
Connexins and Glucose Metabolism in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
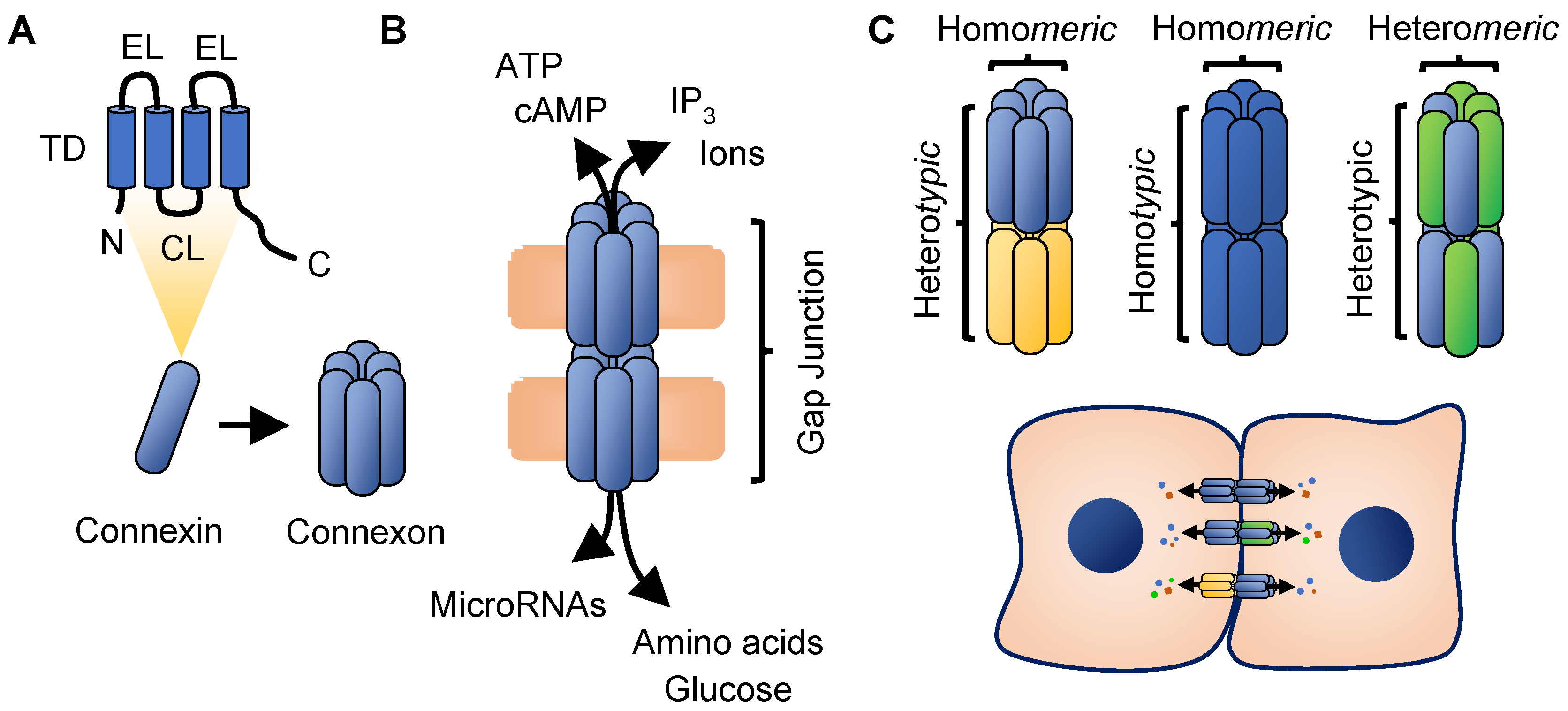
2. Connexin Biology
3. Connexins and Cancer
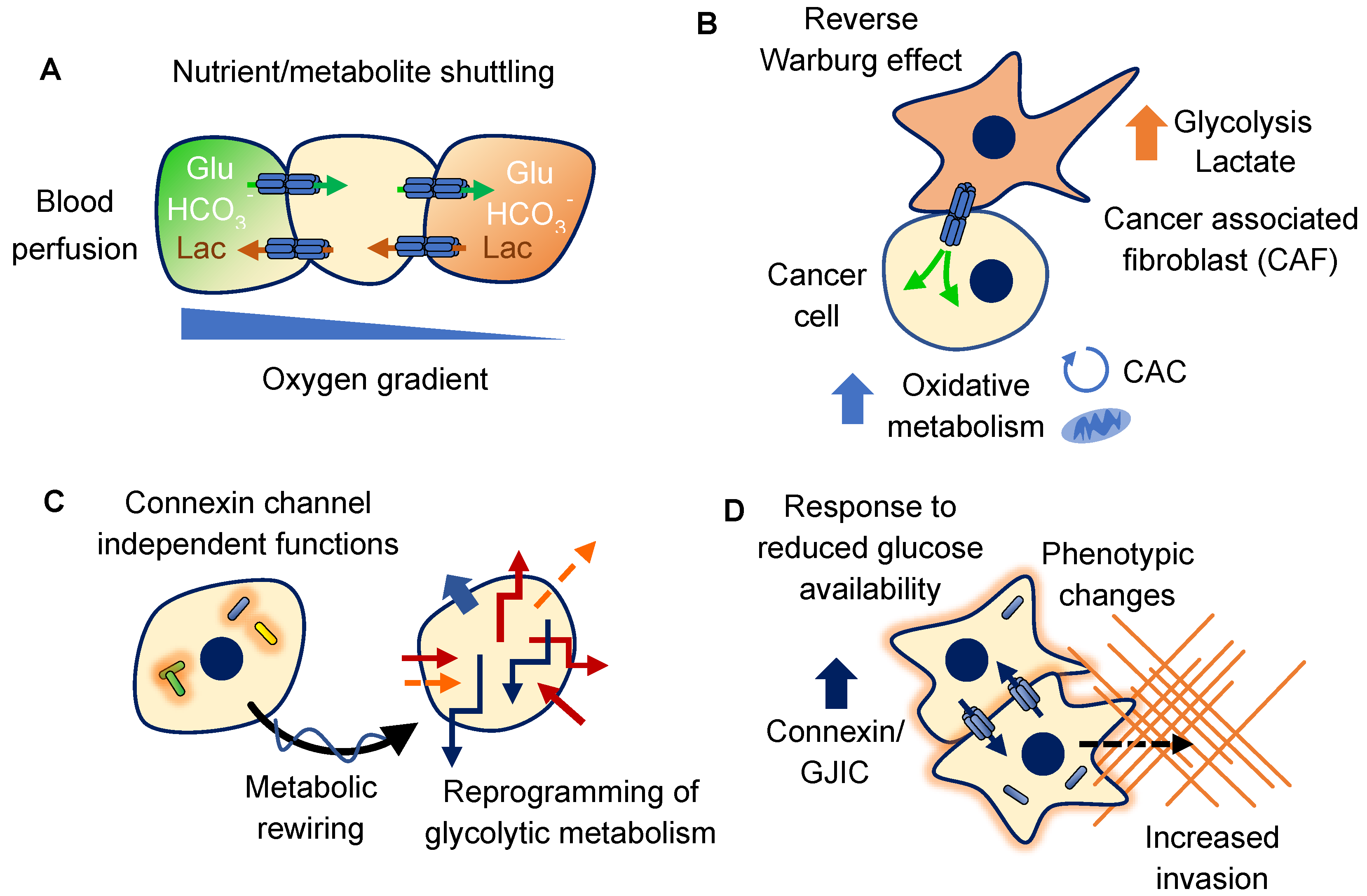
4. Connexins and Glucose
4.1. Metabolic Coupling of Glucose Metabolism
4.2. Metabolic Rewiring
4.3. Response to Glucose Availability
5. Emerging Connections
5.1. Glucose Responsive Degradation Pathways
5.2. Mitochondrial Connexins
5.3. Integration of Growth Pathways
5.4. Hypoxia and HIF1α
5.5. Transcriptional Regulation
5.6. Extracellular Vesicles and Tunneling Nanotubes
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Geissler, A.W.; Lorenz, S. On growth of cancer cells in media in which glucose is replaced by galactose. Hoppe Seyler Z. Physiol. Chem. 1967, 348, 1686–1687. [Google Scholar] [CrossRef] [PubMed]
- Grover-McKay, M.; Walsh, S.A.; Seftor, E.A.; Thomas, P.A.; Hendrix, M.J. Role for glucose transporter 1 protein in human breast cancer. Pathol. Oncol. Res. 1998, 4, 115–120. [Google Scholar] [CrossRef]
- Sakashita, M.; Aoyama, N.; Minami, R.; Maekawa, S.; Kuroda, K.; Shirasaka, D.; Ichihara, T.; Kuroda, Y.; Maeda, S.; Kasuga, M. Glut1 expression in T1 and T2 stage colorectal carcinomas: Its relationship to clinicopathological features. Eur. J. Cancer 2001, 37, 204–209. [Google Scholar] [CrossRef]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Lai, J.-H.; Jan, H.-J.; Liu, L.-W.; Lee, C.-C.; Wang, S.-G.; Hueng, D.-Y.; Cheng, Y.-Y.; Lee, H.-M.; Ma, H.-I. Nodal regulates energy metabolism in glioma cells by inducing expression of hypoxia-inducible factor 1α. Neuro Oncol. 2013, 15, 1330–1341. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364 Pt 1, 309–315. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Vital-Gonzalez, P.A.; Flores-Rodriguez, F.L.; Marin-Hernandez, A.; Ruiz-Azuara, L.; Moreno-Sanchez, R. Control of cellular proliferation by modulation of oxidative phosphorylation in human and rodent fast-growing tumor cells. Toxicol. Appl. Pharmacol. 2006, 215, 208–217. [Google Scholar] [CrossRef]
- Martin, M.; Beauvoit, B.; Voisin, P.J.; Canioni, P.; Guerin, B.; Rigoulet, M. Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. J. Bioenerg. Biomembr. 1998, 30, 565–578. [Google Scholar] [CrossRef]
- Pasdois, P.; Deveaud, C.; Voisin, P.; Bouchaud, V.; Rigoulet, M.; Beauvoit, B. Contribution of the phosphorylable complex I in the growth phase-dependent respiration of C6 glioma cells in vitro. J. Bioenerg. Biomembr. 2003, 35, 439–450. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Gallardo-Perez, J.C.; Aviles-Salas, A.; Marin-Hernandez, A.; Carreno-Fuentes, L.; Maldonado-Lagunas, V.; Moreno-Sanchez, R. Energy metabolism transition in multi-cellular human tumor spheroids. J. Cell. Physiol. 2008, 216, 189–197. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef]
- Smolkova, K.; Bellance, N.; Scandurra, F.; Genot, E.; Gnaiger, E.; Plecita-Hlavata, L.; Jezek, P.; Rossignol, R. Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J. Bioenerg. Biomembr. 2010, 42, 55–67. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3, e02935. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Orlicka-Plocka, M.; Gurda-Wozna, D.; Fedoruk-Wyszomirska, A.; Wyszko, E. Circumventing the Crabtree effect: Forcing oxidative phosphorylation (OXPHOS) via galactose medium increases sensitivity of HepG2 cells to the purine derivative kinetin riboside. Apoptosis 2020, 25, 835–852. [Google Scholar] [CrossRef]
- Redman, E.K.; Brookes, P.S.; Karcz, M.K. Role of p90RSK in regulating the Crabtree effect: Implications for cancer. Biochem. Soc. Trans. 2013, 41, 124–126. [Google Scholar] [CrossRef]
- Dell’Antone, P. Energy metabolism in cancer cells: How to explain the Warburg and Crabtree effects? Med. Hypotheses 2012, 79, 388–392. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Shi, T.; Li, D.; Li, G.; Zhang, Y.; Xu, K.; Lu, L. Modeling and Measurement of Correlation between Blood and Interstitial Glucose Changes. J. Diabetes Res. 2016, 2016, 4596316. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Vikse, C.; Trotter, M.J. Measurement of oxygen diffusion distance in tumor cubes using a fluorescent hypoxia probe. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 397–402. [Google Scholar] [CrossRef]
- Grimes, D.R.; Kannan, P.; Warren, D.R.; Markelc, B.; Bates, R.; Muschel, R.; Partridge, M. Correction to ‘Estimating oxygen distribution from vasculature in three-dimensional tumour tissue’. J. R. Soc. Interface 2016, 13, 20160070. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, P.; Yarani, R.; Dokaneheifard, S.; Mansouri, K. The emerging role of targeting cancer metabolism for cancer therapy. Tumour Biol. 2020, 42, 1010428320965284. [Google Scholar] [CrossRef]
- Goncalves, A.C.; Richiardone, E.; Jorge, J.; Polonia, B.; Xavier, C.P.R.; Salaroglio, I.C.; Riganti, C.; Vasconcelos, M.H.; Corbet, C.; Sarmento-Ribeiro, A.B. Impact of cancer metabolism on therapy resistance—Clinical implications. Drug Resist. Updates 2021, 59, 100797. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef]
- Puebla, C.; Retamal, M.A.; Acuna, R.; Saez, J.C. Regulation of Connexin-Based Channels by Fatty Acids. Front. Physiol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Loewenstein, W.R.; Socolar, S.J.; Higashino, S.; Kanno, Y.; Davidson, N. Intercellular Communication: Renal, Urinary Bladder, Sensory, and Salivary Gland Cells. Science 1965, 149, 295–298. [Google Scholar] [CrossRef]
- Goodenough, D.A. Bulk isolation of mouse hepatocyte gap junctions. Characterization of the principal protein, connexin. J. Cell Biol. 1974, 61, 557–563. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef]
- John, S.A.; Revel, J.P. Connexon integrity is maintained by non-covalent bonds: Intramolecular disulfide bonds link the extracellular domains in rat connexin-43. Biochem. Biophys. Res. Commun. 1991, 178, 1312–1318. [Google Scholar] [CrossRef]
- Majoul, I.V.; Onichtchouk, D.; Butkevich, E.; Wenzel, D.; Chailakhyan, L.M.; Duden, R. Limiting transport steps and novel interactions of Connexin-43 along the secretory pathway. Histochem. Cell Biol. 2009, 132, 263–280. [Google Scholar] [CrossRef]
- Thomas, T.; Jordan, K.; Simek, J.; Shao, Q.; Jedeszko, C.; Walton, P.; Laird, D.W. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 2005, 118 Pt 19, 4451–4462. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Puranam, K.L.; Laird, D.W.; Revel, J.P. Trapping an intermediate form of connexin43 in the Golgi. Exp. Cell Res. 1993, 206, 85–92. [Google Scholar] [CrossRef]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef]
- Laing, J.G.; Beyer, E.C. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 1995, 270, 26399–26403. [Google Scholar] [CrossRef]
- Leithe, E.; Rivedal, E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J. Cell Sci. 2004, 117 Pt 7, 1211–1220. [Google Scholar] [CrossRef]
- Kimura, K.; Nishida, T. Role of the ubiquitin-proteasome pathway in downregulation of the gap-junction protein Connexin43 by TNF-α in human corneal fibroblasts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1943–1947. [Google Scholar] [CrossRef]
- Mollerup, S.; Hofgaard, J.P.; Braunstein, T.H.; Kjenseth, A.; Leithe, E.; Rivedal, E.; Holstein-Rathlou, N.H.; Nielsen, M.S. Norepinephrine inhibits intercellular coupling in rat cardiomyocytes by ubiquitination of connexin43 gap junctions. Cell Commun. Adhes. 2011, 18, 57–65. [Google Scholar] [CrossRef]
- Thomas, M.A.; Zosso, N.; Scerri, I.; Demaurex, N.; Chanson, M.; Staub, O. A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J. Cell Sci. 2003, 116 Pt 11, 2213–2222. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef]
- Laing, J.G.; Tadros, P.N.; Westphale, E.M.; Beyer, E.C. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 1997, 236, 482–492. [Google Scholar] [CrossRef]
- Laing, J.G.; Tadros, P.N.; Green, K.; Saffitz, J.E.; Beyer, E.C. Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc. Res. 1998, 38, 711–718. [Google Scholar] [CrossRef]
- Qin, H.; Shao, Q.; Igdoura, S.A.; Alaoui-Jamali, M.A.; Laird, D.W. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 2003, 278, 30005–30014. [Google Scholar] [CrossRef]
- Fallon, R.F.; Goodenough, D.A. Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 1981, 90, 521–526. [Google Scholar] [CrossRef]
- Laird, D.W.; Puranam, K.L.; Revel, J.P. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991, 273 Pt 1, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Traub, O.; Look, J.; Paul, D.; Willecke, K. Cyclic adenosine monophosphate stimulates biosynthesis and phosphorylation of the 26 kDa gap junction protein in cultured mouse hepatocytes. Eur. J. Cell Biol. 1987, 43, 48–54. [Google Scholar] [PubMed]
- Laird, D.W. The life cycle of a connexin: Gap junction formation, removal, and degradation. J. Bioenerg. Biomembr. 1996, 28, 311–318. [Google Scholar] [CrossRef]
- Segretain, D.; Falk, M.M. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim. Biophys. Acta 2004, 1662, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Gap junction structure: Unraveled, but not fully revealed. F1000Research 2017, 6, 568. [Google Scholar] [CrossRef]
- Kim, N.K.; Santos-Miranda, A.; Chen, H.; Aoyama, H.; Bai, D. Heterotypic docking compatibility of human connexin37 with other vascular connexins. J. Mol. Cell. Cardiol. 2019, 127, 194–203. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef]
- John, S.A.; Kondo, R.; Wang, S.-Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef]
- Li, H.; Liu, T.-F.; Lazrak, A.; Peracchia, C.; Goldberg, G.S.; Lampe, P.D.; Johnson, R.G. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 1996, 134, 1019–1030. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Manolagas, S.C.; Bellido, T. Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 2002, 277, 8648–8657. [Google Scholar] [CrossRef]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Anderson, C.M.; Bergher, J.P.; Swanson, R.A. ATP-induced ATP release from astrocytes. J. Neurochem. 2004, 88, 246–256. [Google Scholar] [CrossRef]
- Anselmi, F.; Hernandez, V.H.; Crispino, G.; Seydel, A.; Ortolano, S.; Roper, S.D.; Kessaris, N.; Richardson, W.; Rickheit, G.; Filippov, M.A.; et al. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. USA 2008, 105, 18770–18775. [Google Scholar] [CrossRef]
- Braet, K.; Vandamme, W.; Martin, P.E.; Evans, W.H.; Leybaert, L. Photoliberating inositol-1,4,5-trisphosphate triggers ATP release that is blocked by the connexin mimetic peptide gap 26. Cell Calcium 2003, 33, 37–48. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef]
- Garcia, M.; Knight, M.M. Cyclic loading opens hemichannels to release ATP as part of a chondrocyte mechanotransduction pathway. J. Orthop. Res. 2010, 28, 510–515. [Google Scholar] [CrossRef]
- McEwan, T.B.; Sophocleous, R.A.; Cuthbertson, P.; Mansfield, K.J.; Sanderson-Smith, M.L.; Sluyter, R. Autocrine regulation of wound healing by ATP release and P2Y2 receptor activation. Life Sci. 2021, 283, 119850. [Google Scholar] [CrossRef]
- Murata, Y.; Yasuo, T.; Yoshida, R.; Obata, K.; Yanagawa, Y.; Margolskee, R.F.; Ninomiya, Y. Action potential-enhanced ATP release from taste cells through hemichannels. J. Neurophysiol. 2010, 104, 896–901. [Google Scholar] [CrossRef]
- Rackauskas, M.; Neverauskas, V.; Skeberdis, V.A. Diversity and properties of connexin gap junction channels. Medicina 2010, 46, 1. [Google Scholar] [CrossRef]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef]
- Ogawa, T.; Hayashi, T.; Kyoizumi, S.; Ito, T.; Trosko, J.E.; Yorioka, N. Up-regulation of gap junctional intercellular communication by hexamethylene bisacetamide in cultured human peritoneal mesothelial cells. Lab. Investig. 1999, 79, 1511–1520. [Google Scholar]
- Martin, D.; Tawadros, T.; Meylan, L.; Abderrahmani, A.; Condorelli, D.F.; Waeber, G.; Haefliger, J.A. Critical role of the transcriptional repressor neuron-restrictive silencer factor in the specific control of connexin36 in insulin-producing cell lines. J. Biol. Chem. 2003, 278, 53082–53089. [Google Scholar] [CrossRef]
- Vinken, M.; Henkens, T.; Vanhaecke, T.; Papeleu, P.; Geerts, A.; van Rossen, E.; Chipman, J.K.; Meda, P.; Rogiers, V. Trichostatin a enhances gap junctional intercellular communication in primary cultures of adult rat hepatocytes. Toxicol. Sci. 2006, 91, 484–492. [Google Scholar] [CrossRef]
- Piechocki, M.P.; Burk, R.D.; Ruch, R.J. Regulation of connexin32 and connexin43 gene expression by DNA methylation in rat liver cells. Carcinogenesis 1999, 20, 401–406. [Google Scholar] [CrossRef]
- Anderson, C.; Catoe, H.; Werner, R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Inose, H.; Ochi, H.; Kimura, A.; Fujita, K.; Xu, R.; Sato, S.; Iwasaki, M.; Sunamura, S.; Takeuchi, Y.; Fukumoto, S.; et al. A microRNA regulatory mechanism of osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 20794–20799. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, S.-S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 Isoform GJA1-20k Promotes Microtubule Dependent Mitochondrial Transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018, 3, e121900. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, I.; Anderson, C.; Werner, R.; Oltra, E. Redefining the structure of the mouse connexin43 gene: Selective promoter usage and alternative splicing mechanisms yield transcripts with different translational efficiencies. Nucleic Acids Res. 2004, 32, 4550–4562. [Google Scholar] [CrossRef][Green Version]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef]
- Princen, F.; Robe, P.; Gros, D.; Jarry-Guichard, T.; Gielen, J.; Merville, M.P.; Bours, V. Rat gap junction connexin-30 inhibits proliferation of glioma cell lines. Carcinogenesis 2001, 22, 507–513. [Google Scholar] [CrossRef]
- Bond, S.L.; Bechberger, J.F.; Khoo, N.K.; Naus, C.C. Transfection of C6 glioma cells with connexin32: The effects of expression of a nonendogenous gap junction protein. Cell Growth Differ. 1994, 5, 179–186. [Google Scholar]
- Kameritsch, P.; Pogoda, K.; Pohl, U. Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys. Acta 2012, 1818, 1993–2001. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Urban-Maldonado, M.; Iacobas, S.; Scemes, E.; Spray, D.C. Array analysis of gene expression in connexin-43 null astrocytes. Physiol. Genom. 2003, 15, 177–190. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Scemes, E.; Spray, D.C. Similar transcriptomic alterations in Cx43 knockdown and knockout astrocytes. Cell Commun. Adhes. 2008, 15, 195–206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Walker, D.L.; Vacha, S.J.; Kirby, M.L.; Lo, C.W. Connexin43 deficiency causes dysregulation of coronary vasculogenesis. Dev. Biol. 2005, 284, 479–498. [Google Scholar] [CrossRef]
- Stains, J.P.; Lecanda, F.; Screen, J.; Towler, D.A.; Civitelli, R. Gap junctional communication modulates gene transcription by altering the recruitment of Sp1 and Sp3 to connexin-response elements in osteoblast promoters. J. Biol. Chem. 2003, 278, 24377–24387. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Pelletier, D.B.; Ao, P.; Boynton, A.L. Connexin43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin-dependent kinases. Cell Growth Differ. 1995, 6, 681–690. [Google Scholar] [PubMed]
- Qiu, X.; Cheng, J.-C.; Klausen, C.; Chang, H.-M.; Fan, Q.; Leung, P.C. EGF-Induced Connexin43 Negatively Regulates Cell Proliferation in Human Ovarian Cancer. J. Cell. Physiol. 2016, 231, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, A.; Kalmykov, E.A.; Polusani, S.R.; Mathis, S.A.; Zucker, S.N.; Nicholson, B.J. Intercellular redistribution of cAMP underlies selective suppression of cancer cell growth by connexin26. PLoS ONE 2013, 8, e82335. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Kaneda, M.; Morita, I. The gap junction-independent tumor-suppressing effect of connexin 43. J. Biol. Chem. 2003, 278, 44852–44856. [Google Scholar] [CrossRef]
- Dang, X.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell. Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef]
- Van Campenhout, R.; Cooreman, A.; Leroy, K.; Rusiecka, O.M.; van Brantegem, P.; Annaert, P.; Muyldermans, S.; Devoogdt, N.; Cogliati, B.; Kwak, B.R.; et al. Non-canonical roles of connexins. Prog. Biophys. Mol. Biol. 2020, 153, 35–41. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J.D. Connexins in Cancer: Jekyll or Hyde? Biomolecules 2020, 10, 1654. [Google Scholar] [CrossRef]
- Yusubalieva, G.M.; Baklaushev, V.P.; Gurina, O.I.; Gulyaev, M.V.; Pirogov, Y.A.; Chekhonin, V.P. Antitumor effects of monoclonal antibodies to connexin 43 extracellular fragment in induced low-differentiated glioma. Bull. Exp. Biol. Med. 2012, 153, 163–169. [Google Scholar] [CrossRef]
- Yusubalieva, G.M.; Baklaushev, V.P.; Gurina, O.I.; Zorkina, Y.A.; Gubskii, I.L.; Kobyakov, G.L.; Golanov, A.V.; Goryainov, S.A.; Gorlachev, G.E.; Konovalov, A.N.; et al. Treatment of poorly differentiated glioma using a combination of monoclonal antibodies to extracellular connexin-43 fragment, temozolomide, and radiotherapy. Bull. Exp. Biol. Med. 2014, 157, 510–515. [Google Scholar] [CrossRef]
- Chekhonin, V.P.; Baklaushev, V.P.; Yusubalieva, G.M.; Belorusova, A.E.; Gulyaev, M.V.; Tsitrin, E.B.; Grinenko, N.F.; Gurina, O.I.; Pirogov, Y.A. Targeted delivery of liposomal nanocontainers to the peritumoral zone of glioma by means of monoclonal antibodies against GFAP and the extracellular loop of Cx43. Nanomedicine 2012, 8, 63–70. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef]
- Trosko, J.E.; Jone, C.; Chang, C.C. Oncogenes, inhibited intercellular communication and tumor promotion. Cell. Interact. Environ. Tumor Promot. 1983, 14, 101–113. [Google Scholar]
- Mehta, P.P.; Hotz-Wagenblatt, A.; Rose, B.; Shalloway, D.; Loewenstein, W.R. Incorporation of the gene for a cell-cell channel protein into transformed cells leads to normalization of growth. J. Membr. Biol. 1991, 124, 207–225. [Google Scholar] [CrossRef]
- King, T.J.; Lampe, P.D. The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res. 2004, 64, 7191–7196. [Google Scholar] [CrossRef]
- Trosko, J.E.; Chang, C.-C.; Upham, B.L.; Tai, M.-H. Ignored hallmarks of carcinogenesis: Stem cells and cell-cell communication. Ann. N. Y. Acad. Sci. 2004, 1028, 192–201. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, H.; McLachlan, E.; Veitch, G.I.; Laird, D.W. Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res. 2005, 65, 2705–2711. [Google Scholar] [CrossRef]
- Wang, W.-K.; Chen, M.-C.; Leong, H.-F.; Kuo, Y.-L.; Kuo, C.-Y.; Lee, C.-H. Connexin 43 suppresses tumor angiogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1α. Int. J. Mol. Sci. 2014, 16, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Caveney, S.; Kidder, G.M.; Naus, C.C. Transfection of C6 glioma cells with connexin 43 cDNA: Analysis of expression, intercellular coupling, and cell proliferation. Proc. Natl. Acad. Sci. USA 1991, 88, 1883–1887. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Elisevich, K.; Zhu, D.; Belliveau, D.J.; Del Maestro, R.F. In vivo growth of C6 glioma cells transfected with connexin43 cDNA. Cancer Res. 1992, 52, 4208–4213. [Google Scholar] [PubMed]
- Eghbali, B.; Kessler, J.A.; Reid, L.M.; Roy, C.; Spray, D.C. Involvement of gap junctions in tumorigenesis: Transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 10701–10705. [Google Scholar] [CrossRef]
- Trosko, J.E. On the potential origin and characteristics of cancer stem cells. Carcinogenesis 2021, 42, 905–912. [Google Scholar] [CrossRef]
- Liang, Q.-L.; Wang, B.-R.; Chen, G.-Q.; Li, G.-H.; Xu, Y.-Y. Clinical significance of vascular endothelial growth factor and connexin43 for predicting pancreatic cancer clinicopathologic parameters. Med. Oncol. 2010, 27, 1164–1170. [Google Scholar] [CrossRef]
- Teleki, I.; Szasz, A.M.; Maros, M.E.; Gyorffy, B.; Kulka, J.; Meggyeshazi, N.; Kiszner, G.; Balla, P.; Samu, A.; Krenacs, T. Correlations of differentially expressed gap junction connexins Cx26, Cx30, Cx32, Cx43 and Cx46 with breast cancer progression and prognosis. PLoS ONE 2014, 9, e112541. [Google Scholar] [CrossRef]
- Liu, X.; Furuya, T.; Li, D.; Xu, J.; Cao, X.; Li, Q.; Xu, J.; Xu, Z.; Sasaki, K.; Liu, X. Connexin 26 expression correlates with less aggressive phenotype of intestinal type-gastric carcinomas. Int. J. Mol. Med. 2010, 25, 709–716. [Google Scholar] [CrossRef]
- Danos, K.; Brauswetter, D.; Birtalan, E.; Pato, A.; Bencsik, G.; Krenacs, T.; Petak, I.; Tamas, L. The Potential Prognostic Value of Connexin 43 Expression in Head and Neck Squamous Cell Carcinomas. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 476–481. [Google Scholar] [CrossRef]
- Du, G.; Yang, Y.; Zhang, Y.; Sun, T.; Liu, W.; Wang, Y.; Li, J.; Zhang, H. Thrombocytosis and immunohistochemical expression of connexin 43 at diagnosis predict survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Cancer Chemother. Pharmacol. 2013, 71, 893–904. [Google Scholar] [CrossRef]
- Sirnes, S.; Bruun, J.; Kolberg, M.; Kjenseth, A.; Lind, G.E.; Svindland, A.; Brech, A.; Nesbakken, A.; Lothe, R.A.; Leithe, E.; et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int. J. Cancer 2012, 131, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Maeda, K.; Noda, E.; Inoue, T.; Fukunaga, S.; Nagahara, H.; Hirakawa, K. Clinical significance of the expression of connexin26 in colorectal cancer. J. Exp. Clin. Cancer Res. 2010, 29, 79. [Google Scholar] [CrossRef] [PubMed]
- Brockmeyer, P.; Jung, K.; Perske, C.; Schliephake, H.; Hemmerlein, B. Membrane connexin 43 acts as an independent prognostic marker in oral squamous cell carcinoma. Int. J. Oncol. 2014, 45, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kimura, M.; Ishiguro, H.; Mizoguchi, K.; Takeyama, H. Connexin 43 expression is associated with poor survival in patients with esophageal squamous cell carcinoma. Mol. Clin. Oncol. 2016, 4, 989–993. [Google Scholar] [CrossRef]
- Poyet, C.; Buser, L.; Roudnicky, F.; Detmar, M.; Hermanns, T.; Mannhard, D.; Hohn, A.; Ruschoff, J.; Zhong, Q.; Sulser, T.; et al. Connexin 43 expression predicts poor progression-free survival in patients with non-muscle invasive urothelial bladder cancer. J. Clin. Pathol. 2015, 68, 819–824. [Google Scholar] [CrossRef]
- Teleki, I.; Krenacs, T.; Szasz, M.A.; Kulka, J.; Wichmann, B.; Leo, C.; Papassotiropoulos, B.; Riemenschnitter, C.; Moch, H.; Varga, Z. The potential prognostic value of connexin 26 and 46 expression in neoadjuvant-treated breast cancer. BMC Cancer 2013, 13, 50. [Google Scholar] [CrossRef]
- Naoi, Y.; Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Arai, T.; Tamaki, Y.; Noguchi, S. Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2007, 106, 11–17. [Google Scholar] [CrossRef]
- Naoi, Y.; Miyoshi, Y.; Taguchi, T.; Kim, S.J.; Arai, T.; Maruyama, N.; Tamaki, Y.; Noguchi, S. Connexin26 expression is associated with aggressive phenotype in human papillary and follicular thyroid cancers. Cancer Lett. 2008, 262, 248–256. [Google Scholar] [CrossRef]
- Ito, A.; Koma, Y.; Uchino, K.; Okada, T.; Ohbayashi, C.; Tsubota, N.; Okada, M. Increased expression of connexin 26 in the invasive component of lung squamous cell carcinoma: Significant correlation with poor prognosis. Cancer Lett. 2006, 234, 239–248. [Google Scholar] [CrossRef]
- Inose, T.; Kato, H.; Kimura, H.; Faried, A.; Tanaka, N.; Sakai, M.; Sano, A.; Sohda, M.; Nakajima, M.; Fukai, Y.; et al. Correlation between connexin 26 expression and poor prognosis of esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009, 16, 1704–1710. [Google Scholar] [CrossRef]
- Bates, D.C.; Sin, W.C.; Aftab, Q.; Naus, C.C. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 2007, 55, 1554–1564. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kumar, A.; Tripathi, R.P.; Chandna, S. Connexin-43 regulates p38-mediated cell migration and invasion induced selectively in tumour cells by low doses of γ-radiation in an ERK-1/2-independent manner. Carcinogenesis 2014, 35, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- Zibara, K.; Awada, Z.; Dib, L.; El-Saghir, J.; Al-Ghadban, S.; Ibrik, A.; El-Zein, N.; El-Sabban, M. Anti-angiogenesis therapy and gap junction inhibition reduce MDA-MB-231 breast cancer cell invasion and metastasis in vitro and in vivo. Sci. Rep. 2015, 5, 12598. [Google Scholar] [CrossRef]
- Lamiche, C.; Clarhaut, J.; Strale, P.O.; Crespin, S.; Pedretti, N.; Bernard, F.X.; Naus, C.C.; Chen, V.C.; Foster, L.J.; Defamie, N.; et al. The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin. Exp. Metastasis 2012, 29, 111–122. [Google Scholar] [CrossRef]
- Ogawa, K.; Pitchakarn, P.; Suzuki, S.; Chewonarin, T.; Tang, M.; Takahashi, S.; Naiki-Ito, A.; Sato, S.; Takahashi, S.; Asamoto, M.; et al. Silencing of connexin 43 suppresses invasion, migration and lung metastasis of rat hepatocellular carcinoma cells. Cancer Sci. 2012, 103, 860–867. [Google Scholar] [CrossRef]
- Tang, B.; Peng, Z.-H.; Yu, P.-W.; Yu, G.; Qian, F.; Zeng, D.-Z.; Zhao, Y.-L.; Shi, Y.; Hao, Y.-X.; Luo, H.-X. Aberrant expression of Cx43 is associated with the peritoneal metastasis of gastric cancer and Cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PLoS ONE 2013, 8, e74527. [Google Scholar] [CrossRef]
- El-Sabban, M.E.; Pauli, B.U. Adhesion-mediated gap junctional communication between lung-metastatatic cancer cells and endothelium. Invasion Metastasis 1994, 14, 164–176. [Google Scholar]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Tabernero, A.; Giaume, C.; Medina, J.M. Endothelin-1 regulates glucose utilization in cultured astrocytes by controlling intercellular communication through gap junctions. Glia 1996, 16, 187–195. [Google Scholar] [CrossRef]
- Giaume, C.; Tabernero, A.; Medina, J.M. Metabolic trafficking through astrocytic gap junctions. Glia 1997, 21, 114–123. [Google Scholar] [CrossRef]
- Matsunami, T.; Suzuki, T.; Hisa, Y.; Takata, K.; Takamatsu, T.; Oyamada, M. Gap junctions mediate glucose transport between GLUT1-positive and -negative cells in the spiral limbus of the rat cochlea. Cell Commun. Adhes. 2006, 13, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Matsunami, T.; Hisa, Y.; Takata, K.; Takamatsu, T.; Oyamada, M. Roles of gap junctions in glucose transport from glucose transporter 1-positive to -negative cells in the lateral wall of the rat cochlea. Histochem. Cell Biol. 2009, 131, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Li, T.; Yi, C.; Huang, N.; Koulakoff, A.; Weng, C.; Li, C.; Zhao, C.-J.; Giaume, C.; Xiao, L. Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J. Cell Sci. 2016, 129, 1902–1914. [Google Scholar] [CrossRef]
- Allard, C.; Carneiro, L.; Grall, S.; Cline, B.H.; Fioramonti, X.; Chretien, C.; Baba-Aissa, F.; Giaume, C.; Penicaud, L.; Leloup, C. Hypothalamic astroglial connexins are required for brain glucose sensing-induced insulin secretion. J. Cereb. Blood Flow Metab. 2014, 34, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Dovmark, T.H.; Saccomano, M.; Hulikova, A.; Alves, F.; Swietach, P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36, 4538–4550. [Google Scholar] [CrossRef]
- Dovmark, T.H.; Hulikova, A.; Niederer, S.A.; Vaughan-Jones, R.D.; Swietach, P. Normoxic cells remotely regulate the acid-base balance of cells at the hypoxic core of connexin-coupled tumor growths. FASEB J. 2018, 32, 83–96. [Google Scholar] [CrossRef]
- Gong, K.; Hong, Q.; Wu, H.; Wang, F.; Zhong, L.; Shen, L.; Xu, P.; Zhang, W.; Cao, H.; Zhan, Y.-Y.; et al. Gap junctions mediate glucose transfer to promote colon cancer growth in three-dimensional spheroid culture. Cancer Lett. 2022, 531, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Whitaker-Menezes, D.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Witkiewicz, A.K.; Heiden, M.G.V.; Migneco, G.; Chiavarina, B.; et al. The reverse Warburg effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010, 9, 1960–1971. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Luo, Y.; Mao, N.; Huang, G.; Teng, C.; Wang, H.; Wu, J.; Liao, X.; Yang, J. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell. Physiol. Biochem. 2018, 51, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Al Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS ONE 2011, 6, e22567. [Google Scholar] [CrossRef] [PubMed]
- Azarnia, R.; Reddy, S.; Kmiecik, T.E.; Shalloway, D.; Loewenstein, W.R. The cellular src gene product regulates junctional cell-to-cell communication. Science 1988, 239, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Hengeveld, T.; Postma, F.R.; Moolenaar, W.H. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J. Biol. Chem. 2001, 276, 8544–8549. [Google Scholar] [CrossRef]
- Toyofuku, T.; Akamatsu, Y.; Zhang, H.; Kuzuya, T.; Tada, M.; Hori, M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J. Biol. Chem. 2001, 276, 1780–1788. [Google Scholar] [CrossRef]
- Herrero-Gonzalez, S.; Gangoso, E.; Giaume, C.; Naus, C.C.; Medina, J.M.; Tabernero, A. Connexin43 inhibits the oncogenic activity of c-Src in C6 glioma cells. Oncogene 2010, 29, 5712–5723. [Google Scholar] [CrossRef]
- Gonzalez-Sanchez, A.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Herrero-Gonzalez, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar] [CrossRef]
- Caner, A.; Asik, E.; Ozpolat, B. SRC Signaling in Cancer and Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 57–71. [Google Scholar]
- Fizazi, K. The role of Src in prostate cancer. Ann. Oncol. 2007, 18, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S. Targeting Src in breast cancer. Ann. Oncol. 2008, 19, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhang, J.; Zhou, L.; Wen, S.; Tang, H.-Y.; Jiang, B.; Zhang, F.; Suleman, M.; Sun, D.; Chen, A.; et al. c-Src Promotes Tumorigenesis and Tumor Progression by Activating PFKFB3. Cell Rep. 2020, 30, 4235–4249.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Jiang, B.; Huang, L.; Ji, Z.; Li, X.; Zhou, H.; Han, A.; Chen, A.; Wu, Y.; et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat. Commun. 2017, 8, 13732. [Google Scholar] [CrossRef] [PubMed]
- Mayoral-Varo, V.; Calcabrini, A.; Sanchez-Bailon, M.P.; Martinez-Costa, O.H.; Gonzalez-Paramos, C.; Ciordia, S.; Hardisson, D.; Aragon, J.J.; Fernandez-Moreno, M.A.; Martin-Perez, J. c-Src functionality controls self-renewal and glucose metabolism in MCF7 breast cancer stem cells. PLoS ONE 2020, 15, e0235850. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Oh, S.; Shin, I. Ablation of CD44 induces glycolysis-to-oxidative phosphorylation transition via modulation of the c-Src-Akt-LKB1-AMPKα pathway. Biochem. J. 2016, 473, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Koc, H.; Koc, E.C. c-Src kinase impairs the expression of mitochondrial OXPHOS complexes in liver cancer. Cell. Signal. 2020, 72, 109651. [Google Scholar] [CrossRef]
- Guedouari, H.; Savoie, M.C.; Jean, S.; Djeungoue-Petga, M.A.; Pichaud, N.; Hebert-Chatelain, E. Multi-omics Reveal that c-Src Modulates the Mitochondrial Phosphotyrosine Proteome and Metabolism According to Nutrient Availability. Cell Physiol. Biochem. 2020, 54, 517–537. [Google Scholar] [PubMed]
- Pelaz, S.G.; Jaraiz-Rodriguez, M.; Alvarez-Vazquez, A.; Talaveron, R.; Garcia-Vicente, L.; Flores-Hernandez, R.; de Cedron, M.G.; Tabernero, M.; de Molina, A.R.; Lillo, C.; et al. Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266-283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef]
- Sanchez-Alvarez, R.; Tabernero, A.; Medina, J.M. The increase in gap junctional communication decreases the rate of glucose uptake in C6 glioma cells by releasing hexokinase from mitochondria. Brain Res. 2005, 1039, 189–198. [Google Scholar] [CrossRef]
- Jothi, J.; Janardhanam, V.A.; Rama, K. Connexin 30 mediated rewiring of glucose metabolism in rat C6 xenograft and grades of glioma. Mol. Cell. Biochem. 2020, 470, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yu, H.; Li, X.; Jin, C.; Zhao, Y.; Xu, S.; Sheng, X. P38 MAPK/miR-1 are involved in the protective effect of EGCG in high glucose-induced Cx43 downregulation in neonatal rat cardiomyocytes. Cell Biol. Int. 2016, 40, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lan, T.; Chang, X.; Huang, K.; Huang, J.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H. Connexin43 mediates NF-κB signalling activation induced by high glucose in GMCs: Involvement of c-Src. Cell Commun. Signal. 2013, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, Y.; Fan, Y.; Wang, M.; Xu, S.; Fu, G. Epigallocatechin-3 gallate, a green tea catechin, attenuated the downregulation of the cardiac gap junction induced by high glucose in neonatal rat cardiomyocytes. Cell. Physiol. Biochem. 2010, 26, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Inoguchi, T.; Umeda, F.; Ueda, F.; Nawata, H. High glucose induces alteration of gap junction permeability and phosphorylation of connexin-43 in cultured aortic smooth muscle cells. Diabetes 1998, 47, 931–936. [Google Scholar] [CrossRef]
- Tien, T.; Barrette, K.F.; Chronopoulos, A.; Roy, S. Effects of high glucose-induced Cx43 downregulation on occludin and ZO-1 expression and tight junction barrier function in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6518–6525. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Yu, H.Y.; Imamura, M.; Kakimoto, M.; Kuroki, T.; Maruyama, T.; Nawata, H. Altered gap junction activity in cardiovascular tissues of diabetes. Med. Electron Microsc. 2001, 34, 86–91. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Ball, K.K.; Cruz, N.F.; Dienel, G.A. Hyperglycaemia and diabetes impair gap junctional communication among astrocytes. ASN Neuro 2010, 2, e00030. [Google Scholar] [CrossRef]
- Wang, G.-Y.; Bi, Y.-G.; Liu, X.-D.; Zhao, Y.; Han, J.-F.; Wei, M.; Zhang, Q.-Y. Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and Connexin43 downregulation in H9c2 cells. Int. J. Med. Sci. 2017, 14, 698–704. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Wu, D.; Liu, W.; Wang, J.; Feng, Z.; Cai, G.; Fu, B.; Hong, Q.; Du, J. Downregulation of connexin 43 expression by high glucose induces senescence in glomerular mesangial cells. J. Am. Soc. Nephrol. 2006, 17, 1532–1542. [Google Scholar] [CrossRef]
- Xie, X.; Chen, C.; Huang, K.; Wang, S.; Hao, J.; Huang, J.; Huang, H. RhoA/rho kinase signaling reduces connexin43 expression in high glucose-treated glomerular mesangial cells with zonula occludens-1 involvement. Exp. Cell Res. 2014, 327, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Haimovici, R.; Kao, R.; Li, A.-F.; Roy, S. Downregulation of connexin 43 expression by high glucose reduces gap junction activity in microvascular endothelial cells. Diabetes 2002, 51, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Manasson, J.; Tien, T.; Moore, C.; Kumar, N.M.; Roy, S. High glucose-induced downregulation of connexin 30.2 promotes retinal vascular lesions: Implications for diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2361–2366. [Google Scholar] [CrossRef]
- Yu, H.; Yang, J.; Zhou, X.; Xiao, Q.; Lu, Y.; Xia, L. High glucose induces dysfunction of airway epithelial barrier through down-regulation of connexin 43. Exp. Cell Res. 2016, 342, 11–19. [Google Scholar] [PubMed]
- Li, A.-F.; Roy, S. High glucose-induced downregulation of connexin 43 expression promotes apoptosis in microvascular endothelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Muto, T.; Tien, T.; Kim, D.; Sarthy, V.P.; Roy, S. High glucose alters Cx43 expression and gap junction intercellular communication in retinal Muller cells: Promotes Muller cell and pericyte apoptosis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4327–4337. [Google Scholar] [CrossRef]
- Li, A.-F.; Sato, T.; Haimovici, R.; Okamoto, T.; Roy, S. High glucose alters connexin 43 expression and gap junction intercellular communication activity in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5376–5382. [Google Scholar] [CrossRef]
- Fernandes, R.; Girao, H.; Pereira, P. High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J. Biol. Chem. 2004, 279, 27219–27224. [Google Scholar] [CrossRef]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Novel role of mitochondrial matrix metalloproteinase-2 in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3832–3841. [Google Scholar] [CrossRef]
- Garvin, J.; Semenikhina, M.; Liu, Q.; Rarick, K.; Isaeva, E.; Levchenko, V.; Staruschenko, A.; Palygin, O.; Harder, D.; Cohen, S. Astrocytic responses to high glucose impair barrier formation in cerebral microvessel endothelial cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R571–R580. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, J.A.; Rohner-Jeanrenaud, F.; Caille, D.; Charollais, A.; Meda, P.; Allagnat, F. Hyperglycemia downregulates Connexin36 in pancreatic islets via the upregulation of ICER-1/ICER-1γ. J. Mol. Endocrinol. 2013, 51, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Platoshyn, O.; Suarez, J.; Yuan, J.-X.; Dillmann, W.H. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am. J. Physiol. Cell Physiol. 2008, 295, C221–C230. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.R.; Carvalho, C.P.; Furtado, A.G.; Barbosa, H.C.; Boschero, A.C.; Collares-Buzato, C.B. Co-expression and regulation of connexins 36 and 43 in cultured neonatal rat pancreatic islets. Can. J. Physiol. Pharmacol. 2005, 83, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.K.; Harik, L.; Gandhi, G.K.; Cruz, N.F.; Dienel, G.A. Reduced gap junctional communication among astrocytes in experimental diabetes: Contributions of altered connexin protein levels and oxidative-nitrosative modifications. J. Neurosci. Res. 2011, 89, 2052–2067. [Google Scholar] [CrossRef]
- Hills, C.E.; Bland, R.; Bennett, J.; Ronco, P.M.; Squires, P.E. TGF-β1 mediates glucose-evoked up-regulation of connexin-43 cell-to-cell communication in HCD-cells. Cell. Physiol. Biochem. 2009, 24, 177–186. [Google Scholar] [CrossRef]
- Hills, C.E.; Bland, R.; Wheelans, D.C.; Bennett, J.; Ronco, P.M.; Squires, P.E. Glucose-evoked alterations in connexin43-mediated cell-to-cell communication in human collecting duct: A possible role in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2006, 291, F1045–F1051. [Google Scholar] [CrossRef][Green Version]
- Mugisho, O.O.; Green, C.R.; Zhang, J.; Binz, N.; Acosta, M.L.; Rakoczy, E.; Rupenthal, I.D. Immunohistochemical Characterization of Connexin43 Expression in a Mouse Model of Diabetic Retinopathy and in Human Donor Retinas. Int. J. Mol. Sci. 2017, 18, 2567. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Valdebenito, S.; Gorska, A.M.; Martinez, A.D.; Bitran, M.; Saez, J.C. Gap junctions coordinate the propagation of glycogenolysis induced by norepinephrine in the pineal gland. J. Neurochem. 2019, 151, 558–569. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-β1 via Nrf2/ARE pathway in glomerular mesangial cells. Free. Radic. Biol. Med. 2017, 102, 77–86. [Google Scholar] [CrossRef]
- Kim, D.; Mouritzen, U.; Larsen, B.D.; Roy, S. Inhibition of Cx43 gap junction uncoupling prevents high glucose-induced apoptosis and reduces excess cell monolayer permeability in retinal vascular endothelial cells. Exp. Eye Res. 2018, 173, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Wang, C.M.; Thrasivoulou, C.; Sutcliffe, J.E.; Gilmartin, D.J.; Tsui, J.; Serena, T.E.; Phillips, A.R.; et al. Overexpression of the gap junction protein Cx43 as found in diabetic foot ulcers can retard fibroblast migration. Cell Biol. Int. 2012, 36, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, X.; Cai, G.-Y.; Lv, Y.; Zhuo, L.; Gao, J.-J.; Cui, S.-Y.; Feng, Z.; Fu, B.; Chen, X.-M. High glucose-induced hypertrophy of mesangial cells is reversed by connexin43 overexpression via PTEN/Akt/mTOR signaling. Nephrol. Dial. Transplant. 2012, 27, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, R.; Chu, Z.; Le, B.; Zeng, H.; Zhang, X.; Wu, Q.; Zhu, G.; Chen, Y.; Liu, Y.; et al. High glucose stimulates proliferative capacity of liver cancer cells possibly via O-GlcNAcylation-dependent transcriptional regulation of GJC1. J. Cell. Physiol. 2018, 234, 606–618. [Google Scholar] [CrossRef]
- Wojciechowska, J.; Krajewski, W.; Bolanowski, M.; Krecicki, T.; Zatonski, T. Diabetes and Cancer: A Review of Current Knowledge. Exp. Clin. Endocrinol. Diabetes 2016, 124, 263–275. [Google Scholar] [CrossRef]
- Han, H.; Zhang, T.; Jin, Z.; Guo, H.; Wei, X.; Liu, Y.; Chen, Q.; He, J. Blood glucose concentration and risk of liver cancer: Systematic review and meta-analysis of prospective studies. Oncotarget 2017, 8, 50164–50173. [Google Scholar] [CrossRef]
- Mori, Y.; Olaru, A.V.; Cheng, Y.; Agarwal, R.; Yang, J.; Luvsanjav, D.; Yu, W.; Selaru, F.M.; Hutfless, S.; Lazarev, M.; et al. Novel candidate colorectal cancer biomarkers identified by methylation microarray-based scanning. Endocr. Relat. Cancer 2011, 18, 465–478. [Google Scholar] [CrossRef]
- Jin, B.; Wang, W.; Du, G.; Huang, G.Z.; Han, L.T.; Tang, Z.Y.; Fan, D.G.; Li, J.; Zhang, S.Z. Identifying hub genes and dysregulated pathways in hepatocellular carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 592–601. [Google Scholar]
- Ma, Z.; Vosseller, K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J. Biol. Chem. 2014, 289, 34457–34465. [Google Scholar] [CrossRef]
- Parker, M.P.; Peterson, K.R.; Slawson, C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers 2021, 13, 1666. [Google Scholar] [CrossRef]
- Hanover, J.A.; Chen, W.; Bond, M.R. O-GlcNAc in cancer: An Oncometabolism-fueled vicious cycle. J. Bioenerg. Biomembr. 2018, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Dai, A.; Han, Y.; Youssef, K.D.; Wang, W.; Donthamsetty, R.; Scott, B.T.; Wang, H.; Dillmann, W.H. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am. J. Physiol. Cell Physiol. 2015, 309, C593–C599. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Chen, C.-C.; Tsai, W.-C.; Lin, H.-T.; Shiao, Y.-L.; Sheu, S.-H.; Wu, B.-N.; Chen, C.-H.; Lai, W.-T. Very-Low-Density Lipoprotein of Metabolic Syndrome Modulates Gap Junctions and Slows Cardiac Conduction. Sci. Rep. 2017, 7, 12050. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Miceli, A.M.; Chaudhry, M.M.; Kaunitz, C.S.; Jai, M.A.; Pancho, R.N.; Lazzar, A.; Taylor, B.S.; Bodempudi, V.; Jain, P.P.; et al. Glucose-limiting conditions induce an invasive population of MDA-MB-231 breast cancer cells with increased connexin 43 expression and membrane localization. J. Cell Commun. Signal. 2021, 15, 223–236. [Google Scholar] [CrossRef]
- Wang, G.; Bi, Y.; Liu, X.; Wei, M.; Zhang, Q. Upregulation of connexin43 by glucose deprivation in H9c2 cells via the extracellular signalregulated kinase/mitogenactivated protein kinase signaling pathway. Mol. Med. Rep. 2018, 17, 729–734. [Google Scholar]
- VanSlyke, J.K.; Deschenes, S.M.; Musil, L.S. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell 2000, 11, 1933–1946. [Google Scholar] [CrossRef][Green Version]
- VanSlyke, J.K.; Musil, L.S. Dislocation and degradation from the ER are regulated by cytosolic stress. J. Cell Biol. 2002, 157, 381–394. [Google Scholar] [CrossRef]
- Li, X.; Su, V.; Kurata, W.E.; Jin, C.; Lau, A.F. A novel connexin43-interacting protein, CIP75, which belongs to the UbL-UBA protein family, regulates the turnover of connexin43. J. Biol. Chem. 2008, 283, 5748–5759. [Google Scholar] [CrossRef]
- Su, V.; Nakagawa, R.; Koval, M.; Lau, A.F. Ubiquitin-independent proteasomal degradation of endoplasmic reticulum-localized connexin43 mediated by CIP75. J. Biol. Chem. 2010, 285, 40979–40990. [Google Scholar] [CrossRef]
- Su, V.; Hoang, C.; Geerts, D.; Lau, A.F. CIP75 (connexin43-interacting protein of 75 kDa) mediates the endoplasmic reticulum dislocation of connexin43. Biochem. J. 2014, 458, 57–67. [Google Scholar] [CrossRef]
- Das, S.; Smith, T.D.; Sarma, J.D.; Ritzenthaler, J.D.; Maza, J.; Kaplan, B.E.; Cunningham, L.A.; Suaud, L.; Hubbard, M.J.; Rubenstein, R.C.; et al. ERp29 restricts Connexin43 oligomerization in the endoplasmic reticulum. Mol. Biol. Cell 2009, 20, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchian, S.; Fang, C.; Hellman, U.; Ingelman-Sundberg, M. A stress-inducible rat liver endoplasmic reticulum protein, ERp29. Eur. J. Biochem. 1998, 251, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, T.; He, Y.; Zhou, S.; Wang, Y.; Zhang, K.; Yu, P. High glucose regulates ERp29 in hepatocellular carcinoma by LncRNA MEG3-miRNA 483-3p pathway. Life Sci. 2019, 232, 116602. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Maes, M.; Cavill, R.; Valkenborg, D.; Ellis, J.K.; Decrock, E.; Leybaert, L.; Staes, A.; Gevaert, K.; Oliveira, A.G.; et al. Proteomic and metabolomic responses to connexin43 silencing in primary hepatocyte cultures. Arch. Toxicol. 2013, 87, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef] [PubMed]
- Iyyathurai, J.; Decuypere, J.P.; Leybaert, L.; D’Hondt, C.; Bultynck, G. Connexins: Substrates and regulators of autophagy. BMC Cell Biol. 2016, 17 (Suppl. 1), 20. [Google Scholar] [CrossRef]
- Lichtenstein, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M. Autophagy: A pathway that contributes to connexin degradation. J. Cell Sci. 2011, 124 Pt 6, 910–920. [Google Scholar] [CrossRef]
- Chen, W.; Guo, Y.; Yang, W.; Zheng, P.; Zeng, J.; Tong, W. Involvement of autophagy in connexin 40 reduction in the late phase of traumatic brain injury in rats. Brain Res. Bull. 2017, 131, 100–106. [Google Scholar]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Goubaeva, F.; Mikami, M.; Giardina, S.; Ding, B.; Abe, J.; Yang, J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar]
- Sankaramoorthy, A.; Roy, S. High Glucose-Induced Apoptosis Is Linked to Mitochondrial Connexin 43 Level in RRECs: Implications for Diabetic Retinopathy. Cells 2021, 10, 3102. [Google Scholar] [CrossRef]
- Wei, X.; Chang, A.C.H.; Chang, H.; Xu, S.; Xue, Y.; Zhang, Y.; Lei, M.; Chang, A.C.Y.; Zhang, Q. Hypoglycemia-Exacerbated Mitochondrial Connexin 43 Accumulation Aggravates Cardiac Dysfunction in Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 800185. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Pridham, K.J.; Shah, F.; Hutchings, K.R.; Sheng, K.L.; Guo, S.; Liu, M.; Kanabur, P.; Lamouille, S.; Lewis, G.; Morales, M.; et al. Connexin 43 confers chemoresistance through activating PI3K. Oncogenesis 2022, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, M.A.; Gu, S.; Hua, R.; Jiang, J.X. Mechanotransduction via the coordinated actions of integrins, PI3K signaling and Connexin hemichannels. Bone Res. 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-P.; Zhou, Y.; Hou, L.-X.; Zhu, X.-X.; Yi, W.; Yang, S.-M.; Lin, T.-Y.; Huang, J.-L.; Zhang, B.; Yin, X.-X. Cx43 deficiency confers EMT-mediated tamoxifen resistance to breast cancer via c-Src/PI3K/Akt pathway. Int. J. Biol. Sci. 2021, 17, 2380–2398. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef]
- Shen, C.; Kim, M.R.; Noh, J.M.; Kim, S.J.; Ka, S.-O.; Kim, J.H.; Park, B.-H.; Park, J.H. Glucocorticoid Suppresses Connexin 43 Expression by Inhibiting the Akt/mTOR Signaling Pathway in Osteoblasts. Calcif. Tissue Int. 2016, 99, 88–97. [Google Scholar] [CrossRef]
- Ishikawa, S.; Kuno, A.; Tanno, M.; Miki, T.; Kouzu, H.; Itoh, T.; Sato, T.; Sunaga, D.; Murase, H.; Miura, T. Role of connexin-43 in protective PI3K-Akt-GSK-3β signaling in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2536–H2544. [Google Scholar] [CrossRef]
- Sabater, A.L.; Andreu, E.J.; Garcia-Guzman, M.; Lopez, T.; Abizanda, G.; Perez, V.L.; Moreno-Montanes, J.; Prosper, F. Combined PI3K/Akt and Smad2 Activation Promotes Corneal Endothelial Cell Proliferation. Investig. Ophthalmol. Vis. Sci. 2017, 58, 745–754. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wu, X.; Li, C.; Huang, Y.; Zhou, H.; Cui, Y. Resveratrol Sensitizes Colorectal Cancer Cells to Cetuximab by Connexin 43 Upregulation-Induced Akt Inhibition. Front. Oncol. 2020, 10, 383. [Google Scholar] [CrossRef]
- Chepied, A.; Daoud-Omar, Z.; Meunier-Balandre, A.C.; Laird, D.W.; Mesnil, M.; Defamie, N. Involvement of the Gap Junction Protein, Connexin43, in the Formation and Function of Invadopodia in the Human U251 Glioblastoma Cell Line. Cells 2020, 9, 117. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Alesutan, I.; Sopjani, M.; Munoz, C.; Fraser, S.; Kemp, B.E.; Foller, M.; Lang, F. Inhibition of connexin 26 by the AMP-activated protein kinase. J. Membr. Biol. 2011, 240, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yao, J.; Gao, K.; Chi, Y.; Mitsui, T.; Ihara, T.; Sawada, N.; Kamiyama, M.; Fan, J.; Takeda, M. AMPK Suppresses Connexin43 Expression in the Bladder and Ameliorates Voiding Dysfunction in Cyclophosphamide-Induced Mouse Cystitis. Sci. Rep. 2016, 6, 19708. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Chen, Q.; Jin, T.; Wang, M.; Ying, H.; Lu, J.; Wang, M.; Zhang, W.; Qiu, F.; Jin, C.; et al. Theaflavin 3,3′-digallate reverses the downregulation of connexin 43 and autophagy induced by high glucose via AMPK activation in cardiomyocytes. J. Cell. Physiol. 2019, 234, 17999–18016. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-N.; Wang, J.-C.; Cai, G.-Y.; Hu, X.; Cui, S.-Y.; Lv, Y.; Yin, Z.; Fu, B.; Hong, Q.; Chen, X.-M. AMPK-mediated downregulation of connexin43 and premature senescence of mesangial cells under high-glucose conditions. Exp. Gerontol. 2014, 51, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, L.; Gao, J.; Bi, X.; Zhang, J.; Xu, S.; Wang, M.; Chen, M.; Qiu, F.; Fu, G. Apelin Ameliorates High Glucose-Induced Downregulation of Connexin 43 via AMPK-Dependent Pathway in Neonatal Rat Cardiomyocytes. Aging Dis. 2018, 9, 66–76. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Gonzalez-Sanchez, A.; Medina, J.M.; Tabernero, A. HIF-1 and c-Src mediate increased glucose uptake induced by endothelin-1 and connexin43 in astrocytes. PLoS ONE 2012, 7, e32448. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Zhang, W.-F.; Wang, Q.; Li, M.; Zhang, C.-B.; Yang, Z.-J.; Tan, R.-J.; Gan, L.-J.; Zhang, L.-L.; Lan, X.-M.; et al. HIF-1α promotes the proliferation and migration of pulmonary arterial smooth muscle cells via activation of Cx43. J. Cell Mol. Med. 2021, 25, 10663–10673. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Dykstra, H.; LaRose, C.; Fisk, C.; Waldhart, A.; Meng, X.; Zhao, G.; Wu, N. TXNIP interaction with GLUT1 depends on PI(4,5)P2. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183757. [Google Scholar] [CrossRef]
- Beg, M.; Zhang, W.; McCourt, A.C.; Enerback, S. ATGL activity regulates GLUT1-mediated glucose uptake and lactate production via TXNIP stability in adipocytes. J. Biol. Chem. 2021, 296, 100332. [Google Scholar]
- Park, J.W.; Lee, S.H.; Woo, G.-H.; Kwon, H.-J.; Kim, D.-Y. Downregulation of TXNIP leads to high proliferative activity and estrogen-dependent cell growth in breast cancer. Biochem. Biophys. Res. Commun. 2018, 498, 566–572. [Google Scholar] [CrossRef]
- Xie, M.; Xie, R.; Xie, S.; Wu, Y.; Wang, W.; Li, X.; Xu, Y.; Liu, B.; Zhou, Y.; Wang, T.; et al. Thioredoxin interacting protein (TXNIP) acts as a tumor suppressor in human prostate cancer. Cell Biol. Int. 2020, 44, 2094–2106. [Google Scholar]
- Tang, J.-Y.; Li, D.-Y.; He, L.; Qiu, X.-S.; Wang, E.-H.; Wu, G.-P. HPV 16 E6/E7 Promote the Glucose Uptake of GLUT1 in Lung Cancer through Downregulation of TXNIP Due to Inhibition of PTEN Phosphorylation. Front. Oncol. 2020, 10, 559543. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e21. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, X.; Yin, H.; Xiao, S.; Yi, S.; Zhang, Y.; Zeng, F. TXNIP induced by MondoA, rather than ChREBP, suppresses cervical cancer cell proliferation, migration and invasion. J. Biochem. 2020, 167, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Gao, K.; Chi, Y.; Zhang, X.; Zhang, H.; Li, G.; Sun, W.; Takeda, M.; Yao, J. A novel TXNIP-based mechanism for Cx43-mediated regulation of oxidative drug injury. J. Cell. Mol. Med. 2015, 19, 2469–2480. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, X.; Gao, K.; Zhang, Z.; Huang, Y.; Yoda, R.; Yao, J. The pivotal role of extracellular signal-regulated kinase in gap junction-mediated regulation of TXNIP. Cell. Signal. 2017, 38, 116–126. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Kim, K.-H.; Rosen, A.; Smyth, J.W.; Sakuma, R.; Delgado-Olguin, P.; Davis, M.; Chi, N.C.; Puviindran, V.; Gaborit, N.; et al. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc. Natl. Acad. Sci. USA 2011, 108, 13576–13581. [Google Scholar] [CrossRef]
- Boztepe, T.; Gulec, S. Investigation of the influence of high glucose on molecular and genetic responses: An in vitro study using a human intestine model. Genes Nutr. 2018, 13, 11. [Google Scholar] [CrossRef]
- Furukawa, M.; Yamada, K.; Kurosawa, M.; Shikama, Y.; Wang, J.; Watanabe, M.; Kanekura, T.; Matsushita, K. High concentration of glucose induces filaggrin-1 expression through AP-1 in skin keratinocytes. J. Dermatol. Sci. 2020, 98, 137–140. [Google Scholar] [CrossRef]
- Lang, J.; Yang, C.; Liu, L.; Li, L.; Wu, L.; Liu, Y.; Luo, H.; Yan, L.; Chen, S.; Ning, J.; et al. High glucose activates ERK1/2 to stabilize AP1 and increase MMP9 expression in diabetic foot ulcers. Exp. Cell Res. 2021, 403, 112550. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-J.; Lin, C.-Y.; Tsai, C.-H.; Huang, Y.-L.; Tang, C.-H. Glucose suppresses IL-1β-induced MMP-1 expression through the FAK, MEK, ERK, and AP-1 signaling pathways. Environ. Toxicol. 2018, 33, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, H.-M.; Mao, Q.-Y.; Cong, X.; Zhang, Y.; Lee, S.-W.; Park, K.; Wu, L.-L.; Xiang, R.-L.; Yu, G.-Y. High Glucose Reduces the Paracellular Permeability of the Submandibular Gland Epithelium via the MiR-22-3p/Sp1/Claudin Pathway. Cells 2021, 10, 3230. [Google Scholar] [CrossRef] [PubMed]
- Oguro, A.; Oida, S.; Imaoka, S. Down-regulation of EPHX2 gene transcription by Sp1 under high-glucose conditions. Biochem. J. 2015, 470, 281–291. [Google Scholar] [CrossRef]
- Deng, R.; Wu, H.; Ran, H.; Kong, X.; Hu, L.; Wang, X.; Su, Q. Glucose-derived AGEs promote migration and invasion of colorectal cancer by up-regulating Sp1 expression. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861 Pt A, 1065–1074. [Google Scholar] [CrossRef]
- Li, T.; Bai, L.; Li, J.; Igarashi, S.; Ghishan, F.K. Sp1 is required for glucose-induced transcriptional regulation of mouse vesicular glutamate transporter 2 gene. Gastroenterology 2008, 134, 1994–2003. [Google Scholar] [CrossRef][Green Version]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef]
- Castano, C.; Kalko, S.; Novials, A.; Parrizas, M. Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12158–12163. [Google Scholar] [CrossRef] [PubMed]
- Agbu, P.; Carthew, R.W. MicroRNA-mediated regulation of glucose and lipid metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 425–438. [Google Scholar] [CrossRef]
- Calderon, J.F.; Retamal, M.A. Regulation of Connexins Expression Levels by MicroRNAs, an Update. Front. Physiol. 2016, 7, 558. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Liu, X.; Wang, F.; Yang, Y.; Tian, X. Hypoxic pancreatic cancer derived exosomal miR-30b-5p promotes tumor angiogenesis by inhibiting GJA1 expression. Int. J. Biol. Sci. 2022, 18, 1220–1237. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O. Posttranscriptional regulation of connexin-43 expression. Arch. Biochem. Biophys. 2012, 524, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, W.; Zhang, X.; Sun, L.; Yang, H.; Wang, Y.; Cao, Y.; Chu, Y.; Liu, G. Downregulation of microRNA-1 attenuates glucose-induced apoptosis by regulating the liver X receptor α in cardiomyocytes. Exp. Ther. Med. 2018, 16, 1814–1824. [Google Scholar] [CrossRef]
- Shan, Z.-X.; Lin, Q.-X.; Deng, C.-Y.; Zhu, J.-N.; Mai, L.-P.; Liu, J.-L.; Fu, Y.-H.; Liu, X.-Y.; Li, Y.-X.; Zhang, Y.-Y.; et al. miR-1/miR-206 regulate Hsp60 expression contributing to glucose-mediated apoptosis in cardiomyocytes. FEBS Lett. 2010, 584, 3592–3600. [Google Scholar] [CrossRef]
- Cao, Y.; Cao, X.; Sun, L.; Li, Y. miR-206 Inhibits Cell Proliferation and Extracellular Matrix Accumulation by Targeting Hypoxia-Inducible Factor 1-alpha (HIF-1α) in Mesangial Cells Treated with High Glucose. Med. Sci. Monit. 2019, 25, 10036–10044. [Google Scholar] [CrossRef]
- El-Lateef, A.E.A.; El-Shemi, A.G.A.; Alhammady, M.S.; Yuan, R.; Zhang, Y. LncRNA NEAT2 Modulates Pyroptosis of Renal Tubular Cells Induced by High Glucose in Diabetic Nephropathy (DN) by via miR-206 Regulation. Biochem. Genet. 2022. [Google Scholar] [CrossRef]
- Jiang, A.; Dong, C.; Li, B.; Zhang, Z.; Chen, Y.; Ning, C.; Wu, W.; Liu, H. MicroRNA-206 regulates cell proliferation by targeting G6PD in skeletal muscle. FASEB J. 2019, 33, 14083–14094. [Google Scholar] [CrossRef]
- Jia, K.-G.; Feng, G.; Tong, Y.-S.; Tao, G.-Z.; Xu, L. miR-206 regulates non-small-cell lung cancer cell aerobic glycolysis by targeting hexokinase 2. J. Biochem. 2020, 167, 365–370. [Google Scholar] [CrossRef]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A novel miR-206/hnRNPA1/PKM2 axis reshapes the Warburg effect to suppress colon cancer growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef]
- Vinod, M.; Patankar, J.V.; Sachdev, V.; Frank, S.; Graier, W.F.; Kratky, D.; Kostner, G.M. MiR-206 is expressed in pancreatic islets and regulates glucokinase activity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E175–E185. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Anjo, S.I.; Manadas, B.; Sluijter, J.P.G.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed]
- Asencio-Barria, C.; Defamie, N.; Saez, J.C.; Mesnil, M.; Godoy, A.S. Direct Intercellular Communications and Cancer: A Snapshot of the Biological Roles of Connexins in Prostate Cancer. Cancers 2019, 11, 1370. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, P.; Hong, T.; Wang, Y.; Liu, N.; He, B.; Zou, S.; Ren, D.; Duan, J.; Zhao, L.; et al. Astrocytes-derived exosomes induce neuronal recovery after traumatic brain injury via delivering gap junction alpha 1–20 k. J. Tissue Eng. Regen. Med. 2020, 14, 412–423. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Zhang, L.-L.; Bi, Q.-C.; Gan, L.-J.; Wei, M.-J.; Hong, T.; Tan, R.-J.; Lan, X.-M.; Liu, L.-H.; Han, X.-J.; et al. Exosomal connexin 43 regulates the resistance of glioma cells to temozolomide. Oncol. Rep. 2021, 45, 44. [Google Scholar] [CrossRef]
- Villamizar, O.; Waters, S.A.; Scott, T.; Grepo, N.; Jaffe, A.; Morris, K.V. Mesenchymal Stem Cell exosome delivered Zinc Finger Protein activation of cystic fibrosis transmembrane conductance regulator. J. Extracell. Vesicles 2021, 10, e12053. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Bi, Q.-C.; Gan, L.-J.; Zhang, L.-L.; Wei, M.-J.; Hong, T.; Liu, R.; Qiu, C.-L.; Han, X.-J.; Jiang, L.-P. Exosomes Derived from Glioma Cells under Hypoxia Promote Angiogenesis through up-Regulated Exosomal Connexin 43. Int. J. Med. Sci. 2022, 19, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Xiong, Y.; Zhang, W.; Wu, X.; Sun, Z.; Zhang, L.; Wu, H.; Tang, Y.; Peng, Y. Astrocytes promote the proliferation of oligodendrocyte precursor cells through connexin 47-mediated LAMB2 secretion in exosomes. Mol. Biol. Rep. 2022, 49, 7263–7273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Cordero Cervantes, D.; Zurzolo, C. Peering into tunneling nanotubes-The path forward. EMBO J. 2021, 40, e105789. [Google Scholar] [CrossRef]
- Valdebenito, S.; Lou, E.; Baldoni, J.; Okafo, G.; Eugenin, E. The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1270. [Google Scholar] [CrossRef]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar]
- Tishchenko, A.; Azorin, D.D.; Vidal-Brime, L.; Munoz, M.J.; Arenas, P.J.; Pearce, C.; Girao, H.; Ramon, Y.C.S.; Aasen, T. Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers 2020, 12, 2798. [Google Scholar] [CrossRef]
- Stephan, J.; Eitelmann, S.; Zhou, M. Approaches to Study Gap Junctional Coupling. Front. Cell. Neurosci. 2021, 15, 640406. [Google Scholar]
- Kumar, A.; Misra, B.B. Challenges and Opportunities in Cancer Metabolomics. Proteomics 2019, 19, e1900042. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, J.C.; Bodenstine, T.M. Connexins and Glucose Metabolism in Cancer. Int. J. Mol. Sci. 2022, 23, 10172. https://doi.org/10.3390/ijms231710172
Jones JC, Bodenstine TM. Connexins and Glucose Metabolism in Cancer. International Journal of Molecular Sciences. 2022; 23(17):10172. https://doi.org/10.3390/ijms231710172
Chicago/Turabian StyleJones, Jennifer C., and Thomas M. Bodenstine. 2022. "Connexins and Glucose Metabolism in Cancer" International Journal of Molecular Sciences 23, no. 17: 10172. https://doi.org/10.3390/ijms231710172
APA StyleJones, J. C., & Bodenstine, T. M. (2022). Connexins and Glucose Metabolism in Cancer. International Journal of Molecular Sciences, 23(17), 10172. https://doi.org/10.3390/ijms231710172