

Hypoxia Triggers TAZ Phosphorylation in Basal A Triple Negative Breast Cancer Cells

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
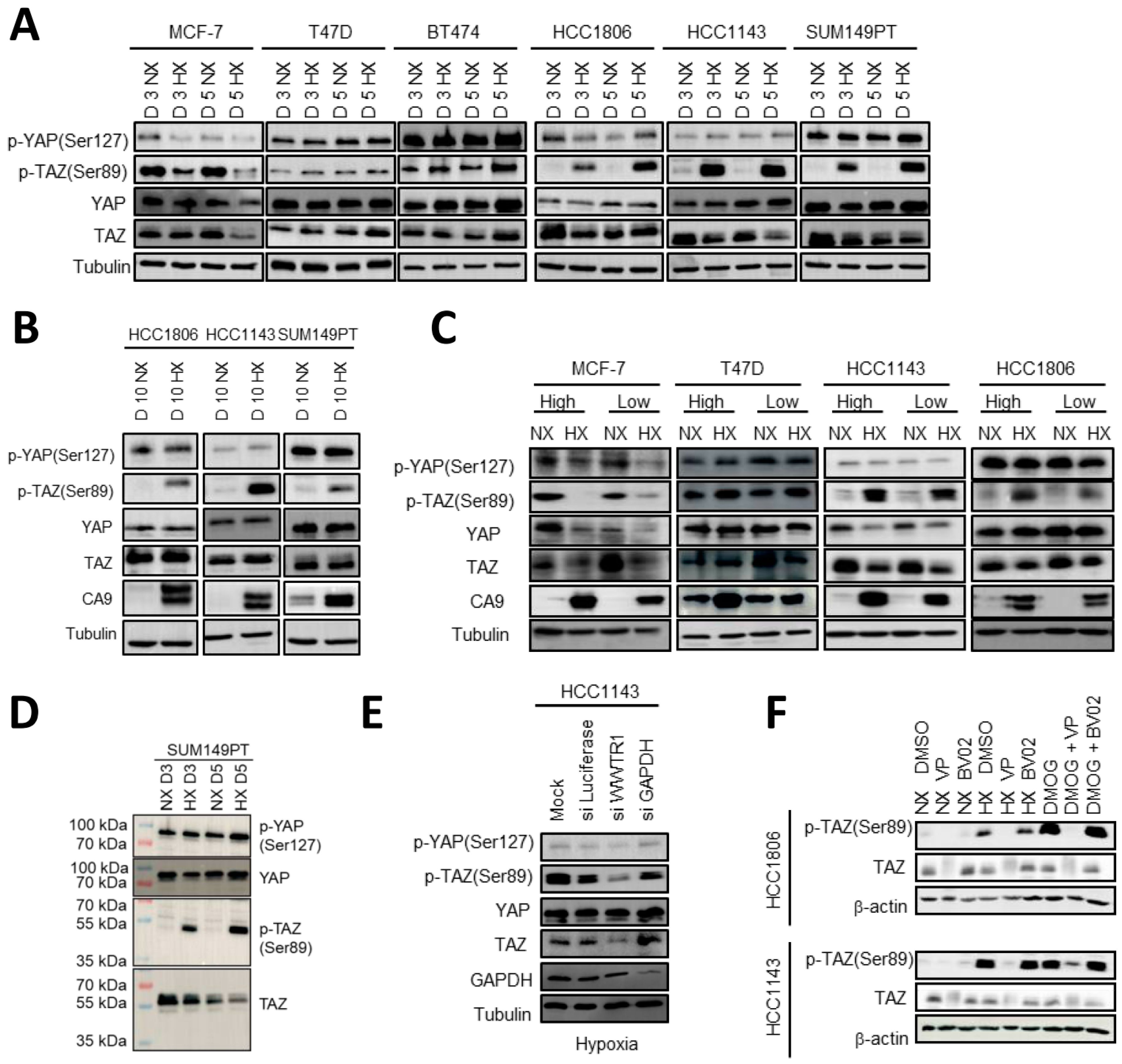
2.1. Shared and Distinct Responses to Hypoxia in Luminal and Basal A Breast Cancer Cells
2.2. Expression of YAP/TAZ/TEAD Complex in Breast Cancer Subtypes and Response to Hypoxia
2.3. Shared and Distinct Changes in Expression of YAP/TAZ/TEAD Target Genes in Response to Hypoxia in Luminal and Basal A Breast Cancer Cells
2.4. HIF1 Mediated TAZ Phosphorylation at Ser89 in Basal A but Not Luminal Breast Cancer Cells
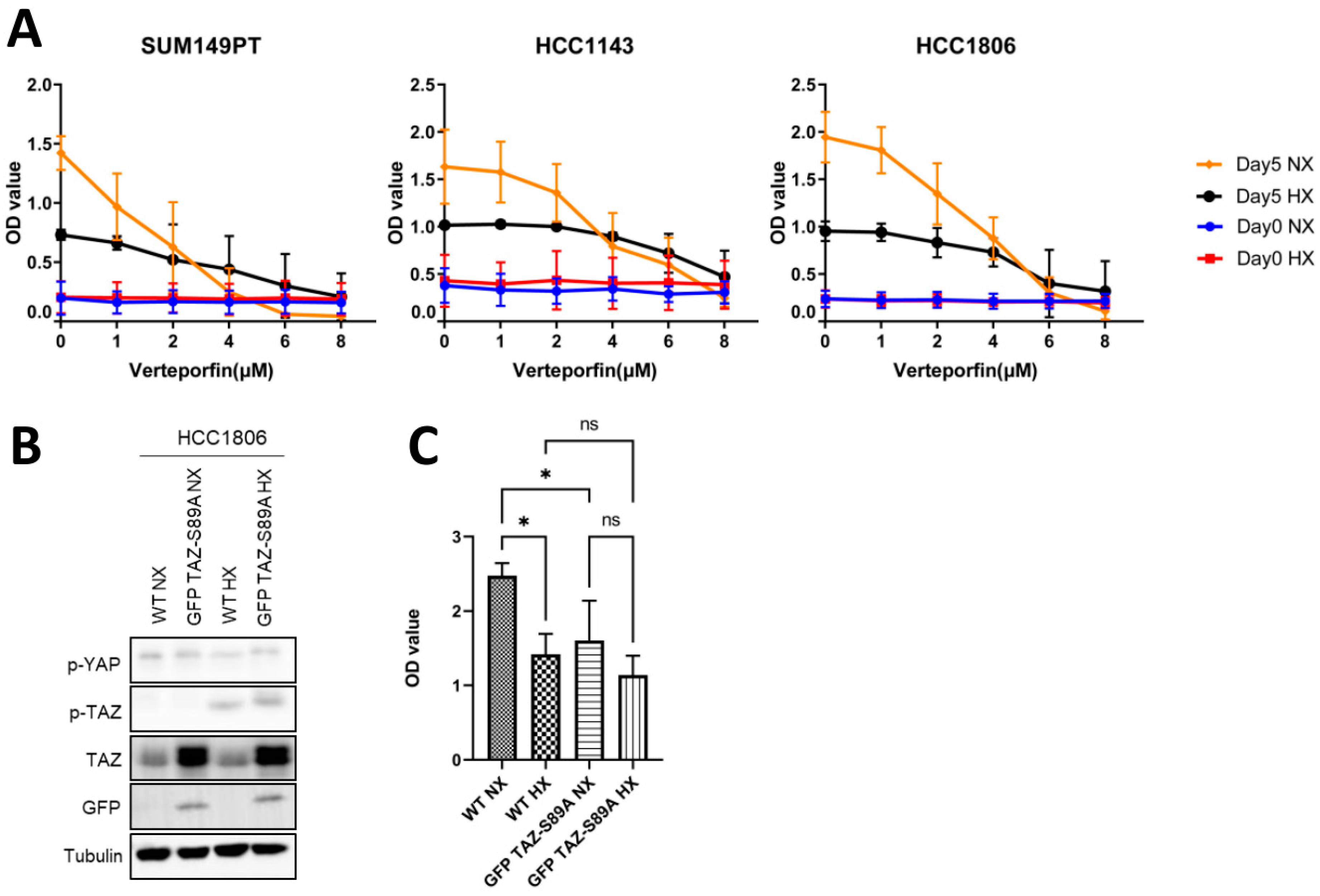
2.5. Expression of a TAZ-S89A Mutant Does Not Prevent Growth Reduction in Basal A Cells under Hypoxia
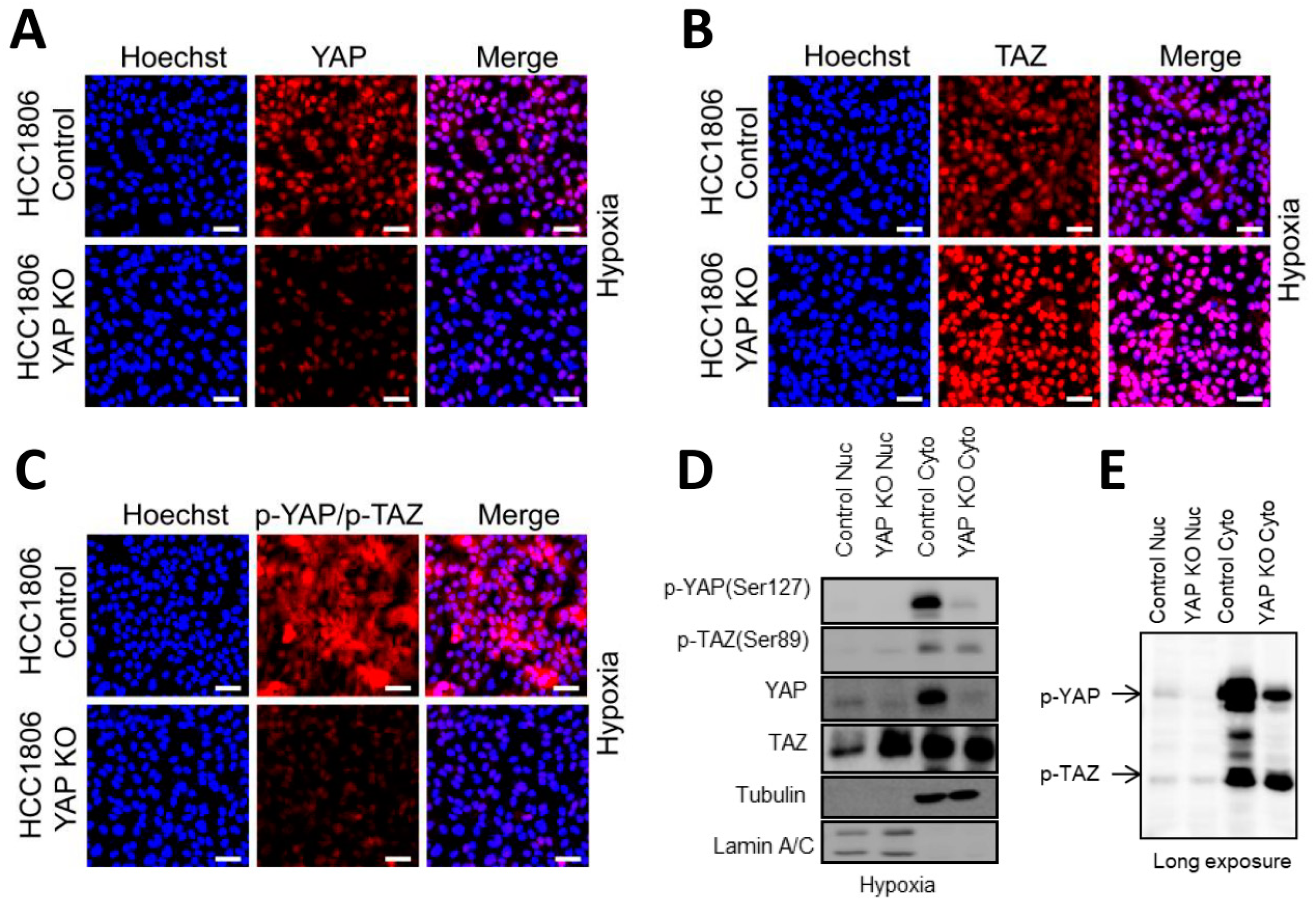
2.6. Basal A Specific TAZ (Ser89) Phosphorylation Is Accompanied by Cytoplasmic Localization in Hypoxia
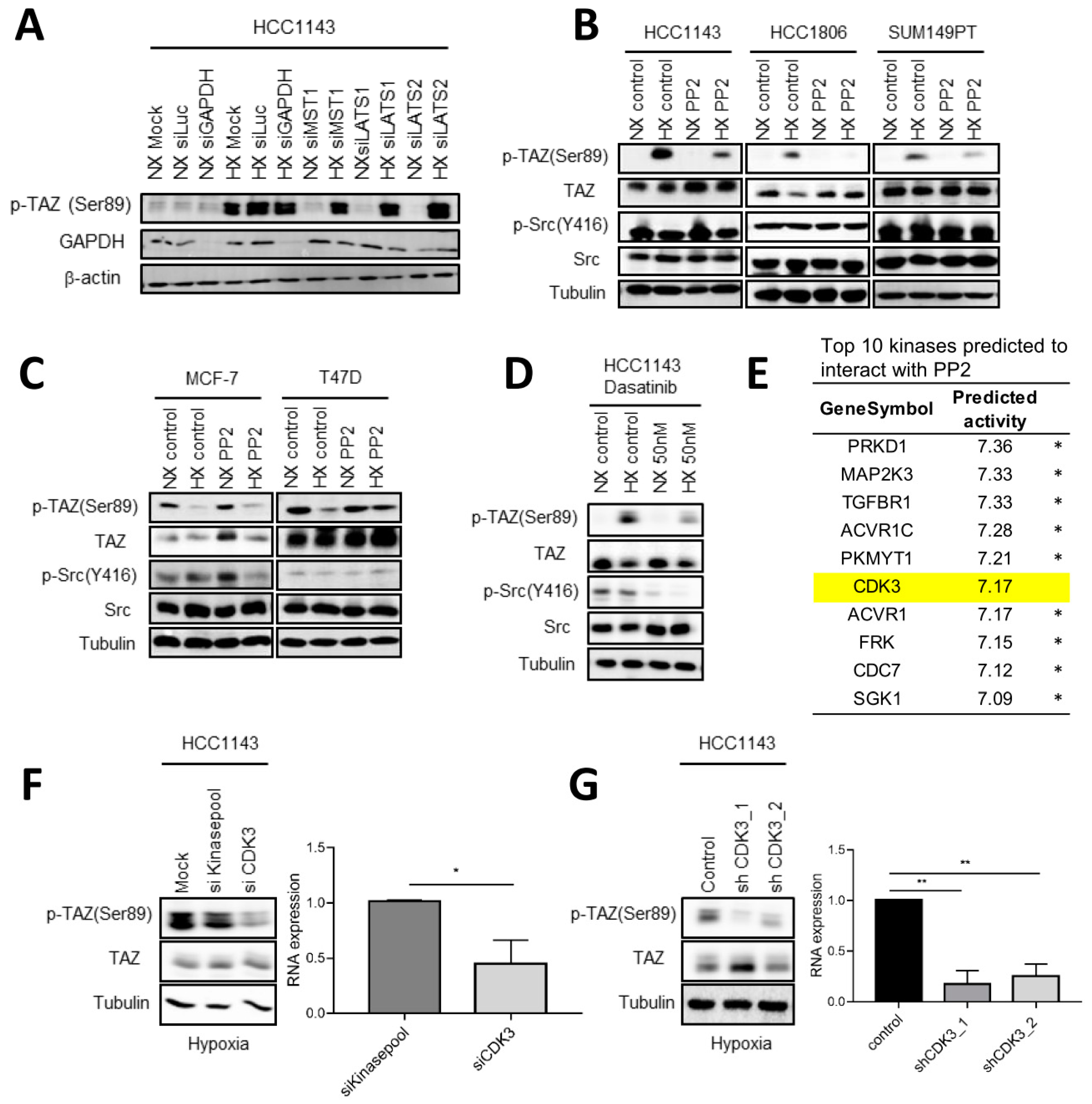
2.7. Involvement of c-Src and CDK3 in TAZ Ser89 Phosphorylation under Hypoxia
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Reagents, and Antibodies
4.2. Sulforhodamine B (SRB) Assay
4.3. Gene Silencing, Deletion, and Ectopic Expression
4.4. Western Blot, Cell Fractionation and Immunofluorescence Microscopy
4.5. TempO-Seq and RT-qPCR
4.6. RNA-Seq and TempO-Seq Data Analysis in Cell Lines and Clinical Samples
4.7. Kinase-Substrate Predictions of PP2 and Dasatinib
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, W.F.; Rosenberg, P.S.; Prat, A.; Perou, C.M.; Sherman, M.E. How many etiological subtypes of breast cancer: Two, three, four, or more? J. Natl. Cancer Inst. 2014, 106, dju165. [Google Scholar] [CrossRef]
- Ahn, H.J.; Jung, S.J.; Kim, T.H.; Oh, M.K.; Yoon, H.-K. Differences in Clinical Outcomes between Luminal A and B Type Breast Cancers according to the St. Gallen Consensus 2013. J. Breast Cancer 2015, 18, 149–159. [Google Scholar] [CrossRef]
- Creighton, C.J. The molecular profile of luminal B breast cancer. Biologics 2012, 6, 289–297. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef]
- Anders, C.; Carey, L.A. Understanding and treating triple-negative breast cancer. Oncology 2008, 22, 1233–1239; discussion 1239–1240, 1243. [Google Scholar]
- Fulford, L.G.; Reis-Filho, J.S.; Ryder, K.; Jones, C.; Gillett, C.E.; Hanby, A.; Easton, D.; Lakhani, S.R. Basal-like grade III invasive ductal carcinoma of the breast: Patterns of metastasis and long-term survival. Breast Cancer Res. 2007, 9, R4. [Google Scholar] [CrossRef]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef]
- Coban, B.; Bergonzini, C.; Zweemer, A.J.M.; Danen, E.H.J. Metastasis: Crosstalk between tissue mechanics and tumour cell plasticity. Br. J. Cancer 2021, 124, 49–57. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Parks, S.K.; Cormerais, Y.; Pouysségur, J. Hypoxia and cellular metabolism in tumour pathophysiology. J. Physiol. 2017, 595, 2439–2450. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- Bos, R.; van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef]
- Dales, J.-P.; Garcia, S.; Meunier-Carpentier, S.; Andrac-Meyer, L.; Haddad, O.; Lavaut, M.-N.; Allasia, C.; Bonnier, P.; Charpin, C. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 2005, 116, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin. Cancer Res. 2006, 12, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Simopoulos, C.; Polychronidis, A.; Gatter, K.C.; Harris, A.L.; Sivridis, E. c-erbB-2 related aggressiveness in breast cancer is hypoxia inducible factor-1alpha dependent. Clin. Cancer Res. 2004, 10, 7972–7977. [Google Scholar] [CrossRef] [PubMed]
- Schindl, M.; Schoppmann, S.F.; Samonigg, H.; Hausmaninger, H.; Kwasny, W.; Gnant, M.; Jakesz, R.; Kubista, E.; Birner, P.; Oberhuber, G. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 2002, 8, 1831–1837. [Google Scholar] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Preisser, F.; Giehl, K.; Rehm, M.; Goppelt-Struebe, M. Inhibitors of oxygen sensing prolyl hydroxylases regulate nuclear localization of the transcription factors Smad2 and YAP/TAZ involved in CTGF synthesis. Biochim. Biophys. Acta 2016, 1863, 2027–2036. [Google Scholar] [CrossRef]
- Yan, L.; Cai, Q.; Xu, Y. Hypoxic conditions differentially regulate TAZ and YAP in cancer cells. Arch. Biochem. Biophys. 2014, 562, 31–36. [Google Scholar] [CrossRef]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Takano, N.; Luo, W.; Lu, H.; Bullen, J.W.; Samanta, D.; Liang, H.; Semenza, G.L. Hypoxia-inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget 2014, 5, 12509–12527. [Google Scholar] [CrossRef]
- Xiang, L.; Gilkes, D.M.; Hu, H.; Luo, W.; Bullen, J.W.; Liang, H.; Semenza, G.L. HIF-1α and TAZ serve as reciprocal co-activators in human breast cancer cells. Oncotarget 2015, 6, 11768–11778. [Google Scholar] [CrossRef]
- Qiu, G.-Z.; Jin, M.-Z.; Dai, J.-X.; Sun, W.; Feng, J.-H.; Jin, W.-L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Zeng, C.; Ye, S.; Dai, X.; He, Q.; Yang, B.; Zhu, H. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): A nexus between hypoxia and cancer. Acta Pharm. Sin. B 2020, 10, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Koedoot, E.; Wolters, L.; Smid, M.; Stoilov, P.; Burger, G.A.; Herpers, B.; Yan, K.; Price, L.S.; Martens, J.W.M.; Le Dévédec, S.E.; et al. Differential reprogramming of breast cancer subtypes in 3D cultures and implications for sensitivity to targeted therapy. Sci. Rep. 2021, 11, 7259. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-K.; Jang, J.-W.; Bae, S.-C. DNA binding partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef]
- Höffken, V.; Hermann, A.; Pavenstädt, H.; Kremerskothen, J. WWC Proteins: Important Regulators of Hippo Signaling in Cancer. Cancers 2021, 13, 306. [Google Scholar] [CrossRef]
- Nayak, A.; Dutta, M.; Roychowdhury, A. Emerging oncogene ATAD2: Signaling cascades and therapeutic initiatives. Life Sci. 2021, 276, 119322. [Google Scholar] [CrossRef]
- Liu, H.; Du, S.; Lei, T.; Wang, H.; He, X.; Tong, R.; Wang, Y. Multifaceted regulation and functions of YAP/TAZ in tumors (Review). Oncol. Rep. 2018, 40, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jho, E.-H. Regulation of the Hippo signaling pathway by ubiquitin modification. BMB Rep. 2018, 51, 143–150. [Google Scholar] [CrossRef]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Han, X.; Chen, J.; Xie, X.; Xu, J.; Zhao, Y.; Shen, J.; Hu, L.; Xu, P.; Song, H.; et al. Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) mediate cell density-dependent proinflammatory responses. J. Biol. Chem. 2018, 293, 18071–18085. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Lu, C.-Y.; Cheng, T.-Y.; Pan, S.-H.; Chen, H.-F.; Chang, N.-S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef]
- Anwar, T.; Sinnett-Smith, J.; Jin, Y.-P.; Reed, E.F.; Rozengurt, E. Ligation of HLA Class I Molecules Induces YAP Activation through Src in Human Endothelial Cells. J. Immunol. 2020, 205, 1953–1961. [Google Scholar] [CrossRef]
- Byun, M.R.; Hwang, J.-H.; Kim, A.R.; Kim, K.M.; Park, J.I.; Oh, H.T.; Hwang, E.S.; Hong, J.-H. SRC activates TAZ for intestinal tumorigenesis and regeneration. Cancer Lett. 2017, 410, 32–40. [Google Scholar] [CrossRef]
- Guillermin, O.; Angelis, N.; Sidor, C.M.; Ridgway, R.; Baulies, A.; Kucharska, A.; Antas, P.; Rose, M.R.; Cordero, J.; Sansom, O.; et al. Wnt and Src signals converge on YAP-TEAD to drive intestinal regeneration. EMBO J. 2021, 40, e105770. [Google Scholar] [CrossRef]
- Kim, J.; Kang, W.; Kang, S.H.; Park, S.H.; Kim, J.Y.; Yang, S.; Ha, S.Y.; Paik, Y.-H. Proline-rich tyrosine kinase 2 mediates transforming growth factor-beta-induced hepatic stellate cell activation and liver fibrosis. Sci. Rep. 2020, 10, 21018. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.-G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, J.; Zhao, X.; Wu, T.; Huang, Z.; Chen, D.; Liu, Y.; Ouyang, G. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep. 2020, 30, 793–806.e6. [Google Scholar] [CrossRef]
- Shanzer, M.; Adler, J.; Ricardo-Lax, I.; Reuven, N.; Shaul, Y. The nonreceptor tyrosine kinase c-Src attenuates SCF(β-TrCP) E3-ligase activity abrogating Taz proteasomal degradation. Proc. Natl. Acad. Sci. USA 2017, 114, 1678–1683. [Google Scholar] [CrossRef]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.P.; Rundsten, C.F.; et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35–49.e7. [Google Scholar] [CrossRef]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Fresques, T.; LaBarge, M.A. Contributions of Yap and Taz dysfunction to breast cancer initiation, progression, and aging-related susceptibility. Aging Cancer 2020, 1, 5–18. [Google Scholar] [CrossRef]
- Tamm, C.; Böwer, N.; Annerén, C. Regulation of mouse embryonic stem cell self-renewal by a Yes-YAP-TEAD2 signaling pathway downstream of LIF. J. Cell Sci. 2011, 124, 1136–1144. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Yeakley, J.M.; Shepard, P.J.; Goyena, D.E.; VanSteenhouse, H.C.; McComb, J.D.; Seligmann, B.E. A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS ONE 2017, 12, e0178302. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Iorio, F.; Matchan, A.; Fonseca, N.; Jaaks, P.; Peat, G.; Pignatelli, M.; Falcone, F.; Benes, C.H.; Dunham, I.; et al. Transcription Factor Activities Enhance Markers of Drug Sensitivity in Cancer. Cancer Res. 2018, 78, 769–780. [Google Scholar] [CrossRef]
- Van der Stel, W.; Yang, H.; Vrijenhoek, N.G.; Schimming, J.P.; Callegaro, G.; Carta, G.; Darici, S.; Delp, J.; Forsby, A.; White, A.; et al. Mapping the cellular response to electron transport chain inhibitors reveals selective signaling networks triggered by mitochondrial perturbation. Arch. Toxicol. 2022, 96, 259–285. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Zhu, Y.; Qiu, P.; Ji, Y. TCGA-assembler: Open-source software for retrieving and processing TCGA data. Nat. Methods 2014, 11, 599–600. [Google Scholar] [CrossRef]
- Koedoot, E.; Smid, M.; Foekens, J.A.; Martens, J.W.M.; Le Dévédec, S.E.; van de Water, B. Co-regulated gene expression of splicing factors as drivers of cancer progression. Sci. Rep. 2019, 9, 5484. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, M.; Rännar, S.; Wieslander, Å. Polypeptide sequence property relationships in Escherichia coli based on auto cross covariances. Chemom. Intell. Lab. Syst. 1995, 29, 295–305. [Google Scholar] [CrossRef]
- Lenselink, E.B.; ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; IJzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef]
- Georgiev, A.G. Interpretable numerical descriptors of amino acid space. J. Comput. Biol. 2009, 16, 703–723. [Google Scholar] [CrossRef]
- van Westen, G.J.; Swier, R.F.; Wegner, J.K.; IJzerman, A.P.; van Vlijmen, H.W.; Bender, A. Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): Comparative study of 13 amino acid descriptor sets. J. Cheminform. 2013, 5, 41. [Google Scholar] [CrossRef]
- Zhou, P.; Zhou, Y.; Wu, S.; Li, B.; Tian, F.; Li, Z. A new descriptor of amino acids based on the three-dimensional vector of atomic interaction field. Chin. Sci. Bull. 2006, 51, 524–529. [Google Scholar] [CrossRef]
- Hellberg, S.; Sjöström, M.; Skagerberg, B.; Wold, S. Peptide quantitative structure-activity relationships, a multivariate approach. J. Med. Chem. 1987, 30, 1126–1135. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost. In Proceedings of the KDD ‘16: The 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Krishnapuram, B., Shah, M., Smola, A., Aggarwal, C., Shen, D., Rastogi, R., Eds.; ACM: New York, NY, USA, 2016; pp. 785–794, ISBN 9781450342322. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Stel, W.v.d.; Noord, V.E.v.d.; Leegwater, H.; Coban, B.; Elbertse, K.; Pruijs, J.T.M.; Béquignon, O.J.M.; Westen, G.v.; Dévédec, S.E.L.; et al. Hypoxia Triggers TAZ Phosphorylation in Basal A Triple Negative Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 10119. https://doi.org/10.3390/ijms231710119
Liu Q, Stel Wvd, Noord VEvd, Leegwater H, Coban B, Elbertse K, Pruijs JTM, Béquignon OJM, Westen Gv, Dévédec SEL, et al. Hypoxia Triggers TAZ Phosphorylation in Basal A Triple Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(17):10119. https://doi.org/10.3390/ijms231710119
Chicago/Turabian StyleLiu, Qiuyu, Wanda van der Stel, Vera E. van der Noord, Hanneke Leegwater, Bircan Coban, Kim Elbertse, Joannes T. M. Pruijs, Olivier J. M. Béquignon, Gerard van Westen, Sylvia E. Le Dévédec, and et al. 2022. "Hypoxia Triggers TAZ Phosphorylation in Basal A Triple Negative Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 17: 10119. https://doi.org/10.3390/ijms231710119
APA StyleLiu, Q., Stel, W. v. d., Noord, V. E. v. d., Leegwater, H., Coban, B., Elbertse, K., Pruijs, J. T. M., Béquignon, O. J. M., Westen, G. v., Dévédec, S. E. L., & Danen, E. H. J. (2022). Hypoxia Triggers TAZ Phosphorylation in Basal A Triple Negative Breast Cancer Cells. International Journal of Molecular Sciences, 23(17), 10119. https://doi.org/10.3390/ijms231710119