
Activity Screening of Fatty Acid Mimetic Drugs Identified Nuclear Receptor Agonists
 ,
,
Abstract
:1. Introduction
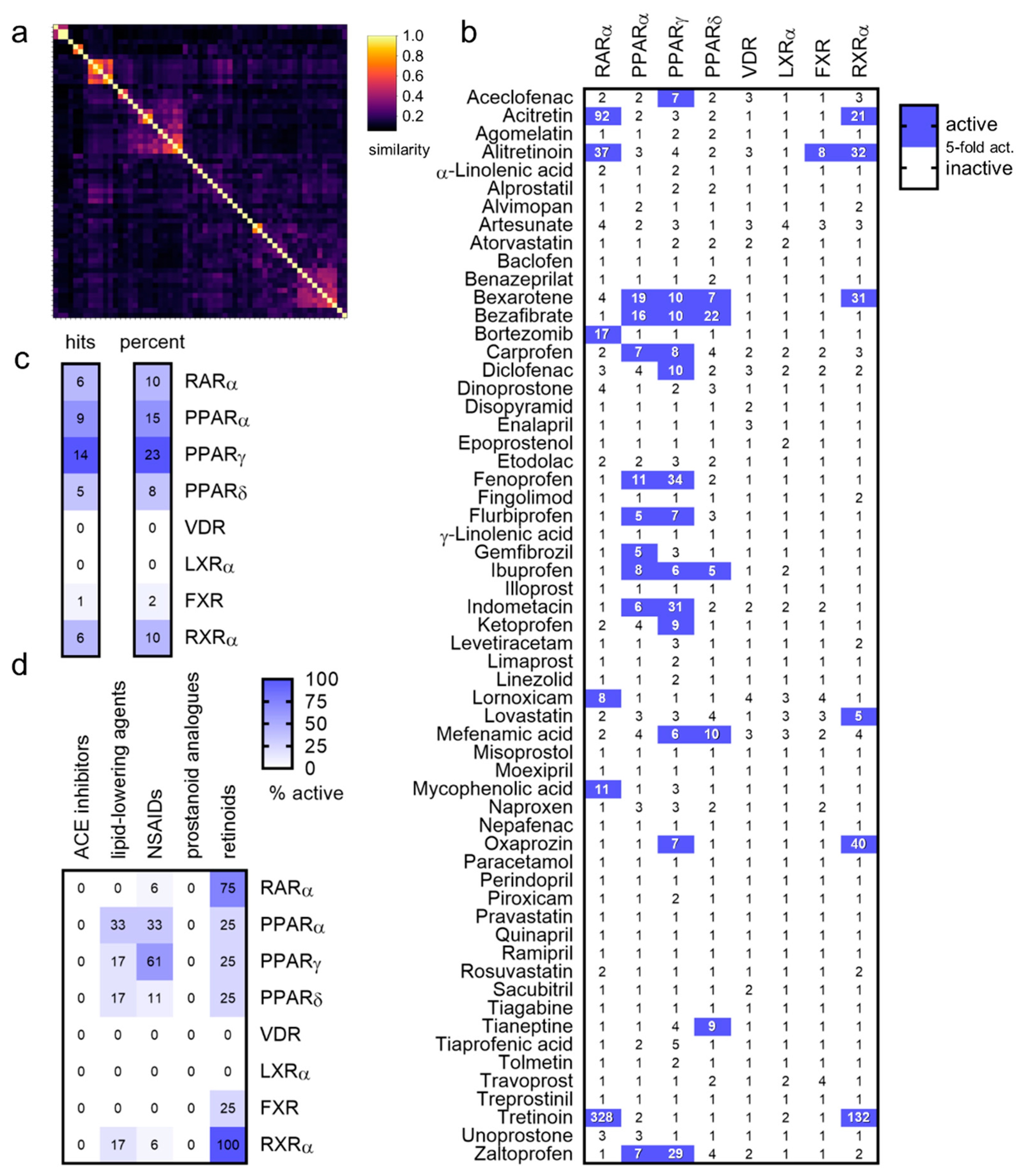
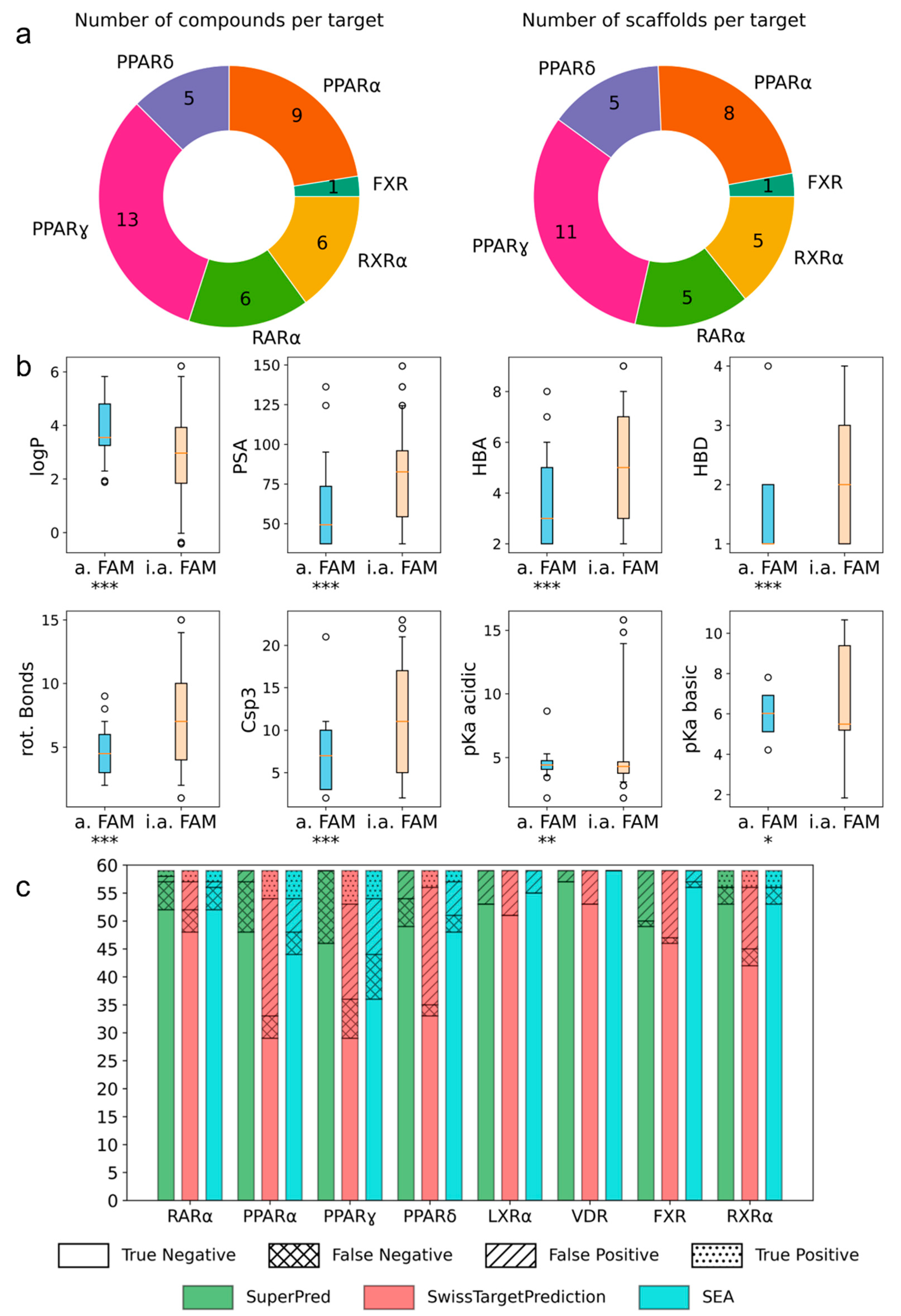
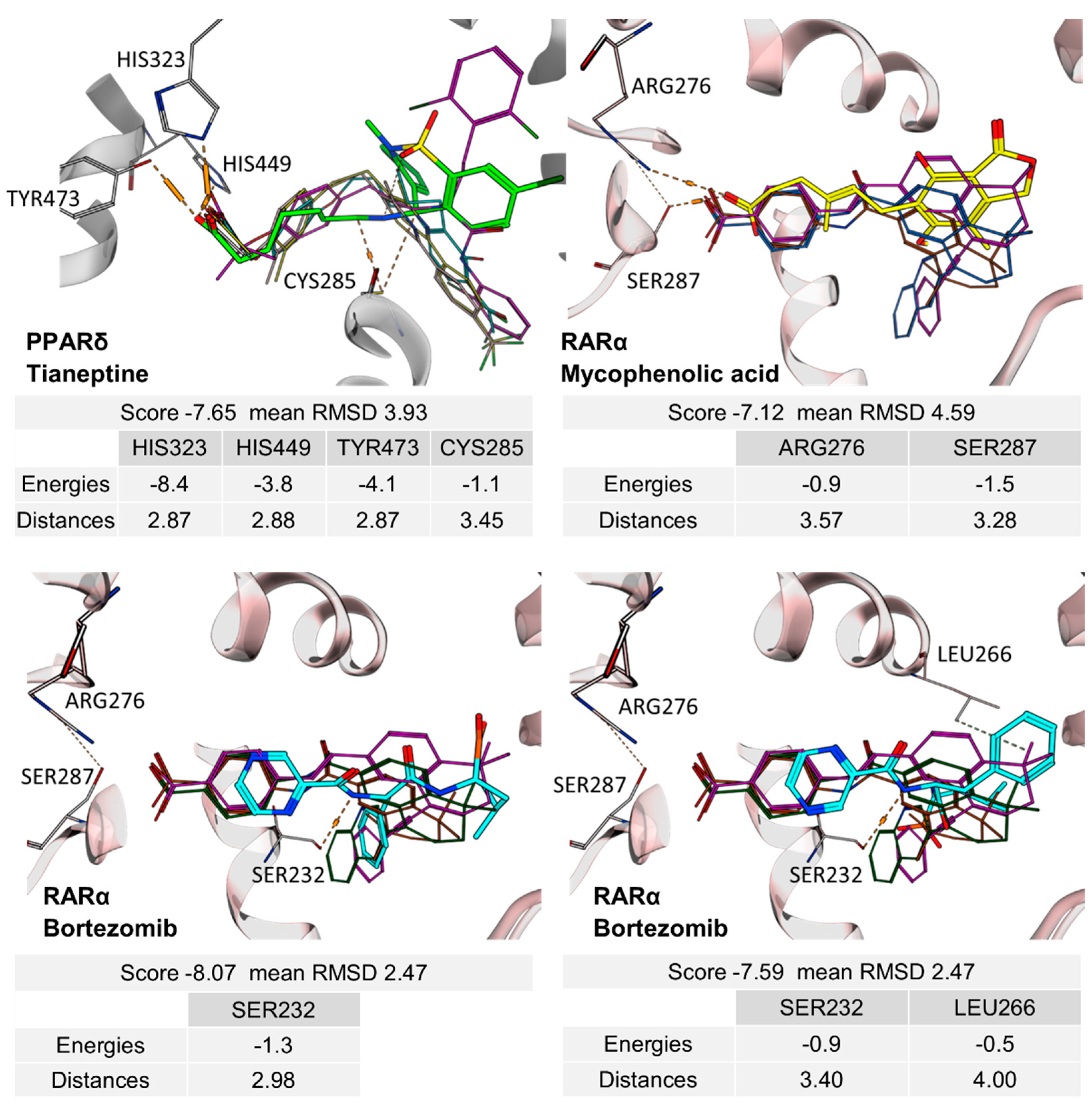
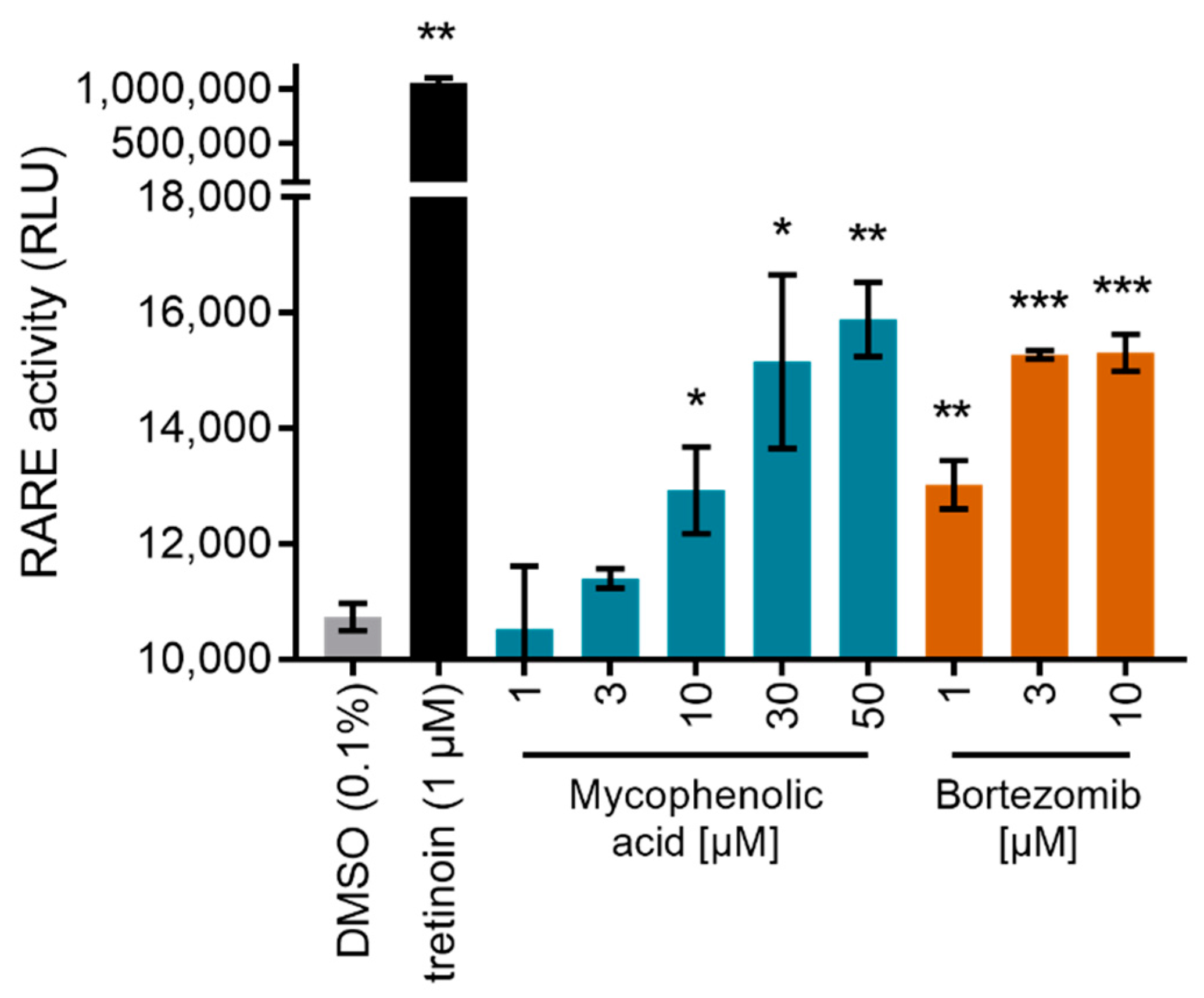
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Heitel, P.; Kalinowsky, L.; Merk, D. Opportunities and Challenges for Fatty Acid Mimetics in Drug Discovery. J. Med. Chem. 2017, 60, 5235–5266. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fairlie, D.P.; Prins, J.B.; Hammock, B.D.; Brown, L. Inflammatory lipid mediators in adipocyte function and obesity. Nat. Rev. Endocrinol. 2010, 6, 71–82. [Google Scholar] [CrossRef]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668, S50–S58. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. Selective optimization of side activities: The SOSA approach. Drug Discov. Today 2006, 11, 160–164. [Google Scholar] [CrossRef]
- Langer, T.; Wermuth, C.-G. Selective Optimization of Side Activities (SOSA): A Promising way for Drug Discovery. In Polypharmacology in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 227–243. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Schierle, S.; Flauaus, C.; Heitel, P.; Willems, S.; Schmidt, J.; Kaiser, A.; Weizel, L.; Goebel, T.; Kahnt, A.S.; Geisslinger, G.; et al. Boosting Anti-Inflammatory Potency of Zafirlukast by Designed Polypharmacology. J. Med. Chem. 2018, 61, 5758–5764. [Google Scholar] [CrossRef]
- Göbel, T.; Diehl, O.; Heering, J.; Merk, D.; Angioni, C.; Wittmann, S.K.; Buscato, E.; Kottke, R.; Weizel, L.; Schader, T.; et al. Zafirlukast Is a Dual Modulator of Human Soluble Epoxide Hydrolase and Peroxisome Proliferator-Activated Receptor γ. Front. Pharmacol. 2019, 10, 263. [Google Scholar] [CrossRef]
- Heitel, P.; Gellrich, L.; Heering, J.; Göbel, T.; Kahnt, A.; Proschak, E.; Schubert-Zsilavecz, M.; Merk, D. Urate transporter inhibitor lesinurad is a selective peroxisome proliferator-activated receptor gamma modulator (sPPARγM) in vitro. Sci. Rep. 2018, 8, 13554. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Heering, J.; Merk, D. Hybrid Reporter Gene Assays: Versatile In Vitro Tools to Characterize Nuclear Receptor Modulators. Methods Mol. Biol. 2019, 1966, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.; Schubert-Zsilavecz, M.; Merk, D. Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): A patent review (2008–present). Expert Opin. Ther. Pat. 2012, 22, 803–841. [Google Scholar] [CrossRef] [PubMed]
- Sameshima, T.; Yukawa, T.; Hirozane, Y.; Yoshikawa, M.; Katoh, T.; Hara, H.; Yogo, T.; Miyahisa, I.; Okuda, T.; Miyamoto, M.; et al. Small-Scale Panel Comprising Diverse Gene Family Targets To Evaluate Compound Promiscuity. Chem. Res. Toxicol. 2020, 33, 154–161. [Google Scholar] [CrossRef]
- Bendels, S.; Bissantz, C.; Fasching, B.; Gerebtzoff, G.; Guba, W.; Kansy, M.; Migeon, J.; Mohr, S.; Peters, J.-U.; Tillier, F.; et al. Safety screening in early drug discovery: An optimized assay panel. J. Pharmacol. Toxicol. Methods 2019, 99, 106609. [Google Scholar] [CrossRef]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures—A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Velkov, T. Thermodynamics of Lipophilic Drug Binding to Intestinal Fatty Acid Binding Protein and Permeation across Membranes. Mol. Pharm. 2009, 6, 557–570. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Keiser, M.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Schierle, S.; Chaikuad, A.; Lillich, F.F.; Ni, X.; Woltersdorf, S.; Schallmayer, E.; Renelt, B.; Ronchetti, R.; Knapp, S.; Proschak, E.; et al. Oxaprozin Analogues as Selective RXR Agonists with Superior Properties and Pharmacokinetics. J. Med. Chem. 2021, 64, 5123–5136. [Google Scholar] [CrossRef] [PubMed]
- Mäder, P. Boron in Medicinal Chemistry: Powerful, but Neglected. Chimia 2019, 73, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular Recognition of Fatty Acids by Peroxisome Proliferator–Activated Receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Le Maire, A.; Teyssier, C.; Erb, C.; Grimaldi, M.; Alvarez, S.; de Lera, A.R.; Balaguer, P.; Gronemeyer, H.; Royer, C.; Germain, P.; et al. A unique secondary-structure switch controls constitutive gene repression by retinoic acid receptor. Nat. Struct. Mol. Biol. 2010, 17, 801–807. [Google Scholar] [CrossRef]
- Curran, M.P.; McKeage, K. Bortezomib: A review of its use in patients with multiple myeloma. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef]
- Otsuki, T.; Sakaguchi, H.; Hatayama, T.; Wu, P.; Takata, A.; Hyodoh, F. Effects of All-trans Retinoic Acid (ATRA) on Human Myeloma Cells. Leuk. Lymphoma 2003, 44, 1651–1656. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Minnema, M.C.; Levin, M.-D.; Broijl, A.; Bos, G.M.J.; Kersten, M.J.; Mutis, T.; Verkleij, C.P.M.; Nijhof, I.S.; Maas-Bosman, P.W.C.; et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5128–5139. [Google Scholar] [CrossRef]
- Palumbo, A.; Battaglio, S.; Napoli, P.; Bruno, B.; Omedè, P.; Boccadoro, M.; Pileri, A. Retinoic acid inhibits the growth of human myeloma cells in vitro. Br. J. Haematol. 1995, 89, 555–560. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, Z.; Wang, Z.; Ye, L.; Xian, M.; Xiao, L.; Su, P.; Bi, E.; Huang, Y.-H.; Qian, J.; et al. RARγ activation sensitizes human myeloma cells to carfilzomib treatment through the OAS-RNase L innate immune pathway. Blood 2022, 139, 59–72. [Google Scholar] [CrossRef]
- Broen, J.C.A.; van Laar, J.M. Mycophenolate mofetil, azathioprine and tacrolimus: Mechanisms in rheumatology. Nat. Rev. Rheumatol. 2020, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Larange, A.; Cheroutre, H. Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu. Rev. Immunol. 2016, 34, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Bullingham, R.E.S.; Nicholls, A.J.; Kamm, B.R. Clinical Pharmacokinetics of Mycophenolate Mofetil. Clin. Pharmacokinet. 1998, 34, 429–455. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical Pharmacokinetics and Pharmacodynamics of Bortezomib. Clin. Pharmacokinet. 2018, 58, 157–168. [Google Scholar] [CrossRef]
- Willems, S.; Marschner, J.A.; Kilu, W.; Faudone, G.; Busch, R.; Duensing-Kropp, S.; Heering, J.; Merk, D. Nurr1 Modulation Mediates Neuroprotective Effects of Statins. Adv. Sci. 2022, 9, 2104640. [Google Scholar] [CrossRef]
- Pollinger, J.; Gellrich, L.; Schierle, S.; Kilu, W.; Schmidt, J.; Kalinowsky, L.; Ohrndorf, J.; Kaiser, A.; Heering, J.; Proschak, E.; et al. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 2019, 62, 2112–2126. [Google Scholar] [CrossRef]
- Rau, O.; Wurglics, M.; Paulke, A.; Zitzkowski, J.; Meindl, N.; Bock, A.; Dingermann, T.; Abdel-Tawab, M.; Schubert-Zsilavecz, M. Carnosic Acid and Carnosol, Phenolic Diterpene Compounds of the Labiate Herbs Rosemary and Sage, are Activators of the Human Peroxisome Proliferator-Activated Receptor Gamma. Planta Medica 2006, 72, 881–887. [Google Scholar] [CrossRef]
- Flesch, D.; Cheung, S.-Y.; Schmidt, J.; Gabler, M.; Heitel, P.; Kramer, J.; Kaiser, A.; Hartmann, M.; Lindner, M.; Lüddens-Dämgen, K.; et al. Nonacidic Farnesoid X Receptor Modulators. J. Med. Chem. 2017, 60, 7199–7205. [Google Scholar] [CrossRef]
- Heitel, P.; Achenbach, J.; Moser, D.; Proschak, E.; Merk, D. DrugBank screening revealed alitretinoin and bexarotene as liver X receptor modulators. Bioorg. Med. Chem. Lett. 2017, 27, 1193–1198. [Google Scholar] [CrossRef]
- Schmidt, J.; Klingler, F.-M.; Proschak, E.; Steinhilber, D.; Schubert-Zsilavecz, M.; Merk, D. NSAIDs Ibuprofen, Indometacin and Diclofenac do not interact with Farnesoid X Receptor. Sci. Rep. 2015, 5, 14782. [Google Scholar] [CrossRef]
- Heitel, P.; Gellrich, L.; Kalinowsky, L.; Heering, J.; Kaiser, A.; Ohrndorf, J.; Proschak, E.; Merk, D. Computer-Assisted Discovery and Structural Optimization of a Novel Retinoid X Receptor Agonist Chemotype. ACS Med. Chem. Lett. 2019, 10, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Garcha, K.; Karamboulas, K.; Cowan, M.F.; Drysdale, L.M.; Horton, W.A.; Underhill, T.M. BMP action in skeletogenesis involves attenuation of retinoid signaling. J. Cell Biol. 2006, 174, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zolfaghari, R.; Mattie, F.J.; Wei, C.-H.; Chisholm, D.R.; Whiting, A.; Ross, A.C. CYP26A1 gene promoter is a useful tool for reporting RAR-mediated retinoid activity. Anal. Biochem. 2019, 577, 98–109. [Google Scholar] [CrossRef]
- Seuter, S.; Väisänen, S.; Rådmark, O.; Carlberg, C.; Steinhilber, D. Functional characterization of vitamin D responding regions in the human 5-Lipoxygenase gene. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
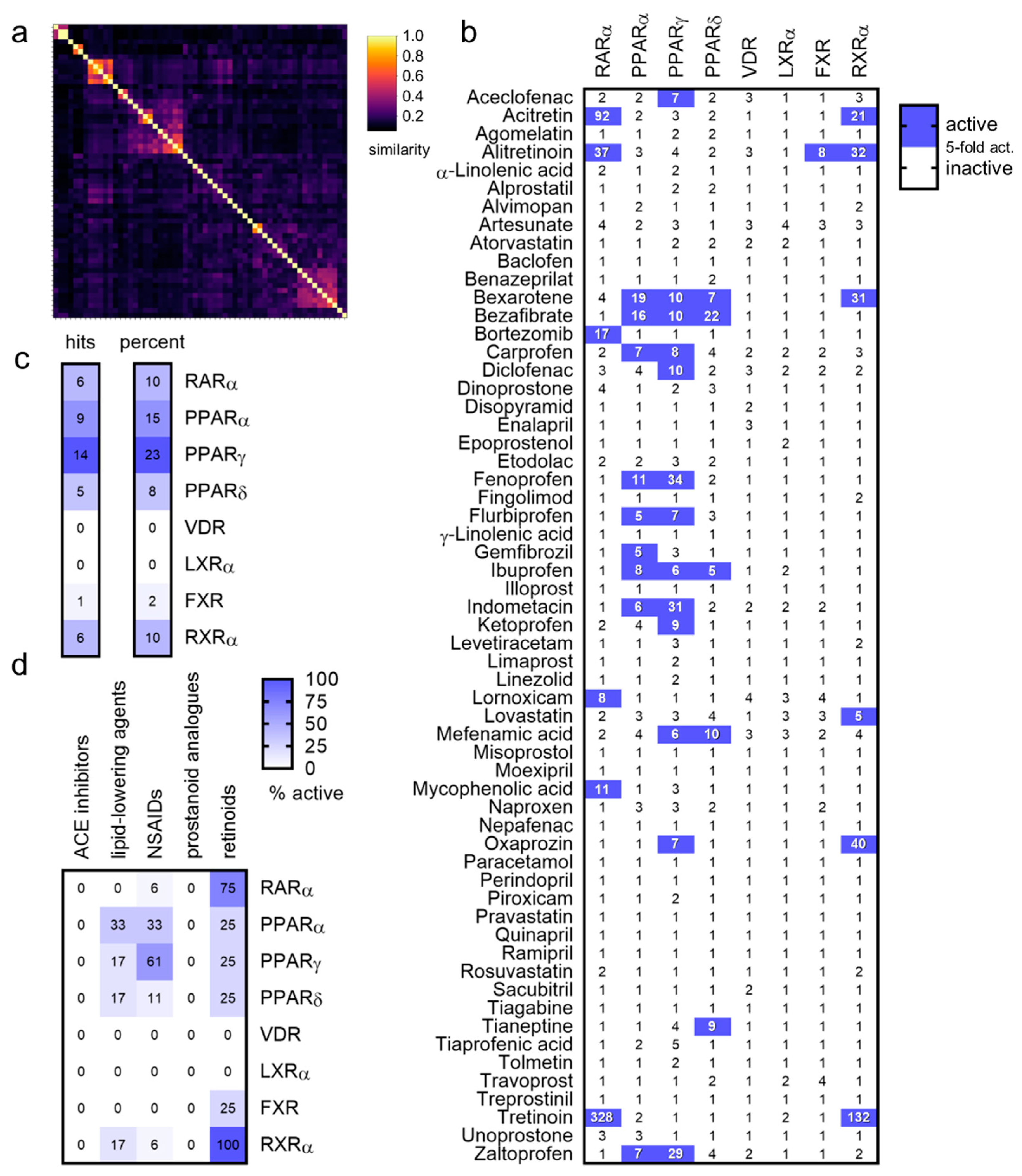
| Drug (Structure, INN) | Activity |
|---|---|
 Oxaprozin | partial RXR agonist [22] EC50 = 16.1 ± 0.6 µM (23 ± 1% max. act.) (RXRα) |
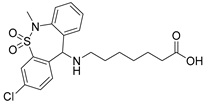 Tianeptine | partial PPARδ agonist EC50 = 28 ± 4 µM (43 ± 4% max. act.) |
 Bortezomib | partial RAR agonist EC50 = 0.8 ± 0.3 µM (<10% max. act.) (RARα) |
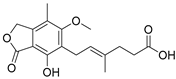 Mycophenolic acid | partial RAR agonist EC50 = 1.5 ± 0.6 µM (<10% max. act.) (RARα) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helmstädter, M.; Schierle, S.; Isigkeit, L.; Proschak, E.; Marschner, J.A.; Merk, D. Activity Screening of Fatty Acid Mimetic Drugs Identified Nuclear Receptor Agonists. Int. J. Mol. Sci. 2022, 23, 10070. https://doi.org/10.3390/ijms231710070
Helmstädter M, Schierle S, Isigkeit L, Proschak E, Marschner JA, Merk D. Activity Screening of Fatty Acid Mimetic Drugs Identified Nuclear Receptor Agonists. International Journal of Molecular Sciences. 2022; 23(17):10070. https://doi.org/10.3390/ijms231710070
Chicago/Turabian StyleHelmstädter, Moritz, Simone Schierle, Laura Isigkeit, Ewgenij Proschak, Julian Aurelio Marschner, and Daniel Merk. 2022. "Activity Screening of Fatty Acid Mimetic Drugs Identified Nuclear Receptor Agonists" International Journal of Molecular Sciences 23, no. 17: 10070. https://doi.org/10.3390/ijms231710070
APA StyleHelmstädter, M., Schierle, S., Isigkeit, L., Proschak, E., Marschner, J. A., & Merk, D. (2022). Activity Screening of Fatty Acid Mimetic Drugs Identified Nuclear Receptor Agonists. International Journal of Molecular Sciences, 23(17), 10070. https://doi.org/10.3390/ijms231710070