
Small RNA-Seq Reveals Similar miRNA Transcriptome in Children and Young Adults with T-ALL and Indicates miR-143-3p as Novel Candidate Tumor Suppressor in This Leukemia

 , , , , , ,
, , , , , ,  , , ,
, , , 
Abstract
:
1. Introduction
2. Results
2.1. Overview of miRNA Transcriptome in AYA Patients
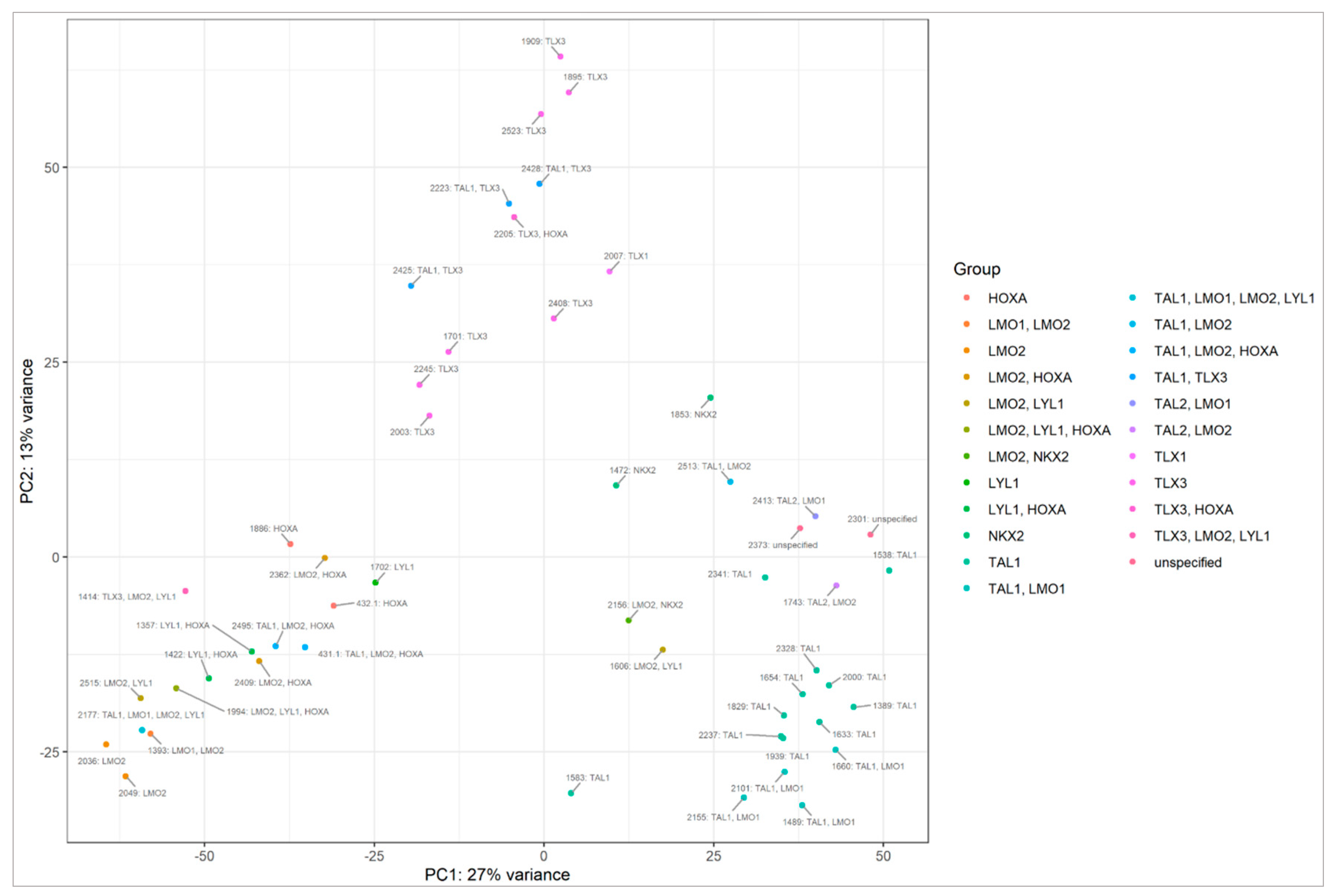
2.2. miRNA Transcriptome Does Not Clearly Discriminate AYA and Pediatric T-ALL
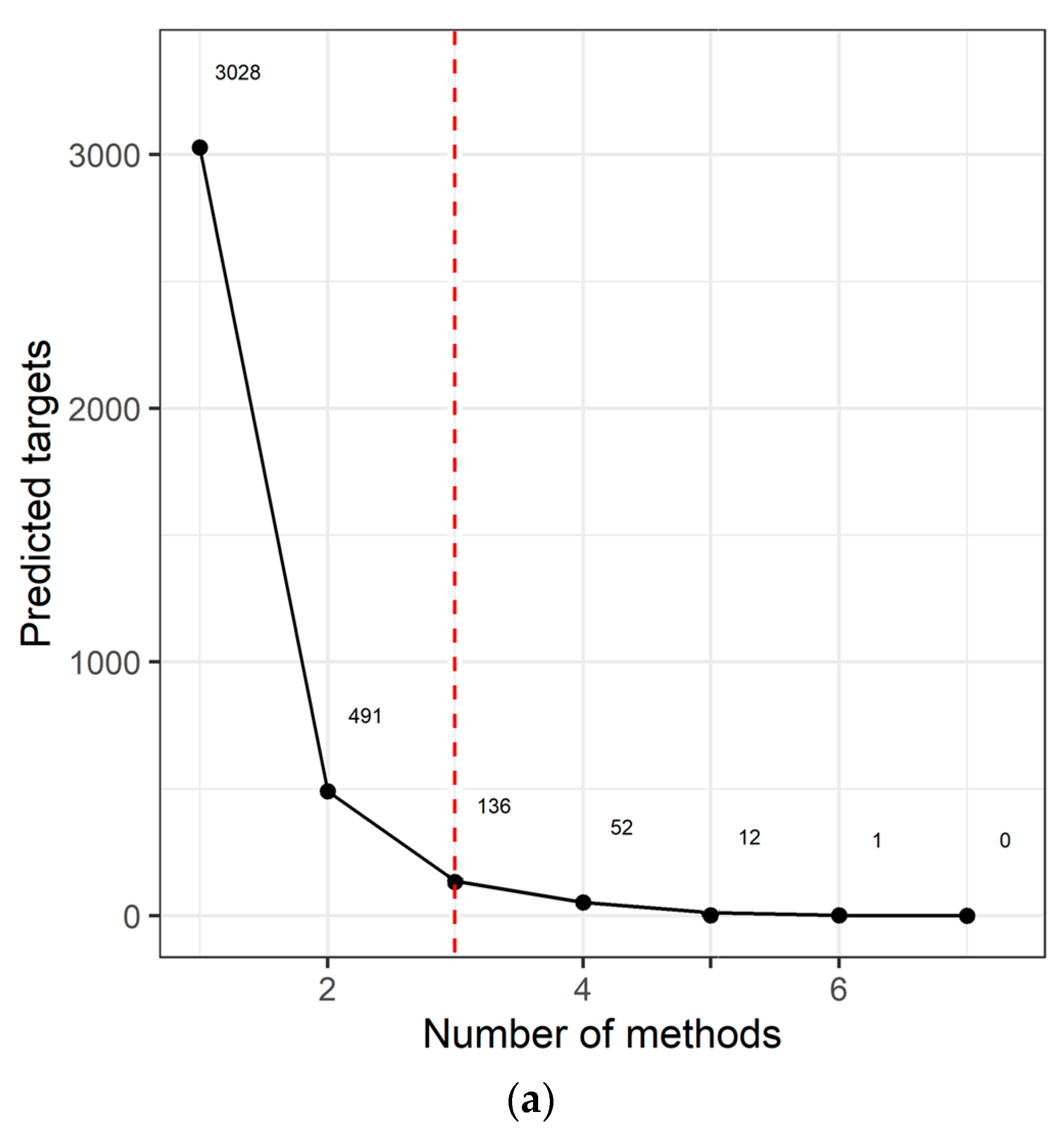
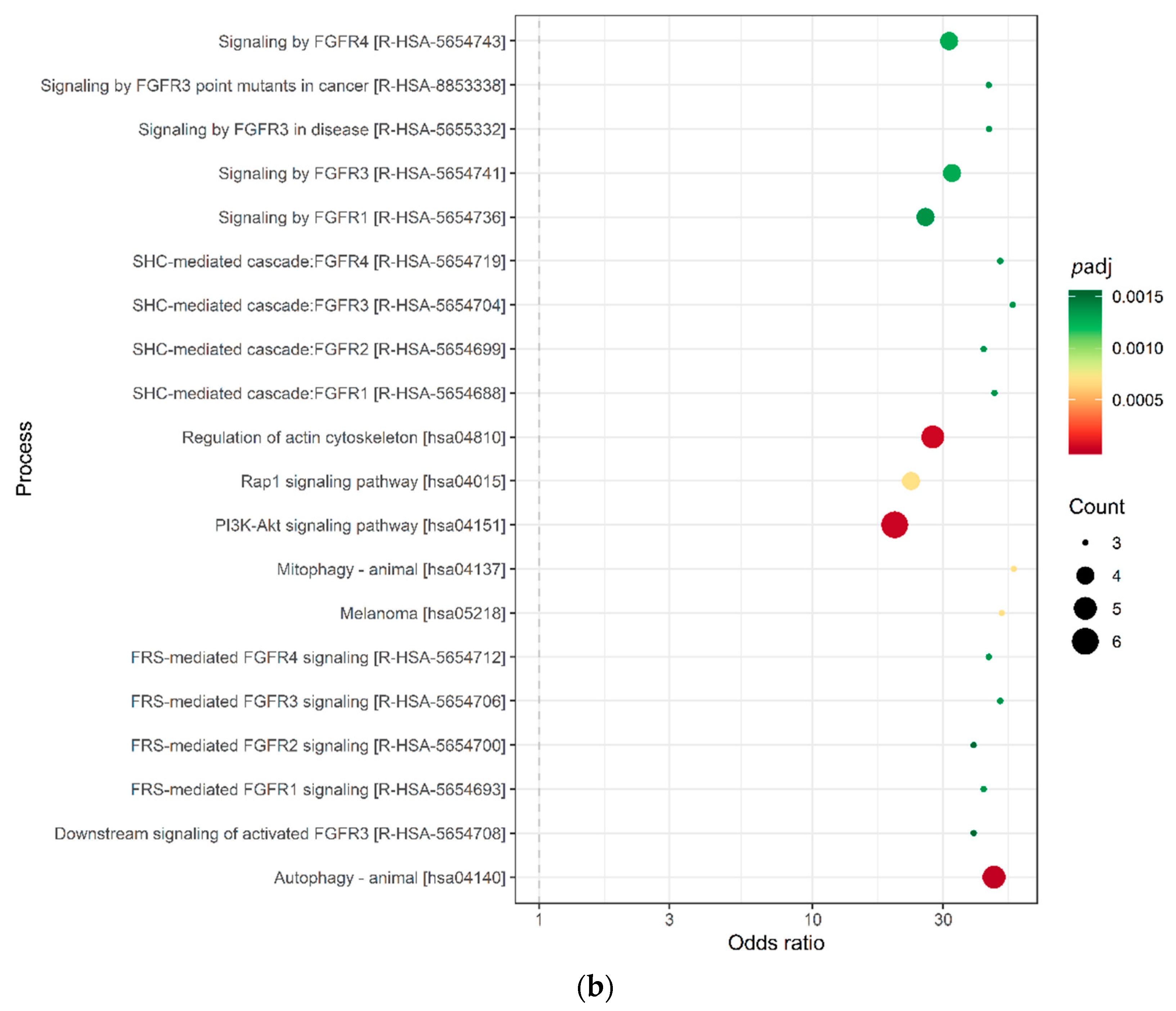
2.3. Target Prediction and Pathway Analysis Indicate Deregulation of Cancer-Related Signaling Pathways
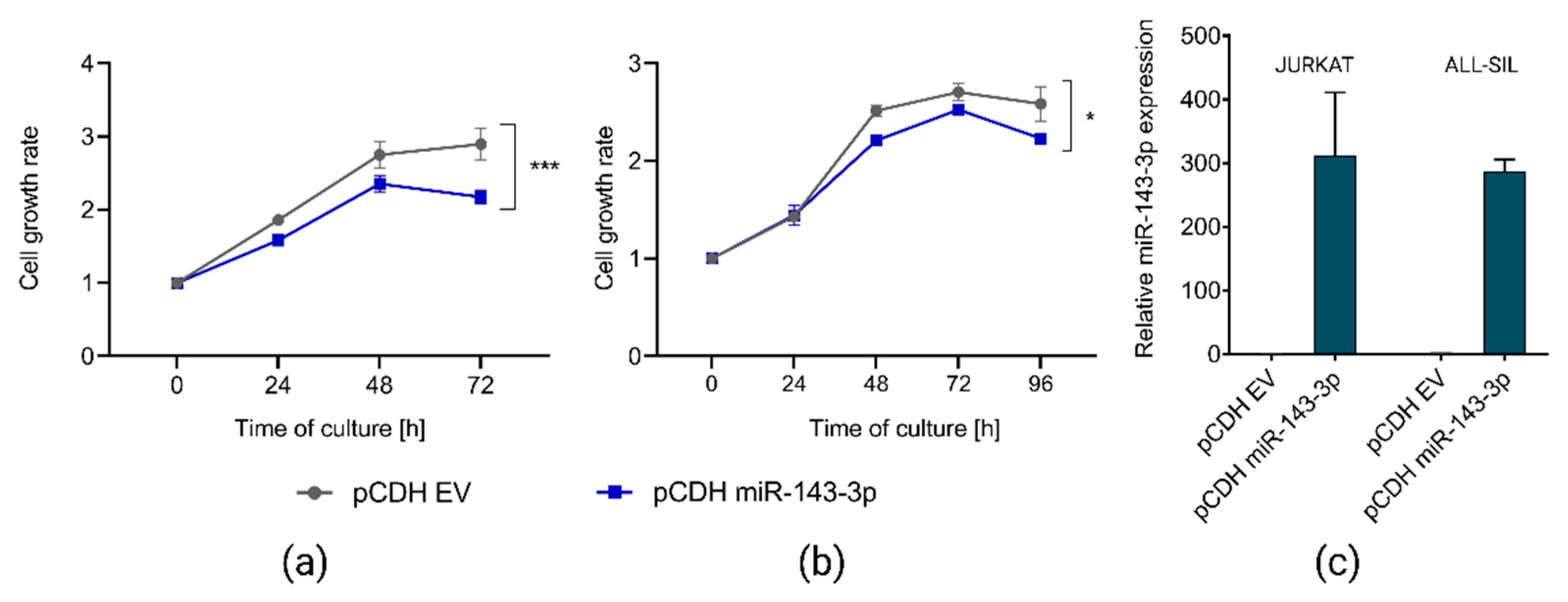
2.4. miR-143-3p Affects Proliferation of T-ALL Cells In Vitro
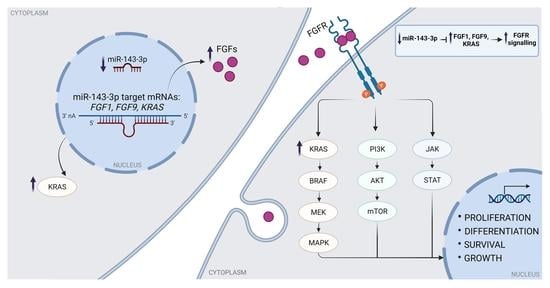
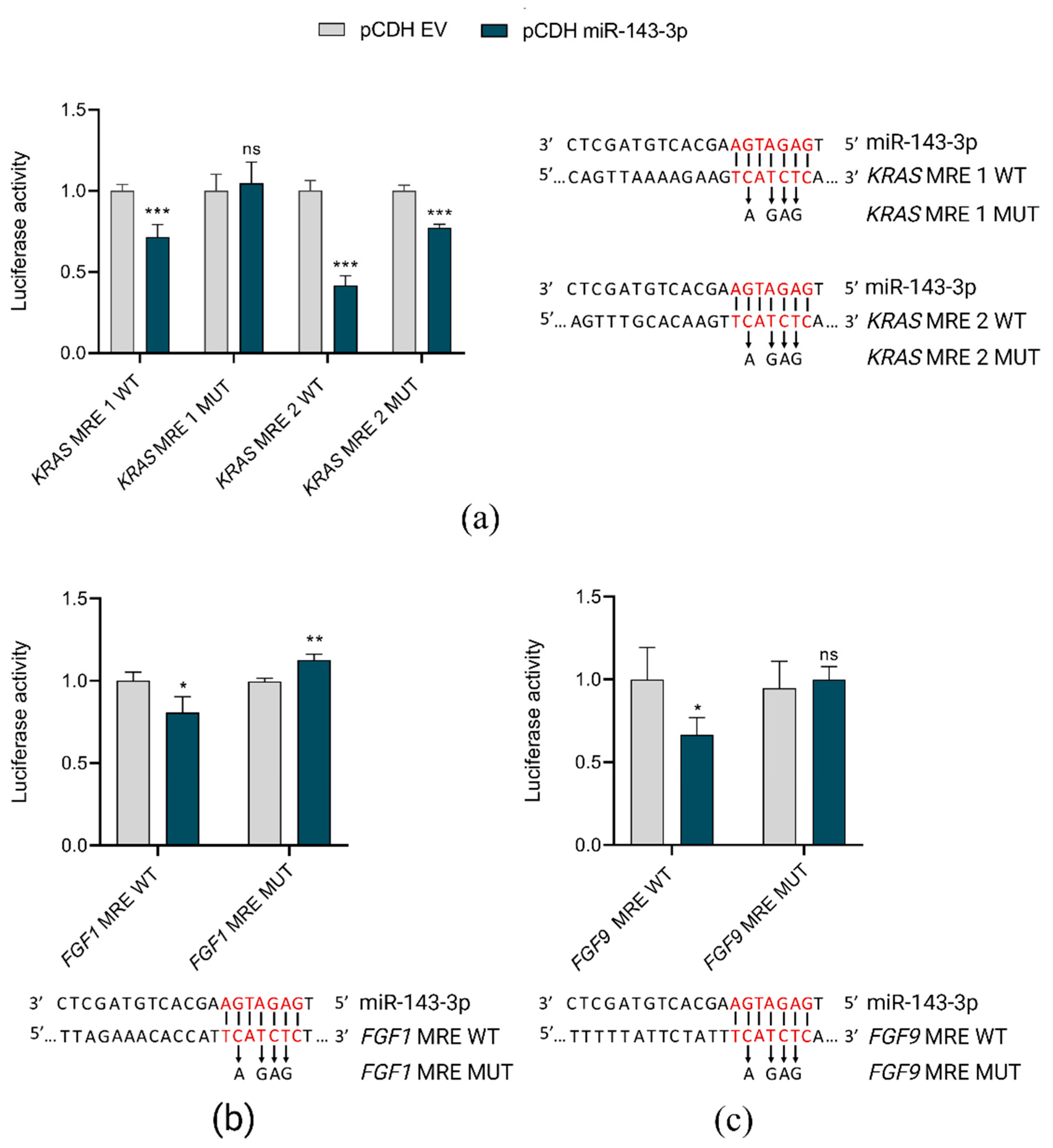
2.5. miR-143-3p Interacts with FGF1, FGF9 and KRAS
3. Discussion
Limitations
4. Materials and Methods
4.1. T-ALL Primary Samples and Controls
4.2. T-ALL Cell Culture In Vitro
4.3. Small RNA Sequencing and Bioinformatics Analyses
4.4. RT-qPCR Validation of miRNAs’ Expression
4.5. miR-143-3p Overexpression in T-ALL Cells In Vitro
4.6. Proliferation Assays
4.7. Dual Luciferase Reporter Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
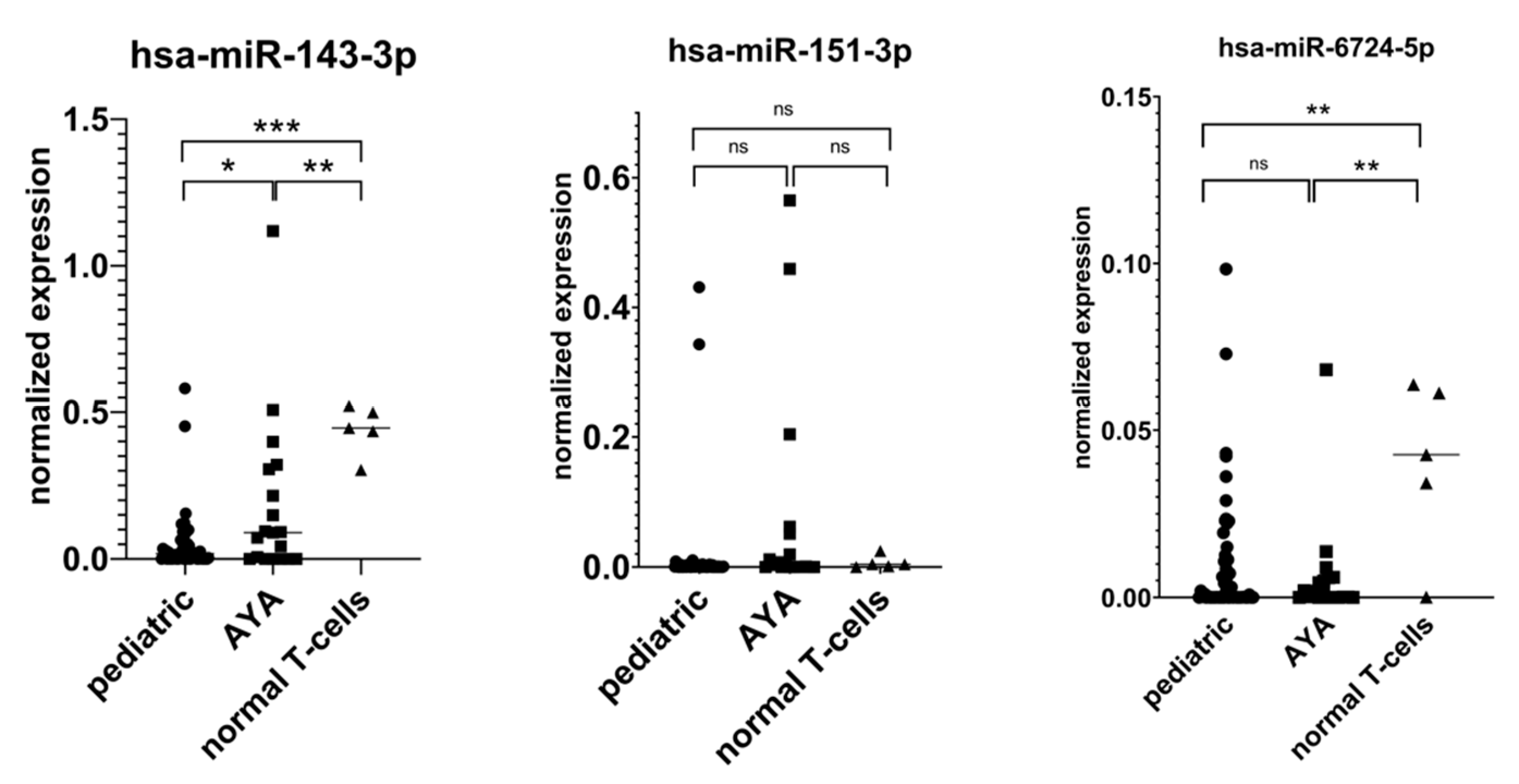
Appendix A. RT-qPCR Validation of miRNA-Seq Results
Appendix B. miRNAs Differentially Expressed in T-ALL Subtypes

References
- Muffly, L.; Alvarez, E.; Lichtensztajn, D.; Abrahão, R.; Gomez, S.L.; Keegan, T. Patterns of care and outcomes in adolescent and young adult acute lymphoblastic leukemia: A population-based study. Blood Adv. 2018, 2, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Testi, A.M.; Canichella, M.; Vitale, A.; Piciocchi, A.; Guarini, A.; Starza, I.D.; Cavalli, M.; De Propris, M.S.; Messina, M.; Elia, L.; et al. Adolescent and young adult acute lymphoblastic leukemia. Final results of the phase II pediatric-like GIMEMA LAL-1308 trial. Am. J. Hematol. 2021, 96, 292–301. [Google Scholar] [CrossRef] [PubMed]
- White, V.M.; Skaczkowski, G.; Pinkerton, R.; Coory, M.; Osborn, M.; Bibby, H.; Nicholls, W.; Orme, L.M.; Conyers, R.; Phillips, M.B.; et al. Clinical management of Australian adolescents and young adults with acute lymphoblastic and myeloid leukemias: A national population-based study. Pediatr. Blood Cancer 2018, 65, e27349. [Google Scholar] [CrossRef] [PubMed]
- Friend, B.D.; Schiller, G.J. Closing the gap: Novel therapies in treating acute lymphoblastic leukemia in adolescents and young adults. Blood Rev. 2018, 32, 122–129. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 186–190. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, L.; Zhong, M.L.; Li, J.F.; Li, B.S.; Peng, L.J.; Dai, Y.T.; Cui, B.W.; Yan, T.Q.; Zhang, W.N.; et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2018, 115, 373–378. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Correia, N.C.; Barata, J.T. MicroRNAs and their involvement in T-ALL: A brief overview. Adv. Biol. Regul. 2019, 74, 100650. [Google Scholar] [CrossRef]
- Dawidowska, M.; Jaksik, R.; Drobna, M.; Szarzyńska-Zawadzka, B.; Kosmalska, M.; Sędek, Ł.; Machowska, L.; Lalik, A.; Lejman, M.; Ussowicz, M.; et al. Comprehensive Investigation of miRNome Identifies Novel Candidate miRNA-mRNA Interactions Implicated in T-Cell Acute Lymphoblastic Leukemia. Neoplasia 2019, 21, 294–310. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Maruo, K.; Yamada, N.; Kitade, Y.; Naoe, T.; Akao, Y. MicroRNA-143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett. 2011, 307, 211–220. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Jou, S.-T.; Lin, S.-W.; Lin, D.-T.; Hu, C.-Y., Jr.; Chang, S.-K.; Yen, C.-T.; Chiou, R.-J.; Lin, K.-H.; Lu, M.-Y.; et al. Down-Regulation of MicroRNA-143 and -145 in Childhood B-Lineage Acute Lymphoblastic Leukemia at Initial Diagnosis and in Relapse but up-Regulated When in Remission. Blood 2008, 112, 4886. [Google Scholar] [CrossRef]
- Hartmann, J.U.; Bräuer-Hartmann, D.; Kardosova, M.; Wurm, A.A.; Wilke, F.; Schödel, C.; Gerloff, D.; Katzerke, C.; Krakowsky, R.; Namasu, C.Y.; et al. MicroRNA-143 targets ERK5 in granulopoiesis and predicts outcome of patients with acute myeloid leukemia. Cell Death Dis. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Z.; Zhang, Y.Y.; Fu, H.Y.; Wu, D.S.; Zhou, H.R. Overexpression of microRNA-143 inhibits growth and induces apoptosis in human leukemia cells. Oncol. Rep. 2014, 31, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Piatopoulou, D.; Avgeris, M.; Drakaki, I.; Marmarinos, A.; Xagorari, M.; Baka, M.; Pourtsidis, A.; Kossiva, L.; Gourgiotis, D.; Scorilas, A. Clinical utility of miR-143/miR-182 levels in prognosis and risk stratification specificity of BFM-treated childhood acute lymphoblastic leukemia. Ann. Hematol. 2018, 97, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.; Meckel, K.; Dougherty, U.; Butun, F.; Mustafi, R.; Lim, J.; Crofton, C.; Chen, X.; Joseph, L.; Bissonnette, M. Tumor suppressors miR-143 and miR-145 and predicted target proteins API5, ERK5, K-RAS, and IRS-1 are differentially expressed in proximal and distal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G179–G187. [Google Scholar] [CrossRef]
- Zhou, L.L.; Dong, J.L.; Huang, G.; Sun, Z.L.; Wu, J. MicroRNA-143 inhibits cell growth by targeting ERK5 and MAP3K7 in breast cancer. Braz. J. Med. Biol. Res. 2017, 50, e5891. [Google Scholar] [CrossRef]
- Szybowska, P.; Kostas, M.; Wesche, J.; Haugsten, E.M.; Wiedlocha, A. Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling. Cells 2021, 10, 1342. [Google Scholar] [CrossRef]
- Xiao, J.F.; Caliri, A.W.; Duex, J.E.; Theodorescu, D. Targetable Pathways in Advanced Bladder Cancer: FGFR Signaling. Cancers 2021, 13, 4891. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Boer, J.M.; Besselink, N.J.M.; Koudijs, M.J.; Pieters, R.; den Boer, M.L. Fibroblast growth factor receptor signaling in pediatric B-cell precursor acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 1875. [Google Scholar] [CrossRef]
- Carll, T.; Patel, A.; Derman, B.; Hyjek, E.; Lager, A.; Wanjari, P.; Segal, J.; Odenike, O.; Fidai, S.; Arber, D. Diagnosis and treatment of mixed phenotype (T-myeloid/lymphoid) acute leukemia with novel ETV6-FGFR2 rearrangement. Blood Adv. 2020, 4, 4924–4928. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e1679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, Y.; Huang, T.; Wu, F.; Pan, Y.; Dong, Y.; Wang, Y.; Chan, A.K.Y.; Liu, L.; Kwan, J.S.H.; et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene 2019, 38, 33–46. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Xia, T.; Li, G.; Tian, T.; Wang, M.; Wang, R.; Zhao, L.; Lan, K.; Zhou, W. MicroRNA-210 promotes cancer angiogenesis by targeting fibroblast growth factor receptor-like 1 in hepatocellular carcinoma. Oncol. Rep. 2016, 36, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef]
- Greulich, H.; Pollock, P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011, 17, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Greco, C.; Grossi, G.; Molinari, A.; Schenone, S. Recent Studies on Ponatinib in Cancers Other Than Chronic Myeloid Leukemia. Cancers 2018, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Padi, S.K.R.; Luevano, L.A.; An, N.; Pandey, R.; Singh, N.; Song, J.H.; Aster, J.C.; Yu, X.Z.; Mehrotra, S.; Kraft, A.S. Targeting the PIM protein kinases for the treatment of a T-cell acute lymphoblastic leukemia subset. Oncotarget 2017, 8, 30199–30216. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Luo, B.; Medeiros, B.C.; Johnson, R.C.; Ewalt, M.D.; Schalkwyk, A.S.; Bangs, C.D.; Cherry, A.M.; Arai, S.; Arber, D.A.; et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia 2016, 30, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.H.; Shin, U.S.; Kim, H.W. Fibroblast growth factors: Biology, function, and application for tissue regeneration. J. Tissue Eng. 2010, 2010, 218142. [Google Scholar] [CrossRef]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribeiro, R.; Viola, S.K.; Edwards, D.K.; Agarwal, A.; Lopez, C.; Jorgens, D.; et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. Elife 2019, 8, e40033. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.; Fahy, L.; Uzan, B.; Pflumio, F. Desperately seeking a home marrow niche for T-cell acute lymphoblastic leukaemia. Adv. Biol. Regul. 2019, 74, 100640. [Google Scholar] [CrossRef]
- Zhu, Z.; Golay, H.G.; Barbie, D.A. Targeting pathways downstream of KRAS in lung adenocarcinoma. Pharmacogenomics 2014, 15, 1507–1518. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Shirvani-Farsani, Z.; Hussen, B.M.; Taheri, M.; Jalili Khoshnoud, R. Emerging role of non-coding RNAs in the regulation of KRAS. Cancer Cell Int. 2022, 22, 68. [Google Scholar] [CrossRef]
- Taghon, T.; Waegemans, E.; Van de Walle, I. Notch signaling during human T cell development. Curr. Top. Microbiol. Immunol. 2012, 360, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Wallaert, A.; Van Loocke, W.; Hernandez, L.; Taghon, T.; Speleman, F.; Van Vlierberghe, P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci. Rep. 2017, 11, 7–7901. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000 Res. 2018, 7, 1338. [Google Scholar] [CrossRef]
- Marcel, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet J. 2012, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Baras, A.S.; Mitchell, C.J.; Myers, J.R.; Gupta, S.; Weng, L.C.; Ashton, J.M.; Cornish, T.C.; Pandey, A.; Halushka, M.K. miRge—A Multiplexed Method of Processing Small RNA-Seq Data to Determine MicroRNA Entropy. PLoS ONE 2015, 10, e0143066. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Drobna, M.; Szarzyńska-Zawadzka, B.; Daca-Roszak, P.; Kosmalska, M.; Jaksik, R.; Witt, M.; Dawidowska, M. Identification of Endogenous Control miRNAs for RT-qPCR in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2018, 19, 2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA ID | Average Number of Normalized Reads | LogFC | p adj |
|---|---|---|---|
| hsa-miR-6724-5p | 19 | −4.411 | 0.013 |
| hsa-miR-143-3p | 6042 | 1.677 | 0.011 |
| hsa-miR-151a-3p | 1979 | 2.482 | 0.002 |
| hsa-miR-4420-3p | 1.64 | 4.461 | 0.005 |
| hsa-miR-4728-3p | 0.26 | 2.486 | 0.046 |
| hsa-miR-4749-3p | 0.67 | 3.787 | 0.005 |
| novel_miRNA_chr17:7306866-7306887 | 0.49 | 2.696 | 0.029 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawidowska, M.; Maćkowska-Maślak, N.; Drobna-Śledzińska, M.; Kosmalska, M.; Jaksik, R.; Szymczak, D.; Jarmuż-Szymczak, M.; Sadowska-Klasa, A.; Wojtaszewska, M.; Sędek, Ł.; et al. Small RNA-Seq Reveals Similar miRNA Transcriptome in Children and Young Adults with T-ALL and Indicates miR-143-3p as Novel Candidate Tumor Suppressor in This Leukemia. Int. J. Mol. Sci. 2022, 23, 10117. https://doi.org/10.3390/ijms231710117
Dawidowska M, Maćkowska-Maślak N, Drobna-Śledzińska M, Kosmalska M, Jaksik R, Szymczak D, Jarmuż-Szymczak M, Sadowska-Klasa A, Wojtaszewska M, Sędek Ł, et al. Small RNA-Seq Reveals Similar miRNA Transcriptome in Children and Young Adults with T-ALL and Indicates miR-143-3p as Novel Candidate Tumor Suppressor in This Leukemia. International Journal of Molecular Sciences. 2022; 23(17):10117. https://doi.org/10.3390/ijms231710117
Chicago/Turabian StyleDawidowska, Małgorzata, Natalia Maćkowska-Maślak, Monika Drobna-Śledzińska, Maria Kosmalska, Roman Jaksik, Donata Szymczak, Małgorzata Jarmuż-Szymczak, Alicja Sadowska-Klasa, Marzena Wojtaszewska, Łukasz Sędek, and et al. 2022. "Small RNA-Seq Reveals Similar miRNA Transcriptome in Children and Young Adults with T-ALL and Indicates miR-143-3p as Novel Candidate Tumor Suppressor in This Leukemia" International Journal of Molecular Sciences 23, no. 17: 10117. https://doi.org/10.3390/ijms231710117
APA StyleDawidowska, M., Maćkowska-Maślak, N., Drobna-Śledzińska, M., Kosmalska, M., Jaksik, R., Szymczak, D., Jarmuż-Szymczak, M., Sadowska-Klasa, A., Wojtaszewska, M., Sędek, Ł., Wróbel, T., Zaucha, J. M., Szczepański, T., Lewandowski, K., Giebel, S., & Witt, M. (2022). Small RNA-Seq Reveals Similar miRNA Transcriptome in Children and Young Adults with T-ALL and Indicates miR-143-3p as Novel Candidate Tumor Suppressor in This Leukemia. International Journal of Molecular Sciences, 23(17), 10117. https://doi.org/10.3390/ijms231710117