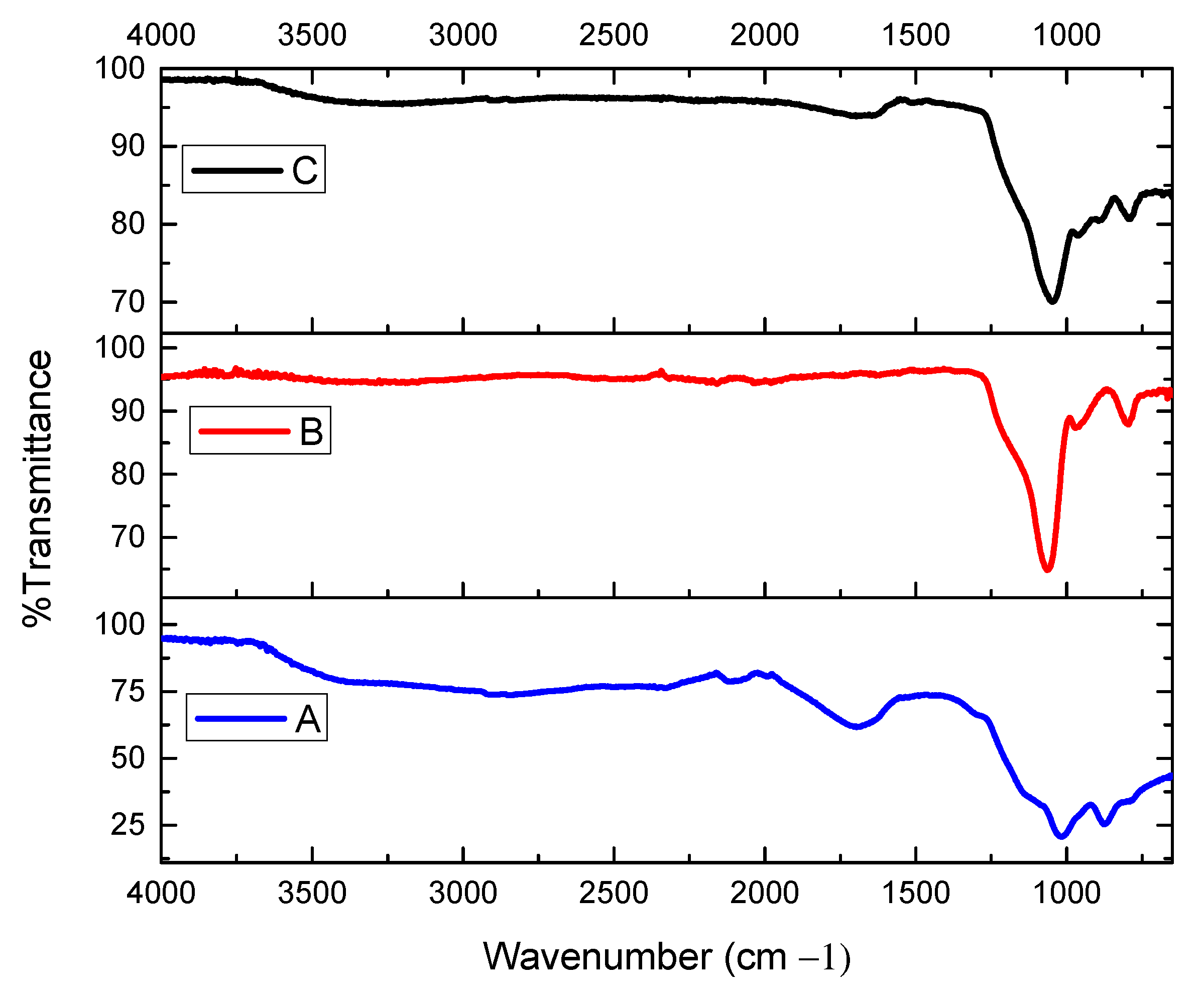
Prepared from 1-isocyano-2-nitrobenzene (0.15 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol,) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (98% yield); mp 112–114 °C; 1H NMR (400 MHz, CDCl3) δ 10.26 (s, 1H, NH), 8.71 (d, J = 8.4 Hz, 1H, Ar-H), 8.51 (s, 1H, Ar-H), 8.15 (d, J = 9.8 Hz, 1H, Ar-H), 8.04 (d, J = 8.4 Hz, 3H, Ar-H), 7.64–7.48 (m, 2H, Ar-H), 7.39 (t, J = 7.7 Hz, 3H, Ar-H), 7.26 (t, J = 7.7 Hz, 1H, Ar-H), 7.14 (t, J = 7.8 Hz, 1H, Ar-H), 6.72 (d, J = 8.4 Hz, 1H, Ar-H), 6.60 (t, J = 7.2 Hz, 1H, (O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 172.40, (NH-CO), 159.83, (O-C=O), 144.69, 136.24, 135.71, 133.88, 133.63, 130.23, 129.33, 128.54, 126.21, 125.91, 124.07, 122.81, 118.77, 116.86. HR-MS (ESI): [M+H+] calculated for C21H17N2O5: 377.1211, found: 377.2603.
- B.
(2-bromophenylcarbamoyl)phenyl)methyl benzoate
Prepared from 1-bromo-2-isocyanobenzene (0.18 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol,) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (76% yield); mp 100–102 °C; IR (NaCl) v(cm−1) 3253 (NH), 1712 (CO) ester, and 1628 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 11.18 (s, 1H), 8.31 (d, J = 8.2 Hz, 1H, NH), 8.13 (d, J = 7.3 Hz, 1H, Ar-H), 8.03 (d, J = 7.2 Hz, 2H, Ar-H), 7.52 (dd, J = 15.8, 8.3 Hz, 1H, Ar-H), 7.44–7.29 (m, 3H, Ar-H), 7.20 (t, J = 7.7 Hz, 1H, Ar-H), 6.89 (t, J = 8.3 Hz, 1H, Ar-H), 6.45 (s, 1H, O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 166.75, (NH-CO), 164.62, (O-C=O), 135.18, 134.84, 134.00, 133.90, 132.28, 130.47, 130.27, 130.04, 129.39, 129.35, 129.07, 128.86, 128.77, 128.62, 128.55, 128.19, 128.16, 127.44, 125.79, 121.67, 113.53, 76.18 (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C21H16BrNO3: 410,0325, found: 410.0331.
- C.
(2-chlorophenylcarbamoyl)(phenyl)methyl benzoate
Prepared from 1-chloro-2-isocyanobenzene (0.14 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (96% yield); mp 143–145 °C; IR (NaCl) v(cm−1) 3300 (NH), 1715 (CO) ester, and 1675 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H, NH), 8.35 (d, J = 8.2 Hz, 1H, Ar-H), 8.12 (d, J = 7.4 Hz, 2H, Ar-H), 7.56 (t, J = 8.0 Hz, 3H, Ar-H), 7.43 (t, J = 7.7 Hz, 2H, Ar-H), 7.37 (s, 1H, Ar-H), 7.33 (dd, J = 9.8, 7.3 Hz, 3H, Ar-H), 7.28 (d, J = 8.0 Hz, 1H, Ar-H), 7.20 (s, 1H, Ar-H), 7.19–7.15 (m, 1H, Ar-H), 6.97 (dd, J = 11.2, 4.3 Hz, 1H, Ar-H), 6.44 (s, 1H, (O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 166.45, (NH-CO), 164.35, (O-C=O), 135.27, 133.92, 133.84, 129.92, 129.32, 129.01, 128.93, 128.75, 127.94, 127.37, 125.16, (Ar-Cl), 122.88, 121.33, 76.18 (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C21H16ClNO3: 366.0831, found: 366.1302.
- D.
(3,4-difluorophenylcarbamoyl)(phenyl)methyl benzoate
Prepared from 1,2-difluoro-4-isocyanobenzene (0.14 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (66% yield); mp 150–151 °C; IR (NaCl) v(cm−1) 3273 (NH), 1732 (CO) ester, and 1671 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H, (NH), 8.17 (d, J = 7.7 Hz, 1H, Ar-H), 8.11 (d, J = 8.3 Hz, 1H, Ar-H), 7.72–7.57 (m, 2H, Ar-H), 7.52 (dd, J = 12.6, 4.8 Hz, 1H, Ar-H), 7.46 (dd, J = 10.9, 5.5 Hz, 2H, Ar-H), 7.15–6.98 (m, 1H, Ar-H), 6.45 (s, 1H, (O-(CH)CO).). 13C NMR (101 MHz, CDCl3) δ 166.46, (NH-CO), 165.40, (O-C=O), 151.01 (NH-R-C-F), 148.91, 134.67, 134.00, 133.71, 130.19, 129.94, 129.47, 129.06, 128.89, 128.78, 128.50, 127.47, 117.31, 117.13, 110.07, 109.85, 76.13, (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C21H15F2NO3: 368,1015, found: 368.1022.
- E.
(3,4-dichlorophenylcarbamoyl)phenyl)methyl benzoate
Prepared from 1,2-dichloro-4-isocyanobenzene (0.17 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow solid (73% yield); mp 152–153 °C; IR (NaCl) v(cm−1) 3273 (NH), 1722 (CO) ester, and 1671 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H, NH), 8.05 (t, J = 8.2 Hz, 3H, Ar-H), 7.65 (s, 1H, Ar-H), 7.54 (t, J = 7.1 Hz, 3H, Ar-H), 7.41 (dd, J = 15.7, 7.9 Hz, 4H, Ar-H), 7.34 (d, J = 6.4 Hz, 2H, Ar-H), 7.27–7.14 (m, 2H, Ar-H), 6.34 (s, 1H, O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 166.75, (NH-CO), 165.68, (O-C=O), 136.54, 134.51, 134.04, 133.75, 132.86, 130.46, 130.22, 129.98, 129.53, 129.33, 129.09, 128.84, 128.79, 128.51, 128.07, 127.50, 121.72, 119.24, 76.25, (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C21H15Cl2NO3: 427.0346, found: 427.1105.
- F.
(3-cyanophenylcarbamoyl)phenyl)methyl benzoate
Prepared from 3-isocyanobenzonitrile (0.13 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (89% yield); mp 136–137 °C; IR (NaCl) v(cm−1) 3350 (NH), 1730 (CO) ester, and 1687 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 11.30 (s, 1H, NH), 8.14–7.82 (m, 2H, Ar-H), 7.66–7.57 (m, 1H, Ar-H), 7.57–7.51 (m, 2H, Ar-H), 7.48 (t, J = 7.9 Hz, 1H, Ar-H), 7.40 (t, J = 7.7 Hz, 2H, Ar-H), 6.36 (s, 1H, O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 172.55, (NH-CO), 167.48, (O-C=O), 133.84, 132.96, 130.77, 130.24, 129.80, 129.40, 128.55, 127.62, 116.65, (C-CN), 113.96, 76.31, (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C22H16N2O3: 357.1172, found: 356.9086.
- G.
(p-tolycarbamoyl)phenyl)methyl benzoate
Prepared from 1-isocyano-4-methylbenzene (0.12 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (94% yield); mp 148–149 °C; IR (NaCl) v(cm−1) 3285 (NH), 1730 (CO) ester, and 1642 (CO) amide. 1H NMR (400 MHz, DMSO) δ 10.44 (s, 1H, NH), 8.07 (s, 2H, Ar-H), 7.96 (s, 1H, Ar-H), 7.71 (s, 3H, Ar-H), 7.57 (s, 3H Ar-H), 7.48 (s, 6H, Ar-H), 7.10 (s, 2H, Ar-H), 6.26 (s, 1H, O-(CH)CO), 2.23 (s, 3H, CH3). 13C NMR (101 MHz, DMSO) δ 167.82 (NH-C=O), 166.82 (O-C=O), 136.42, 135.78, 134.23, 133.31, 133.22, 131.26, 129.93, 129.74, 129.66, 129.56, 129.37, 129.33, 129.17, 129.02, 127.86, 119.81, 76.30, (O-(CH)CO), 20.88 (CH3). HR-MS (ESI): [M+H+] calculated for C22H19NO3: 346.1318, found: 346.1315.
- H.
(m-tolylcarbamoyl)(phenyl)methyl benzoate
Prepared from 1-isocyano-3-methylbenzene (0.12 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (75% yield); IR (NaCl) v(cm−1) 3273 (NH), 1718 (CO) ester, and 1675 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 7.4 Hz, 1H, (NH), 7.80 (s, 1H, Ar-H), 7.55 (dd, J = 14.0, 6.9 Hz, 2H, Ar-H), 7.43 (t, J = 7.7 Hz, 1H, Ar-H), 7.40–7.26 (m, 2H, Ar-H), 7.21 (d, J = 8.1 Hz, 1H, Ar-H), 7.11 (t, J = 7.8 Hz, 1H, Ar-H), 6.86 (d, J = 7.5 Hz, 1H), 6.37 (s, 1H, (O-(CH)CO), 2.23 (s, 2H, CH3), 2.09 (s, 1H, CH3). 13C NMR (101 MHz, CDCl3) δ 166.32, (NH-CO), 165.02, (O-C=O), 139.07, 136.84 (NH-R-CH3), 135.20, 133.83, 129.91, 129.25, 129.13, 128.96, 128.86, 128.75, 127.48, 125.71, 120.71, 117.13, 76.14, (O-(CH)CO), 21.43, (CH3). HR-MS (ESI): [M+H+] calculated for C22H19NO3: 346.1340, found: 346.1836.
- I.
(3,5-dimethylphenylcarbamoyl)(phenyl)methyl benzoate
Prepared from 3,5-dimethyl phenyl isocyanide (0.13 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (94% yield); mp 148–149 °C; 1H NMR (400 MHz, CDCl3) δ 10.45 (s, 1H, NH), 8.03 (dd, J = 14.1, 7.5 Hz, 3H, Ar-H), 7.54 (t, J = 7.4 Hz, 2H, Ar-H), 7.40 (t, J = 7.7 Hz, 3H, Ar-H), 7.28 (d, J = 7.1 Hz, 1H, Ar-H), 6.95–6.84 (m, 1H, Ar-H), 6.82 (t, J = 7.4 Hz, 1H, Ar-H), 6.75 (d, J = 8.1 Hz, 1H, Ar-H), 6.32 (s, 1H, (O-(CH)CO), 4.44 (d, J = 5.9 Hz, 1H, CH3), 3.61 (s, 1H, CH3). 13C NMR (101 MHz, CDCl3) δ 168.04, NH(CO), 164.79, (O-C=O), 135.58, 133.77, 130.24, 129.83, 129.37, 129.10, 128.96, 128.75, 128.60, 128.51, 127.52, 125.52, 120.84, 110.27, 75.90, (O-(CH)CO), 55.11, 40.00. HR-MS (ESI): [M+H+] calculated for C23H21NO3: 361.1522, found: 361.1936.
- J.
(mesitylcarbamoyl)(phenyl)methyl benzoate
Prepared from 2-isocyano-1,3,5-trimethylbenzene (0.15 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a colorless liquid (60% yield); IR (NaCl) v(cm−1) 3257 (NH), 1730 (CO) ester, and 1604 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 8.05 (d, J = 7.5 Hz, 1H), 7.56 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 6.77 (s, 3H), 2.25 (s, 7H), 2.17 (s, 4H). 13C NMR (101 MHz, CDCl3) δ 166.96, 162.35, 138.81, 134.55, 131.32, 130.56, 129.61, 128.86, 128.46, 126.94, 21.17, 18.77. HR-MS (ESI): [M+H+] calculated for C24H23NO3: 374.1655, found: 374.2118.
- K.
(4-methyl-2-nitrophenylcarbamoyl)phenyl)methylbenzoate
Prepared from 4-methyl-2-nitro phenyl isocyanide (0.16 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow solid (92% yield); mp 126–129 °C; IR (NaCl) v(cm−1) 3343 (NH), 1694 (CO) ester, and 1641 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 11.19 (s, 1H, NH), 8.60 (d, J = 8.6 Hz, 1H, Ar-H), 8.19 (d, J = 7.3 Hz, 2H, Ar-H), 7.95 (s, 1H, Ar-H), 7.56 (d, J = 7.7 Hz, 3H Ar-H), 7.35 (d, J = 7.6 Hz, 4H, Ar-H), 7.11 (d, J = 10.2 Hz, 2H, Ar-H), 6.66 (d, J = 8.5 Hz, 2H, Ar-H), 6.43 (s, 1H, O-(CH)CO), 2.42 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 172.76, (NH-CO), 167.52, (O-C=O), 143.91, 142.77, 137.23, 136.37, 135.07, 134.39, 133.87, 131.89, 130.09, 129.56, 129.01, 128.77, 127.15, 126.62, 125.85, 125.71, 125.27, 122.02, 118.80, 76.36, (O-(CH)CO), 21.32. HR-MS (ESI): [M+H+] calculated for C22H18N2O5: 391.1383, found: 391.3062.
- L.
(4-methoxyphenylcarbamoyl)phenyl)methyl benzoate
Prepared from 1-isocyano-4-methoxy benzene (0.13 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (94% yield); IR (NaCl) v(cm−1) 3293 (NH), 1734 (CO) ester, and 1671 (CO) amide. 1H NMR (400 MHz, DMSO) δ 10.42 (s, 1H, NH), 8.08 (d, J = 7.2 Hz, 2H, Ar-NH), 7.70 (d, J = 7.3 Hz, 3H), 7.58 (t, J = 7.7 Hz, 2H), 7.52–7.46 (m, 4H), 7.43 (dd, J = 14.5, 7.4 Hz, 2H), 6.88 (d, J = 9.1 Hz, 2H), 6.22 (s, 1H, O-(CH)CO), 3.70 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO) δ 166.57 (NHC=O), 165.69 (O-C=O), 155.95 (R-C-O-CH3), 135.81, 134.28, 132.01, 129.94, 129.74, 129.50, 129.37, 129.19, 129.03, 127.84, 121.25, 114.39, 76.27 (O-(CH)CO), 55.60 (OCH3). HR-MS (ESI): [M+H+] calculated for C22H19NO4: 362.1346, found: 362.1319.
- M.
(2-methoxybenzylcarbamoyl)phenyl)methyl benzoate
Prepared from 1-(isocyanomethyl)-2-methoxybenzene (0.15 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a colorless liquid (94% yield); IR (NaCl) v(cm−1) 3273 (NH), 1722 (CO) ester, and 1663 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.10–8.04 (m, 1H, NH), 8.02 (dd, J = 8.4, 1.2 Hz, 1H, Ar-H), 7.60–7.49 (m, 1H, Ar-H), 7.48–7.35 (m, 2H, Ar-H), 7.29 (d, J = 7.0 Hz, 1H, Ar-H), 7.23–7.12 (m, 1H, Ar-H), 6.83 (td, J = 7.4, 0.7 Hz, 1H, Ar-H), 6.76 (d, J = 8.2 Hz, 1H, Ar-H), 6.30 (s, 1H, O-(CH)CO)), 4.44 (d, J = 5.9 Hz, 1H, (NH-CH-R), 3.67 (d, J = 6.2 Hz, 1H, (NH-CH-R), 3.63 (s, 1H, O-CH3). 13C NMR (101 MHz, CDCl3) δ 171.76, (NH-CO), 167.97, (O-C=O), 157.61, (C-O-CH3), 135.74, 133.79, 133.59, 130.22, 129.86, 129.81, 129.37, 129.31, 129.07, 128.93, 128.73, 128.60, 128.51, 127.48, 125.57, 120.80, 110.26, 75.90, (O-(CH)CO), 55.10, (O-CH3) 39.95, (NH-(CH2)R. HR-MS (ESI): [M+H+] calculated for C23H21NO4: 376.1435, found: 376.1560.
- N.
(4-methoxybenzylcarbamoyl)phenyl)methyl benzoate
Prepared from 1-(isocyanomethyl)-4-methoxybenzene (0.15 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (98% yield); mp 112–114 °C; 1H NMR (400 MHz, CDCl3) δ 8.72 (s, 1H, NH), 8.09–7.93 (m, 2H, Ar-H), 7.47 (dt, J = 15.6, 9.4 Hz, 1H, Ar-H), 7.43–7.31 (m, 2H, Ar-H), 7.26–7.16 (m, 1H, Ar-H), 7.08 (dd, J = 14.6, 9.1 Hz, 1H, Ar-H), 6.80–6.55 (m, 1H, Ar-H), 6.25 (d, J = 30.0 Hz, 1H, (O-(CH)CO), 4.69 (s, 1H, OCH3), 4.62 (s, 1H), 4.36–4.28 (m, 1H, (NH-CH-R), 3.68–3.62 (m, 1H, (NH-CH-R). 13C NMR (101 MHz, CDCl3) δ 164.35, (NH-CO), 161.83, (O-C=O), 159.17, 143.09, 133.71, 130.46, 130.19, 129.53, 129.26, 129.02, 128.88, 128.49, 127.81, 127.48, 123.36, 114.46, 114.33, 114.18, 114.13, 113.97, 76.08, (O-(CH)CO), 55.26, (OCH3), 41.88, (NH-CH2). HR-MS (ESI): [M+H+] calculated for C23H21NO4: 376.1402, found: 376.2408.
- O.
ethyl 2-[2-(benzoyloxy)-2-phenylacetamido]benzoate
Prepared from ethyl-2 isocyanobenzoate (0.18 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (88% yield); mp 130–132 °C; IR (NaCl) v(cm−1) 3242 (NH), 1730 (CO) ester, and 1683 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, NH), 8.65 (d, J = 8.6 Hz, 1H, Ar-H), 8.27 (d, J = 7.5 Hz, 2H, Ar-H), 7.93 (dd, J = 14.6, 8.6 Hz, 1H, Ar-H), 7.59 (d, J = 7.2 Hz, 2H Ar-H), 7.53 (t, J = 7.4 Hz, 1H, Ar-H), 7.48–7.36 (m, 4H, Ar-H), 7.37–7.24 (m, 3H, Ar-H), 7.01 (t, J = 7.6 Hz, 1H, Ar-H), 6.38 (s, 1H, O-(CH)CO), 4.37 (q, J = 7.1 Hz, 1H, CH), 4.31–4.19 (m, 2H, CH2), 1.29 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 168.12 O-C=O benzoate), 167.48 (NH-CO), 165.09 (O-C=O ester), 140.87, 135.72, 134.55, 133.54, 131.30, 130.75, 129.24, 128.85, 128.37, 127.24, 123.05, 120.54, 115.87, 62.02, 61.44, 14.18. HR-MS (ESI): [M+H+] calculated for C25H21NO6: 431.1317, found: 431.1020.
- P.
(naphthalen-3-ylcarbamoyl)(phenyl)methyl benzoate
Prepared from 1-isocyanonaphthalene (0.15 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white powder (80% yield); mp 171–172 °C; IR (NaCl) v(cm−1) 3422 (NH), 1713 (CO) ester, and 1623 (CO) amide. 1H NMR (400 MHz, DMSO) δ 8.15 (d, J = 7.4 Hz, 1H, (NH), 7.99–7.90 (m, 1H, Ar-H), 7.85 (d, J = 7.3 Hz, 1H, Ar-H), 7.80 (d, J = 8.0 Hz, 1H, Ar-H), 7.77–7.67 (m, 1H, Ar-H), 7.59 (dd, J = 9.7, 5.6 Hz, 2H, Ar-H), 7.57–7.51 (m, 1H, Ar-H), 7.52–7.43 (m, 1H, Ar-H), 6.53 (s, 1H, O-(CH)CO). 13C NMR (101 MHz, DMSO) δ 168.14, (NH-CO), 165.81, (O-C=O), 136.03, 134.25, 134.19, 133.11, 130.03, 129.62, 129.44, 129.34, 129.25, 128.66, 128.63, 127.93, 126.61, 126.49, 126.00, 122.98, 122.83, 76.44, (O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C25H20NO3: 382.1339, found: 382.1316.
- Q.
(2-iodophenylcarbamoyl)phenyl)methyl benzoate
Prepared from 1-iodo-2-isocyanobenzene (0.23 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a green solid (85% yield); mp 104–106 °C; IR (NaCl) v(cm−1) 3249 (NH), 1730 (CO) ester, and 1679 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H, NH), 8.24 (d, J = 8.3 Hz, 1H, Ar-H), 8.18 (d, J = 7.2 Hz, 2H, Ar-H), 7.82 (d, J = 8.0 Hz, 1H, Ar-H), 7.69 (d, J = 6.7 Hz, 2H, Ar-H), 7.57 (t, J = 7.6 Hz, 4H, Ar-H), 7.43 (t, J = 7.7 Hz, 3H, Ar-H), 7.27 (dd, J = 12.1, 4.8 Hz, 2H, Ar-H), 7.08–7.01 (m, 1H, Ar-H), 6.79 (td, J = 7.9, 1.5 Hz, 2H, Ar-H), 6.46 (s, 1H, O-(CH)CO). 13C NMR (101 MHz, CDCl3) δ 166.92, (NH-CO), 164.85, (O-C=O), 139.58, 138.85, 137.32, 135.20, 133.97, 130.48, 130.20, 129.45, 129.35, 129.14, 128.88, 128.69, 127.67, 127.47, 126.43, 121.66, 76.13, O-(CH)CO). HR-MS (ESI): [M+H+] calculated for C21H16INO3: 458.0158, found: 458.0715.
- R.
(benzo[d]thiazol-2-ylcarbamoyl)(phenyl)methyl benzoate
Prepared from 2-isocyanobenzo[d]thiazole (0.16 g, 1 mMol), benzaldehyde (0.11 mL, 1 mMol), and benzoic acid (0.122 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a white solid (63% yield); mp 122–124 °C; IR (NaCl) v(cm−1) 3286 (NH), 1710 (CO) ester, and 1679 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H, NH), 8.09 (d, J = 7.4 Hz, 1H, Ar-H), 7.96 (d, J = 7.4 Hz, 1H, Ar-H), 7.79 (d, J = 7.6 Hz, 1H, Ar-H), 7.61 (t, J = 7.5 Hz, 1H, Ar-H), 7.57–7.50 (m, 1H, Ar-H), 7.45 (dd, J = 16.4, 8.4 Hz, 2H, Ar-H), 7.33 (t, J = 7.6 Hz, 1H, Ar-H), 7.25 (t, J = 9.0 Hz, 1H, Ar-H). 13C NMR (101 MHz, CDCl3) δ 165.18, (NH-CO), 164.31, (O-C=O), 161.31, (Ar-C=S), 133.52, 132.17, 130.96, 130.76, 129.56, 129.23, 128.95, 128.80, 128.06, 127.93, 126.72, 125.26, 123.07, 122.07, 120.68, 120.50, 119.74. HR-MS (ESI): [M+H+] calculated for C22H17N2O3S: 389.0861, found: 389.2442.
- S.
3-(3-methoxyphenylcarbamoyl)-5-bromo-2-oxoindolin-3-yl 4-bromobenzoate
Prepared from 1-isocyano-3-methoxybenzene (1.33 mL, 1 mMol), 5-bromo isatin (0.23 g, 1 mMol), and 4-bromobenzoic acid (0.21 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (90% yield); IR (NaCl) v(cm−1) 3286 (NH), 1710 (CO) ester, and 1679 (CO) amide. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H, (NH-amide)), 8.75 (s, 1H, (NH-indole)), 7.98 (dd, J = 14.9, 7.7 Hz, 3H, (Ar-H), 7.49 (dd, J = 12.7, 7.2 Hz, 2H, (Ar-H)), 7.35 (dd, J = 16.1, 8.2 Hz, 6H, (Ar-H)), 7.16 (dt, J = 23.6, 8.0 Hz, 3H, (Ar-H)), 6.97 (dd, J = 17.5, 8.8 Hz, 2H, (Ar-H)), 6.85 (d, J = 7.8 Hz, 1H, (Ar-H)), 6.62 (dd, J = 8.2, 1.7 Hz, 1H, (Ar-H)), 5.18 (s, NH-indole), 3.68 (s, 3H, (O-CH3)), 2.08 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 173.55, (-CO-indole), 171.03, (-CO-ester), 163.84, (CO-amide), 161.87, (C-OCH3), 160.17, 142.31, 138.36, 134.36, 133.41, 131.12, 130.14, 129.77, 129.61, 128.75, 128.46, 128.08, 124.91, 124.85, 123.44, 112.37, 111.47, 111.22, 105.66, 55.39, 31.00. HR-MS (ESI): [M+H+] calculated for C23H18Br2N2O6: 575.9202, found: 575.6714.
- T.
3-(p-tolylcarbamoyl)-5-bromo-2-oxoindolin-3-yl 4-bromobenzoate
Prepared from 1-isocyano-4-methylbenzene (0.13 mL, 1 mMol) 5-bromo isatin (0.23 g, 1 mMol), and 4-bromobenzoic acid (0.21 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as yellow liquid (76% yield); IR (NaCl) v(cm−1) 3244 (NH), 1718 (CO) ester, and 1682 (CO) amide. 1H NMR (400 MHz, DMSO) δ 11.12 (d, J = 40.1 Hz, 1H, NH-amide), 10.09 (d, J = 23.3 Hz, 1H, NH-indole), 8.71 (d, J = 11.1 Hz, 1H, (Ar-H), 8.23 (s, 1H, Ar-H), 7.87 (d, J = 8.4 Hz, 1H, Ar-H), 7.77–7.61 (m, 1H, Ar-H), 7.47 (d, J = 8.3 Hz, 2H, Ar-H), 7.09 (dd, J = 17.5, 8.0 Hz, 3H, Ar-H), 6.87 (d, J = 8.3 Hz, 1H, Ar-H), 2.25 (s, 3H, CH3). 13C NMR (101 MHz, DMSO) δ 167.13 (CO-indole), 162.97 (CO-amide), 159.82 (CO-ester), 159.49, 140.46, 136.22, 133.16, 133.00, 132.17, 131.77, 130.25, 129.70, 127.37, 119.52, 118.10, 114.71, 20.92. HR-MS (ESI): [M+H+] calculated for C23H16Br2N2O4: 541.9416, found: 541.4483.
- U.
3-(2-nitrophenylcarbamoyl)-5-bromo-2-oxoindolin-3-yl 4-bromobenzoate
Prepared from 1-isocyano-2-nitrobenzene (0.15 g, 1 mMol), 5-bromo isatin (0.23 g, 1 mMol), and 4-bromobenzoic acid (0.21 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (85% yield); IR (NaCl) v(cm−1) 3280 (NH), 1712 (CO) ester, and 1676 (CO) amide. 1H NMR (400 MHz, DMSO) δ 11.07 (s, 1H, NH-amide), 8.20 (d, J = 8.2 Hz, 1H NH-indole), 7.93 (d, J = 8.6 Hz, 3H, Ar-H), 7.88–7.80 (m, 3H, Ar-H), 7.73 (t, J = 7.6 Hz, 1H, Ar-H), 7.66 (d, J = 8.4 Hz, 1H, Ar-H), 7.55 (t, J = 7.7 Hz, 1H, Ar-H), 7.49–7.41 (m, 6H, Ar-H), 7.36 (t, J = 7.7 Hz, 4H, Ar-H), 7.01 (t, J = 6.8 Hz, 4H, Ar-H), 6.87 (t, J = 11.5 Hz, 1H, Ar-H), 6.58 (t, J = 7.7 Hz, 3H, Ar-H). 13C NMR (101 MHz, DMSO) δ 184.86 (-CO-indole), 172.49 (-CO-ester), 167.11 (-CO-amide), 159.84 (-C-NO2), 151.16, 146.68, 144.17, 138.74, 136.10, 135.54, 132.08, 131.71, 131.44, 130.69, 130.43, 130.40, 127.35, 126.12, 125.80, 125.09, 123.15, 119.62, 118.23, 115.85, 112.63. HR-MS (ESI): [M+H+] calculated for C22H13Br2N3O6: 575.9320, found: 575.6714.
- V.
3-(4-chlorophenoxy)-N-(naphthalen-1-yl)-2-oxoindoline-3-carboxamide
Prepared from 1-isocyano-naphthalein (0.15 mL, 1 mMol), isatin (0.147 g, 1 mMol), and 4-chlorophenol (0.13 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (80% yield); IR (NaCl) v(cm−1) 3295 (NH), 1053 (CO), and 1679 (CO) amide. 1H NMR (400 MHz, DMSO) δ 11.06 (s, 2H, NH-amide), 10.34 (s, 1H, NH-indole), 9.71 (s, 1H, Ar-H), 8.59 (d, J = 10.5 Hz, 1H, Ar-H), 8.49 (s, 1H, Ar-H), 8.22–8.05 (m, 1H, Ar-H), 7.98 (dd, J = 25.7, 7.5 Hz, 1H, Ar-H), 7.83 (dd, J = 26.9, 7.8 Hz, 1H, Ar-H), 7.72 (dd, J = 16.1, 7.8 Hz, 1H, Ar-H), 7.58 (t, J = 7.6 Hz, 3H, Ar-H), 7.50 (d, J = 7.3 Hz, 2H, Ar-H), 7.19 (d, J = 8.8 Hz, 2H, Ar-H), 7.06 (t, J = 7.5 Hz, 2H, Ar-H), 6.91 (d, J = 7.9 Hz, 2H, Ar-H), 6.77 (d, J = 8.8 Hz, 2H, Ar-H). 13C NMR (101 MHz, DMSO) δ 184.87 (CO-indole), 160.79 (CO-amide), 159.85, 156.76, 151.15, 138.83, 134.11, 133.03, 129.60, 128.80, 126.59, 126.53, 126.13, 125.25, 125.16, 123.22, 122.76, 122.23, 119.78, 118.29, 117.37, 112.65. HR-MS (ESI): [M+H+] calculated for C22H13Br2N3O6: 573.93, found: 573.80.
- W.
3-(4-chlorophenoxy)-2-oxo-N-p-tolylindoline-3-carboxamide
Prepared from 1-isocyano-4-methylbenzene (0.13 mL, 1 mMol), isatin (0.147 g, 1 mMol), and 4-chlorophenol (0.13 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (87% yield); IR (NaCl) v (cm−1) 3259 (NH), 1048 (CO), and 1680 (CO) amide. 1H NMR (400 MHz, DMSO) δ 11.07 (s, 1H, NH-amide), 10.11 (s, 1H, NH-indole), 9.72 (s, 2H, Ar-H), 8.71 (d, J = 11.1 Hz, 1H, Ar-H), 8.24 (s, 1H, Ar-H), 7.58 (t, J = 7.7 Hz, 1H, Ar-H), 7.55–7.41 (m, 2H, Ar-H), 7.19 (d, J = 8.7 Hz, 4H, Ar-H), 7.14–6.99 (m, 3H, Ar-H), 6.91 (d, J = 7.9 Hz, 1H, Ar-H), 6.77 (d, J = 8.7 Hz, 4H, Ar-H), 2.25 (s, 3H, CH3). 13C NMR (101 MHz, DMSO) δ 184.87 (CO-indole), 162.96 (CO-amide), 159.86, 159.81, 156.76, 151.16, 138.82, 136.22, 133.17, 133.00, 130.25, 129.69, 129.60, 125.16, 123.22, 122.77, 119.54, 118.29, 118.12, 117.37, 112.65, 20.92. HR-MS (ESI): [M+H+] calculated for C22H17ClN2O3: 573.0911, found: 393.2902.
- X.
3-(4-chlorophenoxy)-N-(3,5-dimethylphenyl)-2-oxoindoline-3-carboxamide
Prepared from 1-isocyano-3,5-dimethylbenzene (0.131 mL, 1 mMol), isatin (0.147 g, 1 mMol), and 4-chlorophenol (0.13 g, 1 mMol) according to the general procedure. Purification: column chromatography on silica gel (3:1 DCM/hexane). Isolated as a yellow liquid (87% yield); IR (NaCl) v(cm−1) 3283 (NH), 1050 (CO), and 1679 (CO) amide. 1H NMR (400 MHz, DMSO) δ 11.07 (s, 1H, NH-indole), 10.05 (s, 1H, NH-amide), 9.72 (s, 2H, Ar-H), 8.75 (d, J = 11.0 Hz, 1H, Ar-H), 8.23 (s, 1H, Ar-H), 7.59 (t, J = 7.7 Hz, 1H, Ar-H), 7.50 (d, J = 7.4 Hz, 1H, Ar-H), 7.19 (d, J = 8.8 Hz, 6H, Ar-H), 7.07 (t, J = 7.5 Hz, 1H, Ar-H), 6.91 (d, J = 7.8 Hz, 1H, Ar-H), 6.78 (t, J = 8.7 Hz, 5H, Ar-H), 6.71 (s, 1H, Ar-H), 2.23 (s, 5H, CH3). 13C NMR (101 MHz, DMSO) δ 184.87 (CO-indole), 159.94 (CO-amide), 159.86, 156.76, 151.16, 138.83, 138.57, 138.35, 129.60, 125.60, 125.17, 123.23, 122.77, 118.29, 117.38, 117.27, 115.63, 112.65, 21.52. HR-MS (ESI): [M+H+] calculated for C23H19ClN2O3: 406.1136, found: 406.4155.


 ,
, 

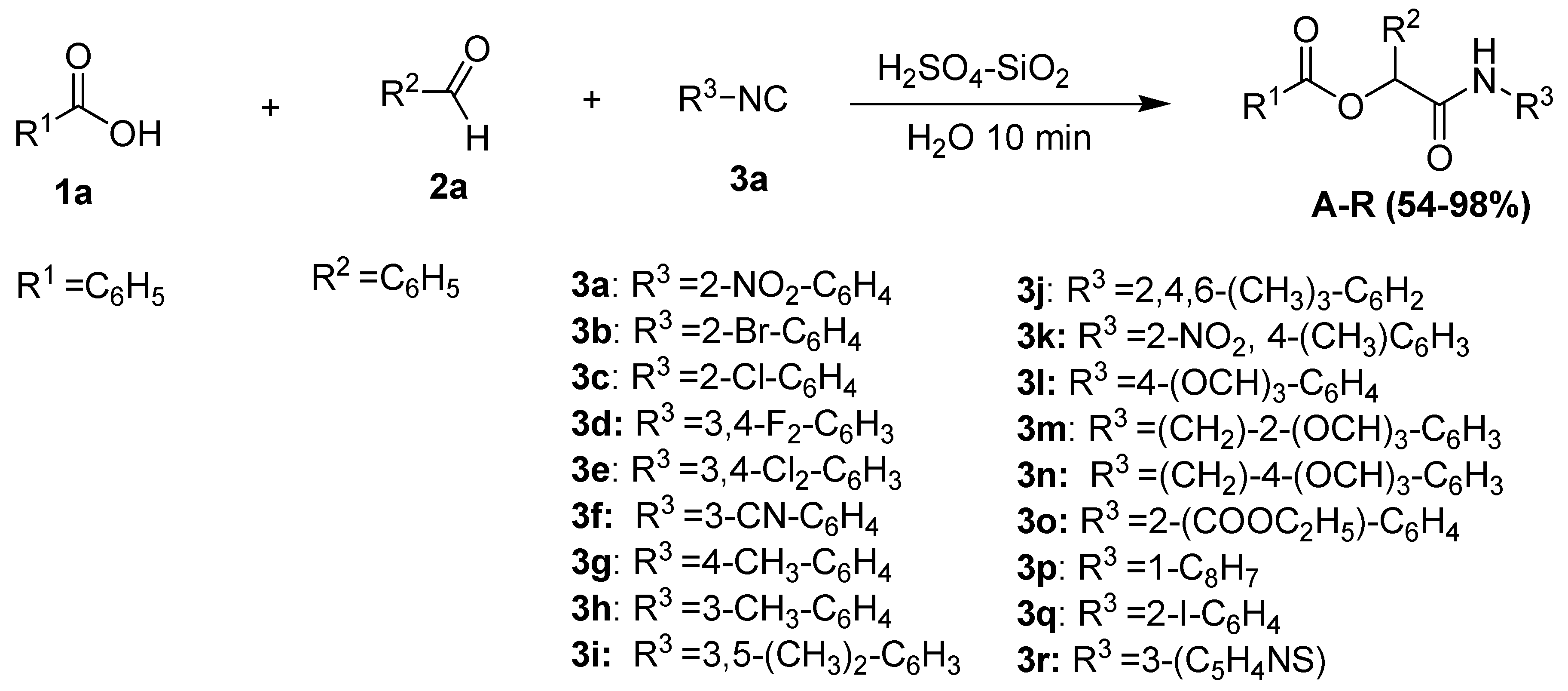


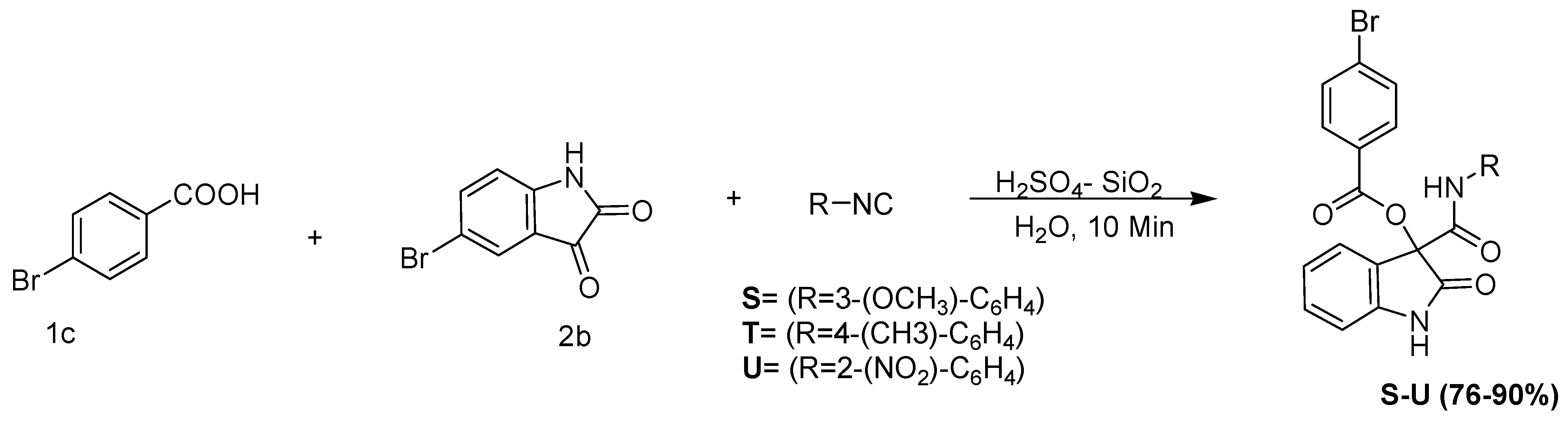
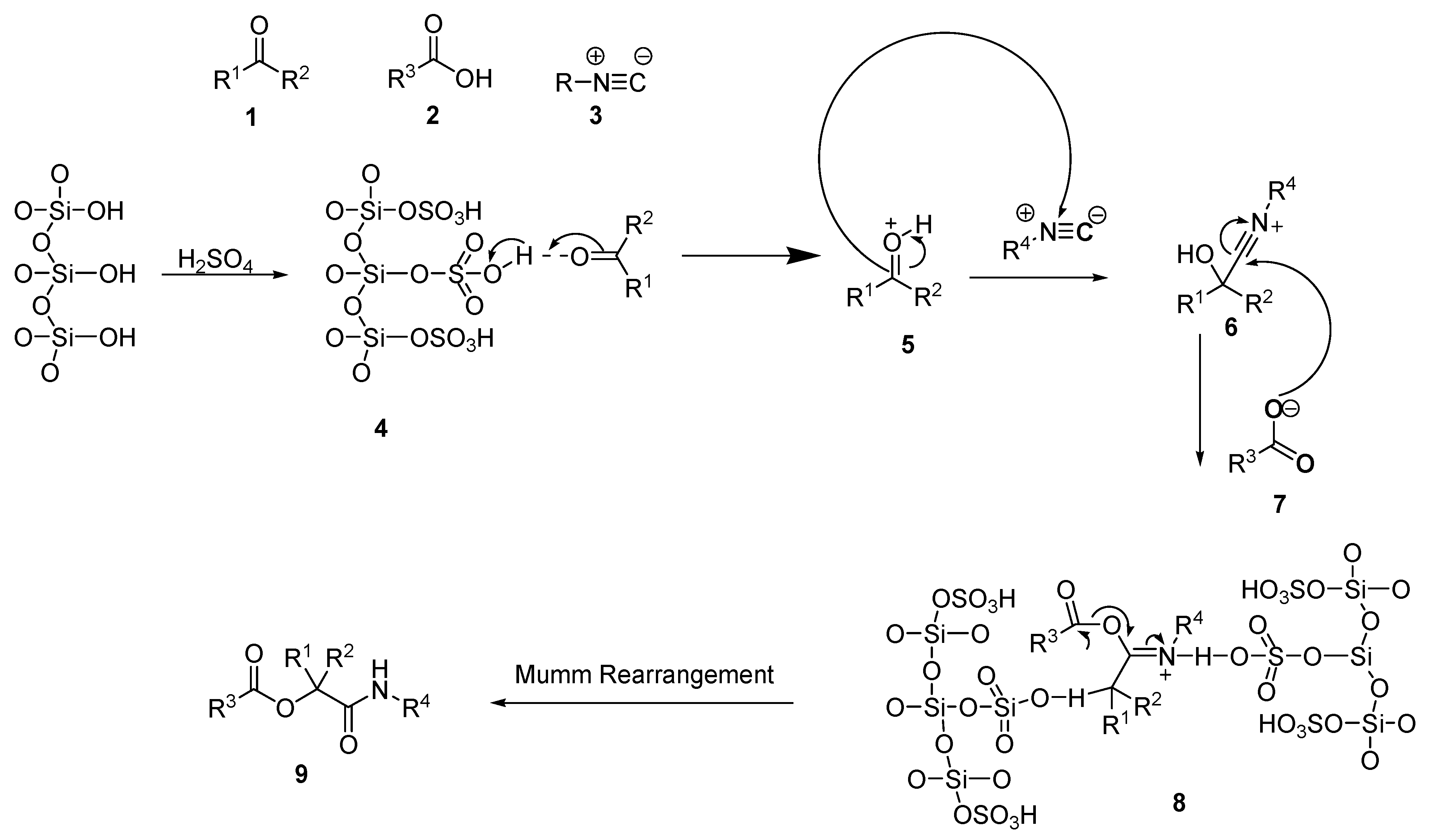

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}