
Sources of Cancer Neoantigens beyond Single-Nucleotide Variants

Abstract
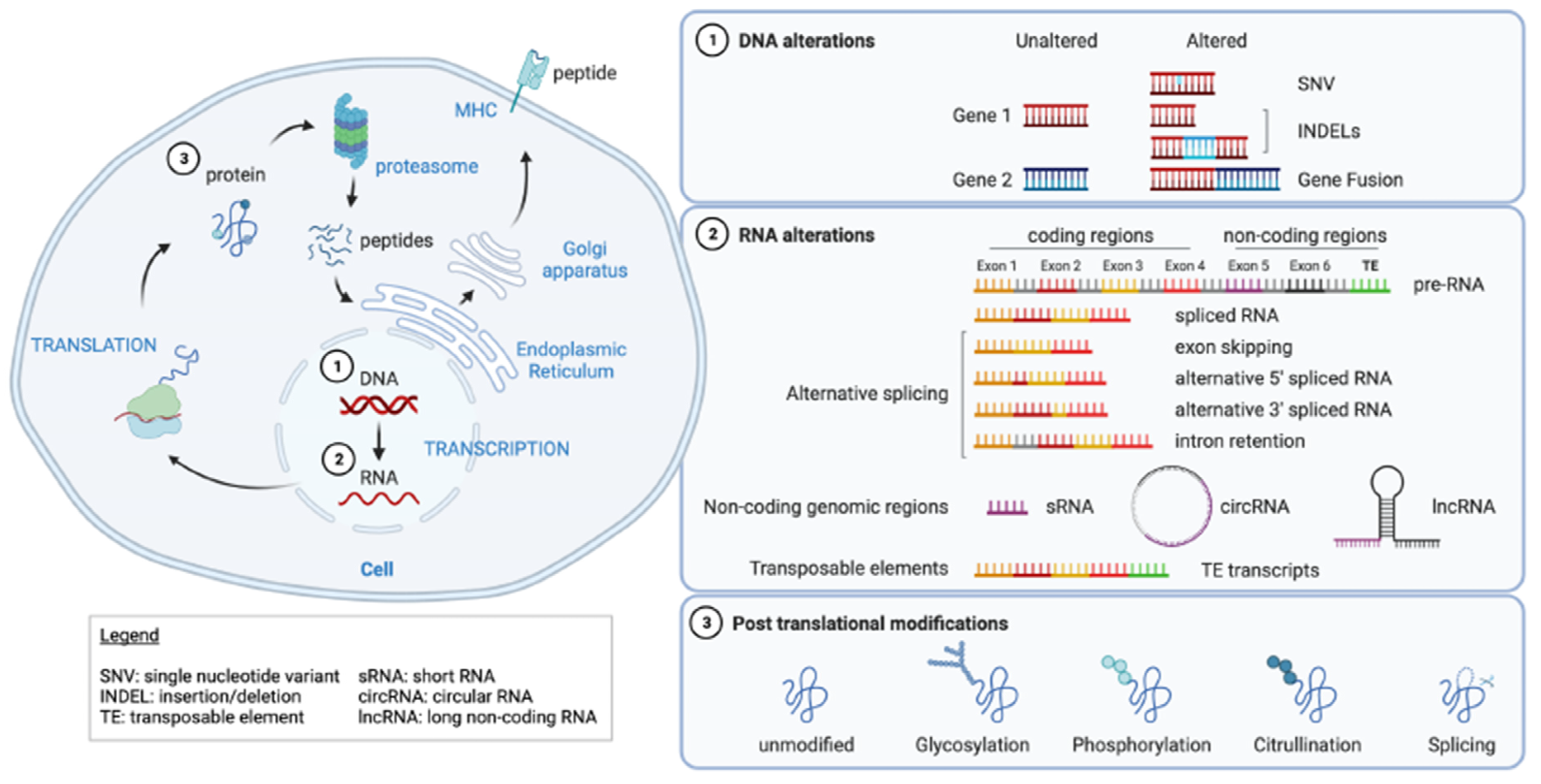
:1. Introduction
2. DNA Alterations
2.1. SNV/Indels
2.2. Gene Fusion
3. RNA Aberrations
3.1. Alternative Splicing
3.2. Non-Coding Genomic Regions
3.3. Transposable Elements
4. Post-Translational Modifications
5. Conclusions

{kind=link}
| Alterations | Presentation | Immunogenicity | Shared between Patients | Tumor-Specificity | Tumor Alteration Burden | Main Challenges |
|---|---|---|---|---|---|---|
| SNV/indels | MHCI, MHCII | CD8, CD4 | Mostly private | Yes | Low to high depending on cancer type | Immunogenicity (similarity to self) |
| Gene fusion | MHCI, MHCII [53,54,55] | CD8, CD4 [53,54,55,56] | Yes | Yes | Low | Identification, prediction |
| Alternative splicing | MHCI, MHCII [76,169] | CD8, CD4 [77,78,79,169] | TBD | TBD | TBD | Identification, tumor-specificity |
| Non-coding genomic regions | MHCI, MHCII [83,86,87,88,89] | CD8 [87] | Yes | Yes | TBD | Immunogenicity, tumor-specificity |
| Transposable Elements | MHCI, MHCII [104,105,106,107,108,109] | CD8, CD4 [104,105,106,107,108,109] | Yes | No | TBD | Identification, tumor-specificity |
| Glycosylation | MHCI [118] | CD8 [118] | Yes | TBD | Low | Identification, prediction, tumor-specificity |
| Phosphorylation | MHCI, MHCII [119,120,121,122] | CD8, CD4 [119,120,121,122] | Yes | TBD | Low | Identification, prediction, tumor-specificity |
| Citrullination | MHCII [134,135,136] | CD4 [134,135,136] | Yes | TBD | Low | Identification, prediction, tumor-specificity |
| Peptide splicing | MHCI | CD8 [137,138,139] | TBD | TBD | TBD | Identification, prediction, tumor-specificity |
| Alterations | Altered Molecule | Identification | Prediction | Most Advanced Development Stage | Example |
|---|---|---|---|---|---|
| SNV/indels | DNA | WES + RNA-seq | Available (many) | Phase 1/1b; several ongoing Phases 2/3 | Immunogenic responses observed in patients receiving peptide/DC/mRNA vaccines; or adoptive T cell therapy in different cancer types [147,148,149,170,171] |
| Gene fusion | DNA | WES + RNA-seq | Available (few) | Phase 2 | Immunogenic response but no clinical efficacy observed in patients with CML following bcr-abl peptide vaccination [172] |
| Alternative splicing | RNA | RNA-seq, Ribo-seq | Available (few) | Preclinical | CD8 T cell recognition of the mutated splicing factor SF3B1 in patients with uveal melanoma [79] |
| Non-coding genomic regions | RNA | RNA-seq, Ribo-seq | NA | Preclinical | Delayed tumor growth of CT26 tumors following cryptic peptide vaccination without proof of specific T cell response [92] |
| Transposable Elements | RNA | WES + RNA-seq, Ribo-seq | Available (few) | Preclinical ongoing Phase 1 | Recognition of HERV antigens by CD8 T cells from patients [108,109] HERV-E TCR Transduced Autologous T Cells in Metastatic Kidney cancer patients (*) |
| Glycosylation | Protein | Mass spectrometry | NA | Phase 3 | No overall survival benefit with L-BLP25 peptide vaccine in NSCLC patients [129]; Improved progression free survival post TG4010 vaccine + chemotherapy in NSCLC patients [130] |
| Phosphorylation | Protein | Mass spectrometry | NA | Phase 1 | Some specific CD8 T cell responses were observed in melanoma patients who received pIRS2 and pBCAR3 peptide vaccines [133] |
| Citrullination | Protein | Mass spectrometry | NA | Preclinical | Delayed B16F1 tumor growth in HLA-DR4 transgenic mice following citrullinated peptide vaccination. Citrullinated-specific CD4 T cell responses also observed in PBMC from ovarian cancer patients [136] |
| Peptide splicing | Protein | Mass spectrometry | NA | Preclinical | Spliced peptide identified and recognized by CD8 T cells in renal cell carcinoma [137] or melanoma [138] patients, and from EBV-B cells [139] |
Author Contributions
Funding
Conflicts of Interest
References
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 Identifies the Patient-Specific CD8+ Tumor-Reactive Repertoire Infiltrating Human Tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef]
- Zou, X.L.; Li, X.B.; Ke, H.; Zhang, G.Y.; Tang, Q.; Yuan, J.; Zhou, C.J.; Zhang, J.L.; Zhang, R.; Chen, W.Y. Prognostic Value of Neoantigen Load in Immune Checkpoint Inhibitor Therapy for Cancer. Front. Immunol. 2021, 12, 689076. [Google Scholar] [CrossRef]
- Santambrogio, L. Molecular Determinants Regulating the Plasticity of the MHC Class II Immunopeptidome. Front. Immunol. 2022, 13, 878271. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Røder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a Method for Quantitative Predictions of Peptide Binding to Any HLA-A and -B Locus Protein of Known Sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Gfeller, D.; Bassani-Sternberg, M. Predicting Antigen Presentation-What Could We Learn from a Million Peptides? Front. Immunol. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Verma, A.; Halder, A.; Marathe, S.; Purwar, R.; Srivastava, S. A Proteogenomic Approach to Target Neoantigens in Solid Tumors. Expert. Rev. Proteom. 2020, 17, 797–812. [Google Scholar] [CrossRef]
- Pollock, S.B.; Rose, C.M.; Darwish, M.; Bouziat, R.; Delamarre, L.; Blanchette, C.; Lill, J.R. Sensitive and Quantitative Detection of MHC-I Displayed Neoepitopes Using a Semiautomated Workflow and TOMAHAQ Mass Spectrometry. Mol. Cell. Proteom. 2021, 20, 100108. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped Sequence Alignment Using Artificial Neural Networks: Application to the MHC Class i System. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Chong, C.; Guillaume, P.; Solleder, M.; Pak, H.S.; Gannon, P.O.; Kandalaft, L.E.; Coukos, G.; Gfeller, D. Deciphering HLA-I Motifs across HLA Peptidomes Improves Neo-Antigen Predictions and Identifies Allostery Regulating HLA Specificity. PLoS Comput. Biol. 2017, 13, e1005725. [Google Scholar] [CrossRef]
- Gfeller, D.; Guillaume, P.; Michaux, J.; Pak, H.-S.; Daniel, R.T.; Racle, J.; Coukos, G.; Bassani-Sternberg, M. The Length Distribution and Multiple Specificity of Naturally Presented HLA-I Ligands. J. Immunol. 2018, 201, 3705–3716. [Google Scholar] [CrossRef]
- Freudenmann, L.K.; Marcu, A.; Stevanović, S. Mapping the Tumour Human Leukocyte Antigen (HLA) Ligandome by Mass Spectrometry. Immunology 2018, 154, 331–345. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep Learning Using Tumor HLA Peptide Mass Spectrometry Datasets Improves Neoantigen Identification. Nat. Biotechnol. 2019, 37, 55–71. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef]
- Boehm, K.M.; Bhinder, B.; Raja, V.J.; Dephoure, N.; Elemento, O. Predicting Peptide Presentation by Major Histocompatibility Complex Class I: An Improved Machine Learning Approach to the Immunopeptidome. BMC Bioinform. 2019, 20, 7. [Google Scholar] [CrossRef]
- de Mattos-Arruda, L.; Vazquez, M.; Finotello, F.; Lepore, R.; Porta, E.; Hundal, J.; Amengual-Rigo, P.; Ng, C.K.Y.; Valencia, A.; Carrillo, J.; et al. Neoantigen Prediction and Computational Perspectives towards Clinical Benefit: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 978–990. [Google Scholar] [CrossRef]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-Allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust Prediction of HLA Class II Epitopes by Deep Motif Deconvolution of Immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Bjerregaard, A.M.; Nielsen, M.; Jurtz, V.; Barra, C.M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. An Analysis of Natural T Cell Responses to Predicted Tumor Neoepitopes. Front. Immunol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best Practices for Bioinformatic Characterization of Neoantigens for Clinical Utility. Genome Med. 2019, 11, 56–66. [Google Scholar] [CrossRef]
- Zhou, C.; Zhu, C.; Liu, Q. Toward in Silico Identification of Tumor Neoantigens in Immunotherapy. Trends Mol. Med. 2019, 25, 980–992. [Google Scholar] [CrossRef]
- Finotello, F.; Rieder, D.; Hackl, H.; Trajanoski, Z. Next-Generation Computational Tools for Interrogating Cancer Immunity. Nat. Rev. Genet. 2019, 20, 724–746. [Google Scholar] [CrossRef]
- Ebrahimi-Nik, H.; Moussa, M.; Englander, R.P.; Singhaviranon, S.; Michaux, J.; Pak, H.S.; Miyadera, H.; Corwin, W.L.; Keller, G.L.J.; Hagymasi, A.T.; et al. Reversion Analysis Reveals the in Vivo Immunogenicity of a Poorly MHC I-Binding Cancer Neoepitope. Nat. Commun. 2021, 12, 6423. [Google Scholar] [CrossRef]
- Chowell, D.; Krishna, S.; Becker, P.D.; Cocita, C.; Shu, J.; Tan, X.; Greenberg, P.D.; Klavinskis, L.S.; Blattman, J.N.; Anderson, K.S. TCR Contact Residue Hydrophobicity Is a Hallmark of Immunogenic CD8+ T Cell Epitopes. Proc. Natl. Acad. Sci. USA 2015, 112, E1754–E1762. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; de Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature 2017, 551, S12–S16. [Google Scholar] [CrossRef]
- Luksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A Neoantigen Fitness Model Predicts Tumour Response to Checkpoint Blockade Immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Duan, F.; Duitama, J.; al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and Bioinformatic Profiling of Mutational Neoepitopes Reveals New Rules to Predict Anticancer Immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef]
- Schmidt, J.; Smith, A.R.; Magnin, M.; Racle, J.; Devlin, J.R.; Bobisse, S.; Cesbron, J.; Bonnet, V.; Carmona, S.J.; Huber, F.; et al. Prediction of Neo-Epitope Immunogenicity Reveals TCR Recognition Determinants and Provides Insight into Immunoediting. Cell Rep. Med. 2021, 2, 100194. [Google Scholar] [CrossRef]
- Capietto, A.H.; Jhunjhunwala, S.; Pollock, S.B.; Lupardus, P.; Wong, J.; Hänsch, L.; Cevallos, J.; Chestnut, Y.; Fernandez, A.; Lounsbury, N.; et al. Mutation Position Is an Important Determinant for Predicting Cancer Neoantigens. J. Exp. Med. 2020, 217, e20190179. [Google Scholar] [CrossRef]
- Gartner, J.J.; Parkhurst, M.R.; Gros, A.; Tran, E.; Jafferji, M.S.; Copeland, A.; Hanada, K.I.; Zacharakis, N.; Lalani, A.; Krishna, S.; et al. A Machine Learning Model for Ranking Candidate HLA Class I Neoantigens Based on Known Neoepitopes from Multiple Human Tumor Types. Nat. Cancer 2021, 2, 563–574. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; al Bakir, M.; et al. Insertion-and-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from Nonsense-Mediated Decay Associates with Anti-Tumor Immunogenicity. Nat. Commun. 2020, 11, 3800. [Google Scholar] [CrossRef]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’Donnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.-L.; Blank, S.V.; et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell 2020, 183, 1634–1649.e17. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia Identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Rabbihs, T.H. Chromosomal Translocations in Human Cancer. Nature 1994, 372, 143–149. [Google Scholar]
- Druker, B.J. Translation of the Philadelphia Chromosome into Therapy for CML. Nature 2008, 372, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; O’Toole, S.A.; Ballinger, M.L.; Gill, D.; Thomas, D.M.; et al. Diagnosis of Fusion Genes Using Targeted RNA Sequencing. Nat. Commun. 2019, 10, 1388. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy Assessment of Fusion Transcript Detection via Read-Mapping and de Novo Fusion Transcript Assembly-Based Methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef]
- Dehghannasiri, R.; Freeman, D.E.; Jordanski, M.; Hsieh, G.L.; Damljanovic, A.; Lehnert, E.; Salzman, J. Improved Detection of Gene Fusions by Applying Statistical Methods Reveals Oncogenic RNA Cancer Drivers. Proc. Natl. Acad. Sci. USA 2019, 116, 15524–15533. [Google Scholar] [CrossRef]
- Kim, P.; Jang, Y.E.; Lee, S. Fusionscan: Accurate Prediction of Fusion Genes from RNA-Seq Data. Genom. Inform. 2019, 17, e26. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and Efficient Detection of Gene Fusions from RNA Sequencing Data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Balan, J.; Jenkinson, G.; Nair, A.; Saha, N.; Koganti, T.; Voss, J.; Zysk, C.; Barr Fritcher, E.G.; Ross, C.A.; Giannini, C.; et al. SeekFusion—A Clinically Validated Fusion Transcript Detection Pipeline for PCR-Based Next-Generation Sequencing of RNA. Front. Genet. 2021, 12, 739054. [Google Scholar] [CrossRef]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.F.; Griffith, M.; Moussavi, A.; Senz, J.; Melnyk, N.; et al. Defuse: An Algorithm for Gene Fusion Discovery in Tumor Rna-Seq Data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Rathe, S.K.; Popescu, F.E.; Johnson, J.E.; Watson, A.L.; Marko, T.A.; Moriarity, B.S.; Ohlfest, J.R.; Largaespada, D.A. Identification of Candidate Neoantigens Produced by Fusion Transcripts in Human Osteosarcomas. Sci. Rep. 2019, 9, 358. [Google Scholar] [CrossRef]
- Cullis, J.; Barrett, A.; Goldman, J.; Lechler, R. Binding of BCR/ABL Junctional Peptides to Major Histocompatibility Complex (MHC) Class I Molecules: Studies in Antigen-Processing Defective Cell Lines. Leukemia 1994, 8, 165–170. [Google Scholar]
- Worley, B.; van den Broeke, L.; Goletz, T.; Pendleton, C.; Daschbach, E.; Thomas, E.; Marincola, F.; Helman, L.; Berzofsky, J. Antigenicity of Fusion Proteins from Sarcoma-Associated Chromosomal Translocations. Cancer Res. 2001, 61, 6868–6875. [Google Scholar]
- van den Broeke, L.T.; Pendleton, C.D.; Mackall, C.; Helman, L.J.; Berzofsky, J.A. Identification and Epitope Enhancement of a PAX-FKHR Fusion Protein Breakpoint Epitope in Alveolar Rhabdomyosarcoma Cells Created by a Tumorigenic Chromosomal Translocation Inducing CTL Capable of Lysing Human Tumors. Cancer Res. 2006, 66, 1818–1823. [Google Scholar] [CrossRef]
- Comoli, P.; Basso, S.; Riva, G.; Barozzi, P.; Guido, I.; Gurrado, A.; Quartuccio, G.; Rubert, L.; Lagreca, I.; Vallerini, D.; et al. Brief Report BCR-ABL-Specific T-Cell Therapy in Ph 1 ALL Patients on Tyrosine-Kinase Inhibitors. Blood J. Am. Soc. Hematol. 2007, 129, 582–586. [Google Scholar] [CrossRef]
- Dagher, R.; Long, L.; Read, E.; Leitman, S.; Carter, C.; Tsokos, M.; Goletz, T.; Avila, N.; Berzofsky, J.; Helman, L.; et al. Pilot Trial of Tumor-Specific Peptide Vaccination and Continuous Infusion Interleukin-2 in Patients with Recurrent Ewing Sarcoma and Alveolar Rhabdomyosarcoma: An Inter-Institute NIH Study. Med. Pediatr. Oncol. 2002, 38, 158–164. [Google Scholar] [CrossRef]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A Pilot Study of Consolidative Immunotherapy in Patients with High-Risk Pediatric Sarcomas. Clin. Cancer Res. 2008, 14, 4850. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of Alternative Splicing and Its Regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting MRNA Processing as an Anticancer Strategy. Nat. Rev. Drug Discov. 2020, 19, 112–129. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Laiho, A.; Venälaïnen, M.S.; McGlinchey, A.J.; Wang, N.; Elo, L.L. Systematic Evaluation of Differential Splicing Tools for RNA-Seq Studies. Brief. Bioinform. 2020, 21, 2052–2065. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, L. Alternative Splicing: Human Disease and Quantitative Analysis from High-Throughput Sequencing. Comput. Struct. Biotechnol. J. 2021, 19, 183–195. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’grady, G.L.; et al. Improving Genetic Diagnosis in Mendelian Disease with Transcriptome Sequencing Genotype-Tissue Expression Consortium. Sci. Transl. Med. 2017, 386, eaal5209. [Google Scholar] [CrossRef]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic Diagnosis of Mendelian Disorders via RNA Sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of Rare-Disease Genes Using Blood Transcriptome Sequencing and Large Control Cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Ferraro, N.M.; Strober, B.J.; Einson, J.; Abell, N.S.; Aguet, F.; Barbeira, A.N.; Bonazzola, R.; Brown, A.; Castel, S.E.; Jo, B.; et al. Transcriptomic Signatures across Human Tissues Identify Functional Rare Genetic Variation. Science 2020, 369, eaaz5900. [Google Scholar] [CrossRef]
- Jenkinson, G.; Li, Y.I.; Basu, S.; Cousin, M.A.; Oliver, G.R.; Klee, E.W. LeafCutterMD: An Algorithm for Outlier Splicing Detection in Rare Diseases. Bioinformatics 2020, 36, 4609–4615. [Google Scholar] [CrossRef]
- Suo, C.; Hrydziuszko, O.; Lee, D.; Pramana, S.; Saputra, D.; Joshi, H.; Calza, S.; Pawitan, Y. Integration of Somatic Mutation, Expression and Functional Data Reveals Potential Driver Genes Predictive of Breast Cancer Survival. Bioinformatics 2015, 31, 2607–2613. [Google Scholar] [CrossRef]
- Shen, S.; Wang, Y.; Wang, C.; Wu, Y.N.; Xing, Y. SURVIV for Survival Analysis of MRNA Isoform Variation. Nat. Commun. 2016, 7, 11548. [Google Scholar] [CrossRef]
- Li, Y.; Sahni, N.; Pancsa, R.; McGrail, D.J.; Xu, J.; Hua, X.; Coulombe-Huntington, J.; Ryan, M.; Tychhon, B.; Sudhakar, D.; et al. Revealing the Determinants of Widespread Alternative Splicing Perturbation in Cancer. Cell Rep. 2017, 21, 798–812. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Yong, L. Systematic Profiling of Alternative Splicing Signature Reveals Prognostic Predictor for Ovarian Cancer. Gynecol. Oncol. 2018, 148, 368–374. [Google Scholar] [CrossRef]
- Bjørklund, S.S.; Panda, A.; Kumar, S.; Seiler, M.; Robinson, D.; Gheeya, J.; Yao, M.; Alnæs, G.I.G.; Toppmeyer, D.; Riis, M.; et al. Widespread Alternative Exon Usage in Clinically Distinct Subtypes of Invasive Ductal Carcinoma. Sci. Rep. 2017, 7, 798–812. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef]
- Marcelino Meliso, F.; Hubert, C.G.; Favoretto Galante, P.A.; Penalva, L.O. RNA Processing as an Alternative Route to Attack Glioblastoma. Hum. Genet. 2017, 136, 1129–1141. [Google Scholar] [CrossRef]
- Kahles, A.; van Lehmann, K.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Caesar-Johnson, S.J.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Lupetti, R.; Pisarra, P.; Verrecchia, A.; Farina, C.; Nicolini, G.; Anichini, A.; Bordignon, C.; Sensi, M.; Parmiani, G.; Traversari, C. Translation of a Retained Intron in Tyrosinase-Related Protein (TRP) 2 MRNA Generates a New Cytotoxic T Lymphocyte (CTL)-Defined and Shared Human Melanoma Antigen Not Expressed in Normal Cells of the Melanocytic Lineage. J. Exp. Med. 1998, 188, 1005–1016. [Google Scholar] [CrossRef]
- Slager, E.H.; van der Minne, C.E.; Krüse, M.; Krueger, D.D.; Griffioen, M.; Osanto, S. Identification of Multiple HLA-DR-Restricted Epitopes of the Tumor-Associated Antigen CAMEL by CD4 + Th1/Th2 Lymphocytes. J. Immunol. 2004, 172, 5095–5102. [Google Scholar] [CrossRef]
- Bigot, J.; Lalanne, A.I.; Lucibello, F.; Gueguen, P.; Houy, A.; Dayot, S.; Ganier, O.; Gilet, J.; Tosello, J.; Nemati, F.; et al. Splicing Patterns in Sf3b1mutated Uveal Melanoma Generate Shared Immunogenic Tumor-Specific Neoepitopes. Cancer Discov. 2021, 11, 1938–1951. [Google Scholar] [CrossRef]
- Qin, T.; Li, J.; Zhang, K.Q. Structure, Regulation, and Function of Linear and Circular Long Non-Coding RNAs. Front. Genet. 2020, 11, 150. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.S.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Brunner, A.-D.; Cogan, Z.; Nuñez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; Leonetti, M.D.; et al. Pervasive Functional Translation of Noncanonical Human Open Reading Frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Marranci, A.; Pandolfi, P.P. Pseudogenes in Human Cancer. Front. Med. 2015, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Lill, J.R.; Mathews, W.R.; Rose, C.M.; Schirle, M. Proteomics in the Pharmaceutical and Biotechnology Industry: A Look to the next Decade. Expert. Rev. Proteom. 2021, 18, 503–526. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Dölken, L.; Schilling, B.; Schlosser, A. Identification of the Cryptic HLA-I Immunopeptidome. Cancer Immunol. Res. 2020, 8, 1018–1026. [Google Scholar] [CrossRef]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated Proteogenomic Deep Sequencing and Analytics Accurately Identify Non-Canonical Peptides in Tumor Immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef]
- Ruiz Cuevas, M.V.; Hardy, M.P.; Hollý, J.; Bonneil, É.; Durette, C.; Courcelles, M.; Lanoix, J.; Côté, C.; Staudt, L.M.; Lemieux, S.; et al. Most Non-Canonical Proteins Uniquely Populate the Proteome or Immunopeptidome. Cell Rep. 2021, 34, 108815. [Google Scholar] [CrossRef]
- Ouspenskaia, T.; Law, T.; Clauser, K.R.; Klaeger, S.; Sarkizova, S.; Aguet, F.; Li, B.; Christian, E.; Knisbacher, B.A.; Le, P.M.; et al. Unannotated Proteins Expand the MHC-I-Restricted Immunopeptidome in Cancer. Nat. Biotechnol. 2022, 40, 209–217. [Google Scholar] [CrossRef]
- Xiang, R.; Ma, L.; Yang, M.; Zheng, Z.; Chen, X.; Jia, F.; Xie, F.; Zhou, Y.; Li, F.; Wu, K.; et al. Increased Expression of Peptides from Non-Coding Genes in Cancer Proteomics Datasets Suggests Potential Tumor Neoantigens. Commun. Biol. 2021, 4, 3236384. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding Regions Are the Main Source of Targetable Tumor-Specific Antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef]
- Tokita, S.; Kanaseki, T.; Torigoe, T. Therapeutic Potential of Cancer Vaccine Based on MHC Class I Cryptic Peptides Derived from Non-Coding Regions. Immuno 2021, 1, 424–431. [Google Scholar] [CrossRef]
- Senft, A.D.; Macfarlan, T.S. Transposable Elements Shape the Evolution of Mammalian Development. Nat. Rev. Genet. 2021, 22, 691–711. [Google Scholar] [CrossRef]
- Lander, S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar]
- Vargiu, L.; Rodriguez-Tomé, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and Characterization of Human Endogenous Retroviruses Mosaic Forms Are Common. Retrovirology 2016, 13, 7. [Google Scholar] [CrossRef]
- Pradhan, R.K.; Ramakrishna, W. Transposons: Unexpected Players in Cancer. Gene 2022, 808, 145975. [Google Scholar] [CrossRef]
- Burns, K.H. Our Conflict with Transposable Elements and Its Implications for Human Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 51–70. [Google Scholar] [CrossRef]
- Vendrell-Mir, P.; Barteri, F.; Merenciano, M.; González, J.; Casacuberta, J.M.; Castanera, R. A Benchmark of Transposon Insertion Detection Tools Using Real Data. Mob. DNA 2019, 10, 53. [Google Scholar] [CrossRef]
- Chu, C.; Borges-Monroy, R.; Viswanadham, V.V.; Lee, S.; Li, H.; Lee, E.A.; Park, P.J. Comprehensive Identification of Transposable Element Insertions Using Multiple Sequencing Technologies. Nat. Commun. 2021, 12, 3836. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable Element Expression in Tumors Is Associated with Immune Infiltration and Increased Antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous Retroviral Signatures Predict Immunotherapy Response in Clear Cell Renal Cell Carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J., III; Perry-Lalley, D.; Dudley, M.E.; Rosenberg, S.A.; Yang, J.C. High Avidity CTLs for Two Self-Antigens Demonstrate Superior In Vitro and In Vivo Antitumor Efficacy. J. Immunol. 1999, 162, 989–994. [Google Scholar]
- Kershaw, M.H.; Hsu, C.; Mondesire, W.; Parker, L.L.; Wang, G.; Overwijk, W.W.; Lapointe, R.; Yang, J.C.; Wang, R.-F.; Restifo, N.P.; et al. Immunization against Endogenous Retroviral Tumor-Associated Antigens 1. Cancer Res. 2001, 61, 7920–7924. [Google Scholar]
- Mullins, C.S.; Linnebacher, M. Endogenous Retrovirus Sequences as a Novel Class of Tumor-Specific Antigens: An Example of HERV-H Env Encoding Strong CTL Epitopes. Cancer Immunol. Immunother. 2012, 61, 1093–1100. [Google Scholar] [CrossRef]
- Cherkasova, E.; Scrivani, C.; Doh, S.; Weisman, Q.; Takahashi, Y.; Harashima, N.; Yokoyama, H.; Srinivasan, R.; Linehan, W.M.; Lerman, M.I.; et al. Detection of an Immunogenic HERV-E Envelope with Selective Expression in Clear Cell Kidney Cancer. Cancer Res. 2016, 76, 2177–2185. [Google Scholar] [CrossRef]
- Schiavetti, F.; Thonnard, J.; Colau, D.; Boon, T.; Coulie, P.G. A Human Endogenous Retroviral Sequence Encoding an Antigen Recognized on Melanoma by Cytolytic T Lymphocytes 1. Cancer Res. 2002, 62, 5510–5516. [Google Scholar]
- Wang-Johanning, F.; Radvanyi, L.; Rycaj, K.; Plummer, J.B.; Yan, P.; Sastry, K.J.; Piyathilake, C.J.; Hunt, K.K.; Johanning, G.L. Human Endogenous Retrovirus K Triggers an Antigen-Specific Immune Response in Breast Cancer Patients. Cancer Res. 2008, 68, 5869–5877. [Google Scholar] [CrossRef]
- Takahashi, Y.; Harashima, N.; Kajigaya, S.; Yokoyama, H.; Cherkasova, E.; McCoy, J.P.; Hanada, K.I.; Mena, O.; Kurlander, R.; Abdul, T.; et al. Regression of Human Kidney Cancer Following Allogeneic Stem Cell Transplantation Is Associated with Recognition of an HERV-E Antigen by T Cells. J. Clin. Investig. 2008, 118, 1099–1109. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Shraibman, B.; Kadosh, D.M.; Barnea, E.; Admon, A. Human Leukocyte Antigen (HLA) Peptides Derived from Tumor Antigens Induced by Inhibition of DNA Methylation for Development of Drug-Facilitated Immunotherapy. Mol. Cell. Proteom. 2016, 15, 3058–3070. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Leung, S.Y. Abstract PR06: Genomic Characterization of Immune Escape Pathways in Gastric Cancer. Cancer Immunol. Res. 2015, 3, PR06. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic Silencing by SETDB1 Suppresses Tumour Intrinsic Immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Zhang, S.M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B Promotes Immune Evasion by Recruiting SETDB1 to Silence Retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.P. Recent Developments in Epigenetic Cancer Therapeutics: Clinical Advancement and Emerging Trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef]
- Malaker, S.A.; Penny, S.A.; Steadman, L.G.; Myers, P.T.; Loke, J.C.; Raghavan, M.; Bai, D.L.; Shabanowitz, J.; Hunt, D.F.; Cobbold, M. Identification of Glycopeptides as Posttranslationally Modified Neoantigens in Leukemia. Cancer Immunol. Res. 2017, 5, 376–384. [Google Scholar] [CrossRef]
- Zarling, A.L.; Polefrone, J.M.; Evans, A.M.; Mikesh, L.M.; Shabanowitz, J.; Lewis, S.T.; Engelhard, V.H.; Hunt, D.F. Identification of Class I MHC-Associated Phosphopeptides as Targets for Cancer Immunotherapy. Proc. Natl. Acad. Sci. USA 2006, 103, 14889–14894. [Google Scholar] [CrossRef]
- Depontieu, F.R.; Qian, J.; Zarling, A.L.; Mcmiller, T.L.; Salay, T.M.; Norris, A.; English, A.M.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; et al. Identification of Tumor-Associated, MHC Class II-Restricted Phosphopeptides as Targets for Immunotherapy. Proc. Natl. Acad. Sci. USA 2009, 106, 12073–12078. [Google Scholar] [CrossRef]
- Penny, S.A.; Abelin, J.G.; Malaker, S.A.; Myers, P.T.; Saeed, A.Z.; Steadman, L.G.; Bai, D.L.; Ward, S.T.; Shabanowitz, J.; Hunt, D.F.; et al. Tumor Infiltrating Lymphocytes Target HLA-I Phosphopeptides Derived From Cancer Signaling in Colorectal Cancer. Front. Immunol. 2021, 12, 723566. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, M.; De, H.; Peña, L.; Norris, A.; Polefrone, J.M.; Qian, J.; English, A.M.; Cummings, K.L.; Penny, S.; Turner, J.E.; et al. MHC Class I-Associated Phosphopeptides Are the Targets of Memory-like Immunity in Leukemia. Sci. Transl. Med. 2013, 5, 203ra125. [Google Scholar] [CrossRef] [PubMed]
- Brentville, V.A.; Atabani, S.; Cook, K.; Durrant, L.G. Novel Tumour Antigens and the Development of Optimal Vaccine Design. Ther. Adv. Vaccines Immunother. 2018, 6, 31–47. [Google Scholar] [CrossRef]
- Kudelka, M.R.; Ju, T.; Heimburg-Molinaro, J.; Cummings, R.D. Simple Sugars to Complex Disease-Mucin-Type O-Glycans in Cancer. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 126, pp. 53–135. [Google Scholar]
- Peixoto, A.; Relvas-Santos, M.; Azevedo, R.; Lara Santos, L.; Ferreira, J.A. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front. Oncol. 2019, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Cen, Q.; Lei, H. A Review on Development of MUC1-Based Cancer Vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.E.; Bosquée, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus Placebo after Chemoradiotherapy for Stage III Non-Small-Cell Lung Cancer (START): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Quoix, E.; Lena, H.; Losonczy, G.; Forget, F.; Chouaid, C.; Papai, Z.; Gervais, R.; Ottensmeier, C.; Szczesna, A.; Kazarnowicz, A.; et al. TG4010 Immunotherapy and First-Line Chemotherapy for Advanced Non-Small-Cell Lung Cancer (TIME): Results from the Phase 2b Part of a Randomised, Double-Blind, Placebo-Controlled, Phase 2b/3 Trial. Lancet Oncol. 2016, 17, 212–223. [Google Scholar] [CrossRef]
- Mohammed, F.; Cobbold, M.; Zarling, A.L.; Salim, M.; Barrett-Wilt, G.A.; Shabanowitz, J.; Hunt, D.F.; Engelhard, V.H.; Willcox, B.E. Phosphorylation-Dependent Interaction between Antigenic Peptides and MHC Class I: A Molecular Basis for the Presentation of Transformed Self. Nat. Immunol. 2008, 9, 1236–1243. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-Restricted Phosphopeptide Antigens: Preclinical Validation and First-in-Humans Clinical Trial in Participants with High-Risk Melanoma. J. Immunother. Cancer 2020, 8, e000262. [Google Scholar] [CrossRef] [PubMed]
- Feitsma, A.L.; van der Voort, E.I.H.; Franken, K.L.M.C.; el Bannoudi, H.E.; Elferink, B.G.; Drijfhout, J.W.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M.; Ioan-Facsinay, A. Identification of Citrullinated Vimentin Peptides as T Cell Epitopes in HLA-DR4-Positive Patients with Rheumatoid Arthritis. Arthritis Rheum. 2010, 62, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, C.; Dubnovitsky, A.; Sandin, C.; Kozhukh, G.; Uchtenhagen, H.; James, E.A.; Rönnelid, J.; Ytterberg, A.J.; Pieper, J.; Reed, E.; et al. Functional and Structural Characterization of a Novel HLA-DRB1*04: 01-Restricted α-Enolase T Cell Epitope in Rheumatoid Arthritis. Front. Immunol. 2016, 7, 494. [Google Scholar] [CrossRef]
- Brentville, V.A.; Metheringham, R.L.; Gunn, B.; Symonds, P.; Daniels, I.; Gijon, M.; Cook, K.; Xue, W.; Durrant, L.G. Citrullinated Vimentin Presented on MHC-II in Tumor Cells Is a Target for CD4+ T-Cell-Mediated Antitumor Immunity. Cancer Res. 2016, 76, 548–560. [Google Scholar] [CrossRef]
- Brentville, V.A.; Metheringham, R.L.; Daniels, I.; Atabani, S.; Symonds, P.; Cook, K.W.; Vankemmelbeke, M.; Choudhury, R.; Vaghela, P.; Gijon, M.; et al. Combination Vaccine Based on Citrullinated Vimentin and Enolase Peptides Induces Potent CD4-Mediated Anti-Tumor Responses. J. Immunother. Cancer 2020, 8, e000560. [Google Scholar] [CrossRef]
- Hanada, K.-I.; Yewdell, J.W.; Yang, J.C. Immune Recognition of a Human Renal Cancer Antigen through Post-Translational Protein Splicing. Nature 2004, 427, 252–256. [Google Scholar] [CrossRef]
- Vigneron, N.; Stroobant, V.; Chapiro, J.; Ooms, A.; Degiovanni, G.; Morel, S.; van der Bruggen, P.; Boon, T.; van den Eynde, B.J. An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science 2004, 304, 587–590. [Google Scholar] [CrossRef]
- Warren, E.H.; Vigneron, N.J.; Gavin, M.A.; Coulie, P.G.; Stroobant, V.; Dalet, A.; Tykodi, S.S.; Xuereb, S.M.; Mito, J.K.; Riddell, S.R.; et al. An Antigen Produced by Splicing of Noncontiguous Peptides in the Reverse Order. Science 2006, 313, 1444–1447. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A Large Fraction of HLA Class Iligands Are Proteasome-Generatedspliced Peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef]
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the Contribution of Proteasomal Spliced Peptides to the HLA-I Ligandome. Mol. Cell. Proteom. 2018, 17, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Liepe, J.; Mishto, M.; Textoris-Taube, K.; Janek, K.; Keller, C.; Henklein, P.; Kloetzel, P.M.; Zaikin, A. The 20S Proteasome Splicing Activity Discovered by SpliceMet. PLoS Comput. Biol. 2010, 6, e1000830. [Google Scholar] [CrossRef] [PubMed]
- Specht, G.; Roetschke, H.P.; Mansurkhodzhaev, A.; Henklein, P.; Textoris-Taube, K.; Urlaub, H.; Mishto, M.; Liepe, J. Large Database for the Analysis and Prediction of Spliced and Non-Spliced Peptide Generation by Proteasomes. Sci. Data 2020, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, Z.; Solntsev, S.K.; Shortreed, M.R.; Frey, B.L.; Smith, L.M. Global Identification of Post-Translationally Spliced Peptides with Neo-Fusion. J. Proteom. Res. 2019, 18, 349–358. [Google Scholar] [CrossRef]
- Dalet, A.; Robbins, P.F.; Stroobant, V.; Vigneron, N.; Li, Y.F.; El-Gamil, M.; Hanada, K.I.; Yang, J.C.; Rosenberg, S.A.; van den Eyndea, B.J. An Antigenic Peptide Produced by Reverse Splicing and Double Asparagine Deamidation. Proc. Natl. Acad. Sci. USA 2011, 108, E323–E331. [Google Scholar] [CrossRef]
- Michaux, A.; Larrieu, P.; Stroobant, V.; Fonteneau, J.-F.; Jotereau, F.; van den Eynde, B.J.; Moreau-Aubry, A.; Vigneron, N. A Spliced Antigenic Peptide Comprising a Single Spliced Amino Acid Is Produced in the Proteasome by Reverse Splicing of a Longer Peptide Fragment Followed by Trimming. J. Immunol. 2014, 192, 1962–1971. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Lopez, J.S.; Camidge, R.; Iafolla, M.; Rottey, S.; Schuler, M.; Hellmann, M.; Balmanoukian, A.; Dirix, L.; Gordon, M.; Sullivan, R.; et al. Abstract CT301: A Phase Ib Study to Evaluate RO7198457, an Individualized Neoantigen Specific ImmunoTherapy (INeST), in Combination with Atezolizumab in Patients with Locally Advanced or Metastatic Solid Tumors. Cancer Res. 2020, 80, CT301. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef]
- Claeys, A.; Luijts, T.; Marchal, K.; van den Eynden, J. Low Immunogenicity of Common Cancer Hot Spot Mutations Resulting in False Immunogenic Selection Signals. PLoS Genet. 2021, 17, e1009368. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological Ignorance Is an Enabling Feature of the Oligo-Clonal T Cell Response to Melanoma Neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Vormehr, M.; Reinhard, K.; Blatnik, R.; Josef, K.; Beck, J.D.; Salomon, N.; Suchan, M.; Selmi, A.; Vascotto, F.; Zerweck, J.; et al. A Non-Functional Neoepitope Specific CD8 + T-Cell Response Induced by Tumor Derived Antigen Exposure in Vivo. Oncoimmunology 2019, 8, 1553478. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Al-Bakir, M.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Lo, A.A.; Wallace, A.; Oreper, D.; Lounsbury, N.; Havnar, C.; Pechuan-Jorge, X.; Wu, T.D.; Bourgon, R.; Jones, R.; Krogh, K.; et al. Indication-Specific Tumor Evolution and Its Impact on Neoantigen Targeting and Biomarkers for Individualized Cancer Immunotherapies. J. Immunother. Cancer 2021, 9, e003001. [Google Scholar] [CrossRef] [PubMed]
- Sarivalasis, A.; Boudousquié, C.; Balint, K.; Stevenson, B.J.; Gannon, P.O.; Iancu, E.M.; Rossier, L.; Martin Lluesma, S.; Mathevet, P.; Sempoux, C.; et al. A Phase I/II Trial Comparing Autologous Dendritic Cell Vaccine Pulsed Either with Personalized Peptides (PEP-DC) or with Tumor Lysate (OC-DC) in Patients with Advanced High-Grade Ovarian Serous Carcinoma. J. Transl. Med. 2019, 17, 391. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma—A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen Identification Strategies Enable Personalized Immunotherapy in Refractory Solid Tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II Neoantigens Shape Tumour Immunity and Response to Immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Congdon, K.L.; Nair, S.K.; Li, Q.J.; Herndon, J.E.; Suryadevara, C.M.; Riccione, K.A.; Archer, G.E.; Norberg, P.K.; Sanchez-Perez, L.A.; et al. A Conjoined Universal Helper Epitope Can Unveil Antitumor Effects of a Neoantigen Vaccine Targeting an MHC Class I-Restricted Neoepitope. NPJ Vaccines 2021, 6, 12. [Google Scholar] [CrossRef]
- Vauchy, C.; Gamonet, C.; Ferrand, C.; Daguindau, E.; Galaine, J.; Beziaud, L.; Chauchet, A.; Henry Dunand, C.J.; Deschamps, M.; Rohrlich, P.S.; et al. CD20 Alternative Splicing Isoform Generates Immunogenic CD4 Helper T Epitopes. Int. J. Cancer 2015, 137, 116–126. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Cathcart, K.; Pinilla-Ibarz, J.; Korontsvit, T.; Schwartz, J.; Zakhaleva, V.; Papadopoulos, E.B.; Scheinberg, D.A. A Multivalent Bcr-Abl Fusion Peptide Vaccination Trial in Patients with Chronic Myeloid Leukemia. Blood 2004, 103, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capietto, A.-H.; Hoshyar, R.; Delamarre, L. Sources of Cancer Neoantigens beyond Single-Nucleotide Variants. Int. J. Mol. Sci. 2022, 23, 10131. https://doi.org/10.3390/ijms231710131
Capietto A-H, Hoshyar R, Delamarre L. Sources of Cancer Neoantigens beyond Single-Nucleotide Variants. International Journal of Molecular Sciences. 2022; 23(17):10131. https://doi.org/10.3390/ijms231710131
Chicago/Turabian StyleCapietto, Aude-Hélène, Reyhane Hoshyar, and Lélia Delamarre. 2022. "Sources of Cancer Neoantigens beyond Single-Nucleotide Variants" International Journal of Molecular Sciences 23, no. 17: 10131. https://doi.org/10.3390/ijms231710131
APA StyleCapietto, A.-H., Hoshyar, R., & Delamarre, L. (2022). Sources of Cancer Neoantigens beyond Single-Nucleotide Variants. International Journal of Molecular Sciences, 23(17), 10131. https://doi.org/10.3390/ijms231710131