
Functional Prediction of trans-Prenyltransferases Reveals the Distribution of GFPPSs in Species beyond the Brassicaceae Clade

 , , , and
, , , and
Abstract
:
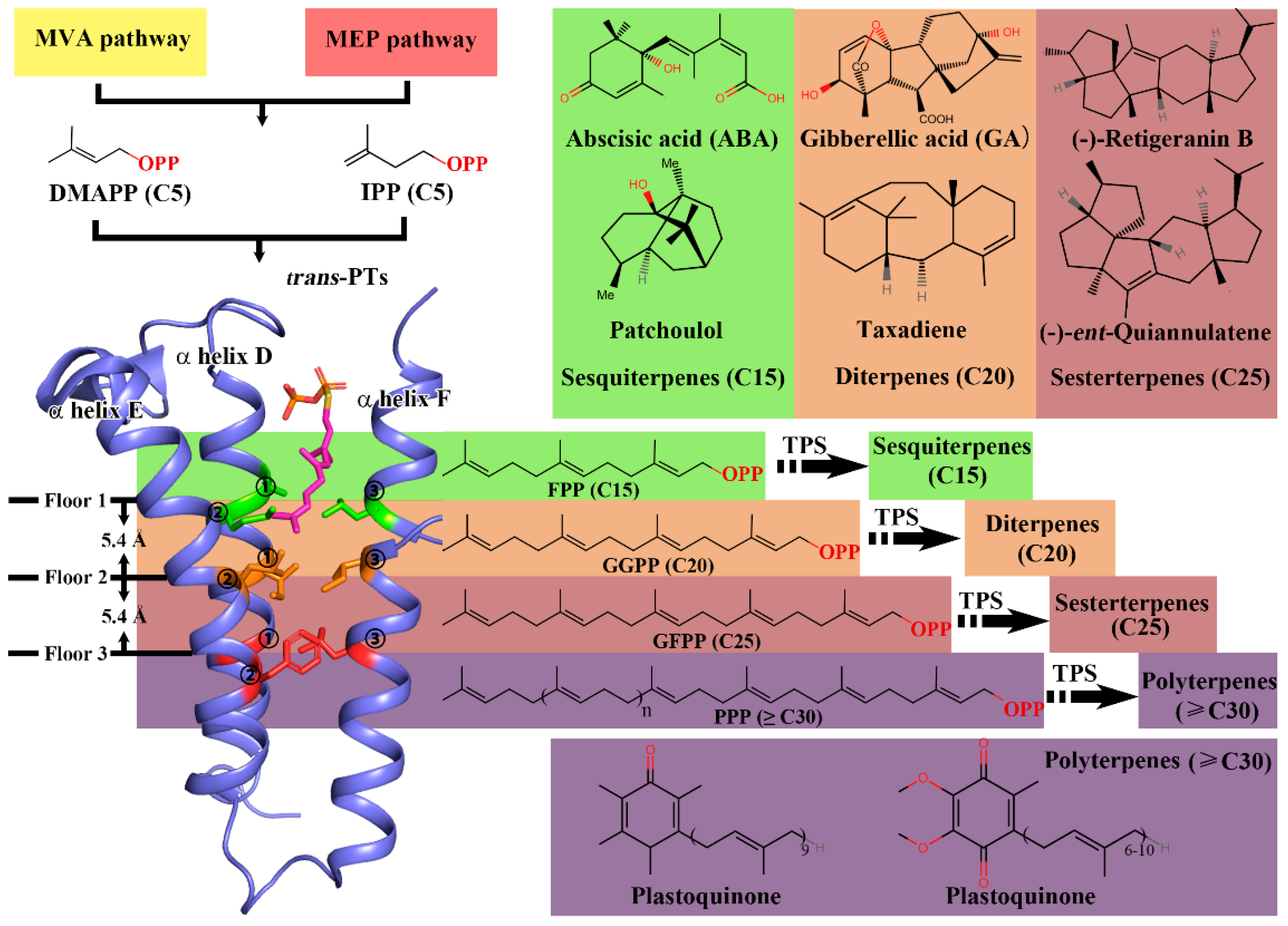
1. Introduction
2. Results
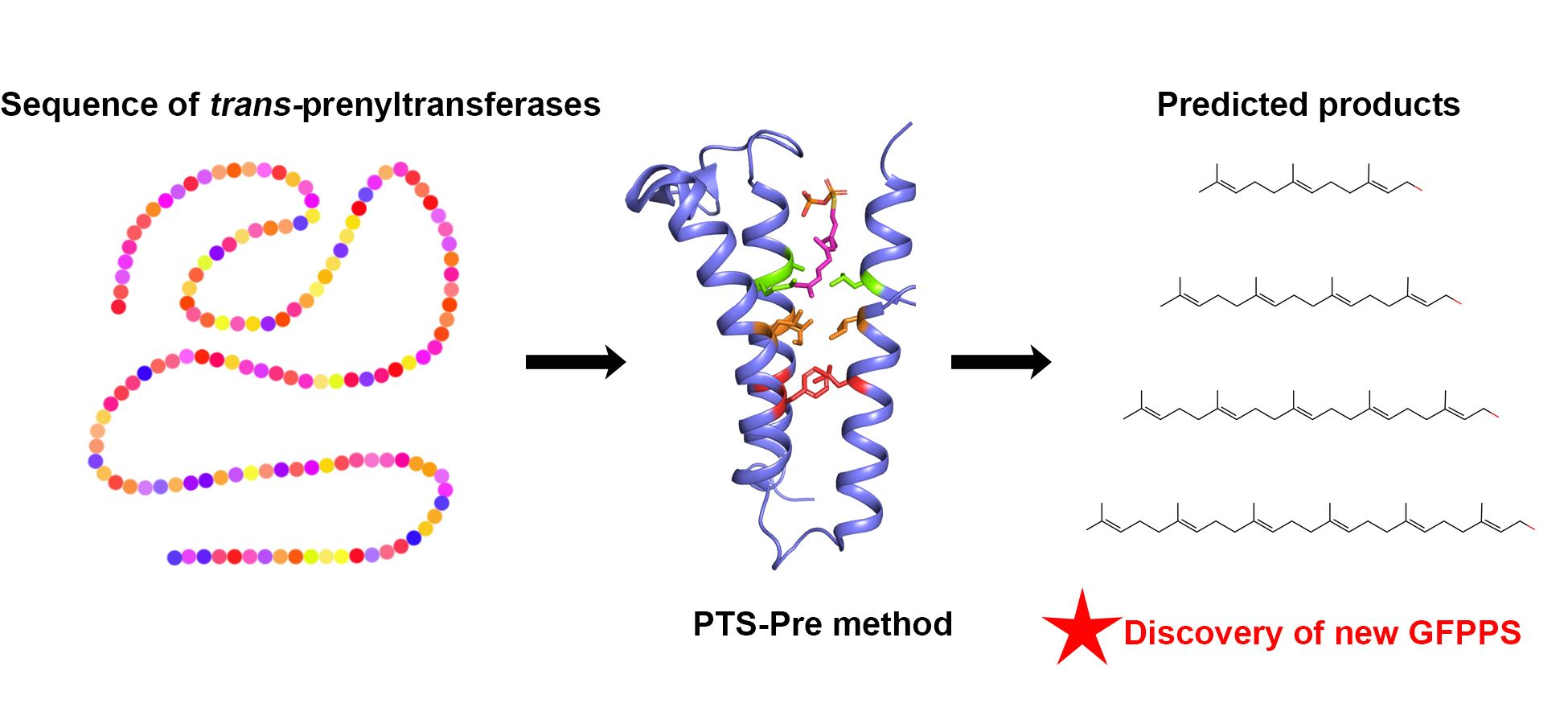
2.1. Data Collection and Establishment of PTS-Pre for the Prediction of trans-Prenyltransferases
2.2. Validation of PTS-Pre Prediction Using Experimentally Determined trans-PTs Sequences
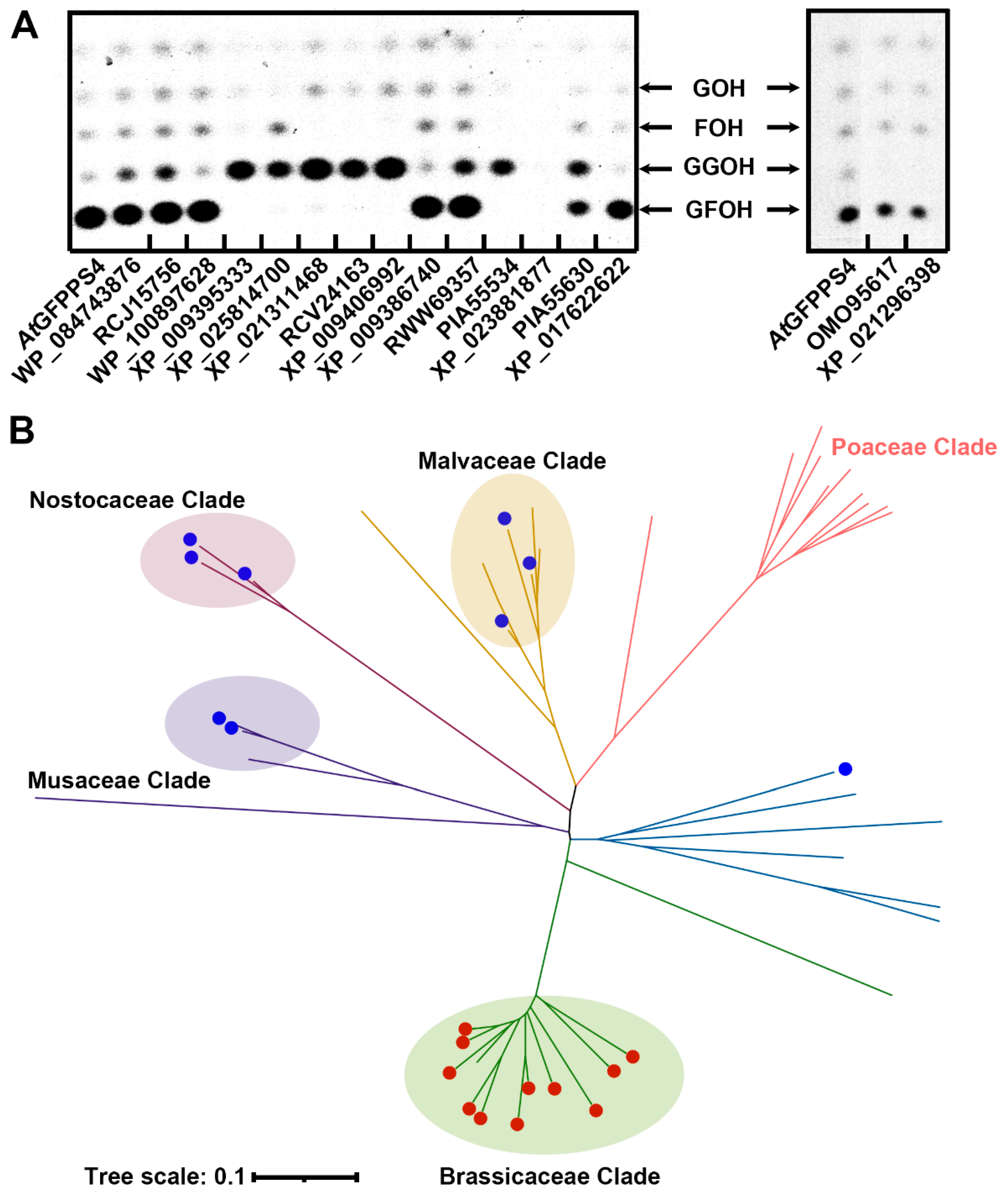
2.3. Genome Mining of the trans-PT Gene Family Identified More GFPPSs beyond the Brassicaceae Clade
3. Discussion
4. Materials and Methods
4.1. Sequence Similarity Network and Phylogenetic Analysis
4.2. Gene cloning, Expression, and Protein Purification
4.3. In Vitro trans-Prenyltransferase Activity Assays
4.4. Computational Function Annotation Design of PTS-Pre
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christianson, D.W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarvey, D.J.; Croteau, R. Terpenoid metabolism. Plant Cell 1995, 7, 1015–1026. [Google Scholar] [PubMed] [Green Version]
- Vranova, E.; Coman, D.; Gruissem, W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avalos, M.; Garbeva, P.; Vader, L.; van Wezel, G.P.; Dickschat, J.S.; Ulanova, D. Biosynthesis, evolution and ecology of microbial terpenoids. Nat. Prod. Rep. 2022, 39, 249–272. [Google Scholar] [CrossRef]
- Zhou, F.; Pichersky, E. More is better: The diversity of terpene metabolism in plants. Curr. Opin. Plant Biol. 2020, 55, 1–10. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.H.; Ko, T.P.; Wang, A.H.J. Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 2002, 269, 3339–3354. [Google Scholar] [CrossRef]
- Vandermoten, S.; Haubruge, E.; Cusson, M. New insights into short-chain prenyltransferases: Structural features, evolutionary history and potential for selective inhibition. Cell Mol. Life Sci. 2009, 66, 3685–3695. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Q.; Fan, D.; Li, J.; Wang, G.; Zhang, P. Structural Analyses of Short-Chain Prenyltransferases Identify an Evolutionarily Conserved GFPPS Clade in Brassicaceae Plants. Mol. Plant 2016, 9, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Nagel, R.; Gershenzon, J.; Schmidt, A. Nonradioactive assay for detecting isoprenyl diphosphate synthase activity in crude plant extracts using liquid chromatography coupled with tandem mass spectrometry. Anal. Biochem. 2012, 422, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.D.; Dixon, R.A. Heterodimeric geranyl(geranyl)diphosphate synthase from hop (Humulus lupulus) and the evolution of monoterpene biosynthesis. Proc. Natl. Acad. Sci. USA 2009, 106, 9914–9919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarshis, L.C.; Proteau, P.J.; Kellogg, B.A.; Sacchettini, J.C.; Poulter, C.D. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. USA 1996, 93, 15018–15023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallrapp, F.H.; Pan, J.J.; Ramamoorthy, G.; Almonacid, D.E.; Hillerich, B.S.; Seidel, R.; Patskovsky, Y.; Babbitt, P.C.; Almo, S.C.; Jacobson, M.P.; et al. Prediction of function for the polyprenyl transferase subgroup in the isoprenoid synthase superfamily. Proc. Natl. Acad. Sci. USA 2013, 110, E1196–E1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Chen, Q.; Li, J.; Liu, Z.; Mitsuhashi, T.; Zhang, Y.; Liu, H.; Ma, Y.; He, J.; Shinada, T.; Sato, T.; et al. Molecular Basis for Sesterterpene Diversity Produced by Plant Terpene Synthases. Plant Commun. 2020, 1, 100051. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, T.; Liu, Y.X.; Liu, H.; Zhao, T.; Liu, Z.; Gan, X.; Hallab, A.; Wang, X.; He, J.; et al. Recently duplicated sesterterpene (C25) gene clusters in Arabidopsis thaliana modulate root microbiota. Sci. China Life Sci. 2019, 62, 947–958. [Google Scholar] [CrossRef]
- Shao, J.; Chen, Q.W.; Lv, H.J.; He, J.; Liu, Z.F.; Lu, Y.N.; Liu, H.L.; Wang, G.D.; Wang, Y. (+)-Thalianatriene and (−)-Retigeranin B Catalyzed by Sesterterpene Synthases from Arabidopsis thaliana. Org. Lett. 2017, 19, 1816–1819. [Google Scholar] [CrossRef]
- Huang, A.C.C.; Kautsar, S.A.; Hong, Y.J.; Medema, M.H.; Bond, A.D.; Tantillo, D.J.; Osbourn, A. Unearthing a sesterterpene biosynthetic repertoire in the Brassicaceae through genome mining reveals convergent evolution. Proc. Natl. Acad. Sci. USA 2017, 114, E6005–E6014. [Google Scholar] [CrossRef] [Green Version]
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef]
- Pegg, S.C.H.; Brown, S.D.; Ojha, S.; Seffernick, J.; Meng, E.C.; Morris, J.H.; Chang, P.J.; Huang, C.C.; Ferrin, T.E.; Babbitt, P.C. Leveraging enzyme structure-function relationships for functional inference and experimental design: The structure-function linkage database. Biochemistry 2006, 45, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zamyatnin, A.A. Protein volume in solution. Prog. Biophys. Mol. Biol. 1972, 24, 107–123. [Google Scholar] [CrossRef]
- Han, X.; Chen, C.C.; Kuo, C.J.; Huang, C.H.; Zheng, Y.Y.; Ko, T.P.; Zhu, Z.; Feng, X.X.; Wang, K.; Oldfield, E.; et al. Crystal structures of ligand-bound octaprenyl pyrophosphate synthase from Escherichia coli reveal the catalytic and chain-length determining mechanisms. Proteins 2015, 83, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Luo, S.H.; Schmidt, A.; Wang, G.D.; Sun, G.L.; Grant, M.; Kuang, C.; Yang, M.J.; Jing, S.X.; Li, C.H.; et al. A Geranylfarnesyl Diphosphate Synthase Provides the Precursor for Sesterterpenoid (C25) Formation in the Glandular Trichomes of the Mint Species Leucosceptrum canum. Plant Cell 2016, 28, 804–822. [Google Scholar] [CrossRef] [Green Version]
- Nagel, R.; Bernholz, C.; Vranova, E.; Kosuth, J.; Bergau, N.; Ludwig, S.; Wessjohann, L.; Gershenzon, J.; Tissier, A.; Schmidt, A. Arabidopsis thaliana isoprenyl diphosphate synthases produce the C-25 intermediate geranylfarnesyl diphosphate. Plant J. 2015, 84, 847–859. [Google Scholar] [CrossRef]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702. [Google Scholar] [CrossRef]
- Akhtar, T.A.; Matsuba, Y.; Schauvinhold, I.; Yu, G.; Lees, H.A.; Klein, S.E.; Pichersky, E. The tomato cis-prenyltransferase gene family. Plant J. 2013, 73, 640–652. [Google Scholar] [CrossRef]
- Takahashi, S.; Koyama, T. Structure and function of cis-prenyl chain elongating enzymes. Chem. Rec. 2006, 6, 194–205. [Google Scholar] [CrossRef]
- Li, D.S.; Hua, J.; Luo, S.H.; Liu, Y.C.; Chen, Y.G.; Ling, Y.; Guo, K.; Liu, Y.; Li, S.H. An extremely promiscuous terpenoid synthase from the Lamiaceae plant Colquhounia coccinea var. mollis catalyzes the formation of sester-/di-/sesqui-/mono-terpenoids. Plant Commun. 2021, 2, 100233. [Google Scholar] [CrossRef]
- Karunanithi, P.S.; Zerbe, P. Terpene Synthases as Metabolic Gatekeepers in the Evolution of Plant Terpenoid Chemical Diversity. Front. Plant Sci. 2019, 10, 1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.R.; Bhat, W.W.; Sadre, R.; Miller, G.P.; Garcia, A.S.; Hamberger, B. Promiscuous terpene synthases from Prunella vulgaris highlight the importance of substrate and compartment switching in terpene synthase evolution. New Phytol. 2019, 223, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, R.; Thomas, J.A.; Adekunle, F.A.; Mann, F.M.; Peters, R.J. Arginine in the FARM and SARM: A Role in Chain-Length Determination for Arginine in the Aspartate-Rich Motifs of Isoprenyl Diphosphate Synthases from Mycobacterium tuberculosis. Molecules 2018, 23, 2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.; Liu, Y.; Li, S.H. The untapped potential of plant sesterterpenoids: Chemistry, biological activities and biosynthesis. Nat. Prod. Rep. 2021, 38, 2293–2314. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Shang, C.H.; Xu, X.L.; Yuan, Z.H.; Wang, Z.M.; Hu, L.; Alam, M.A.; Xie, J. Cloning and differential expression analysis of geranylgeranyl diphosphate synthase gene from Dunaliella parva. J. Appl. Phycol. 2016, 28, 2397–2405. [Google Scholar] [CrossRef]
- Qi, Q.; Li, R.; Gai, Y.; Jiang, X.N. Cloning and functional identification of farnesyl diphosphate synthase from Pinus massoniana Lamb. J. Plant Biochem. Biotechnol. 2017, 26, 132–140. [Google Scholar] [CrossRef]
- Yang, L.E.; Huang, X.Q.; Lu, Q.Q.; Zhu, J.Y.; Lu, S. Cloning and characterization of the geranylgeranyl diphosphate synthase (GGPS) responsible for carotenoid biosynthesis in Pyropia umbilicalis. J. Appl. Phycol. 2016, 28, 671–678. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Protein ID | Best Template | Sequence Identity (%) | PTS-Pre Predicted | Experimentally Determined |
|---|---|---|---|---|---|
| GaGGPPSL-1 | XP_017641950 | 1FPS | 45.4 | C15 | C15 |
| GaGGPPSL-2 | XP_017620811 | 5E8H | 67.5 | C20 | C20 |
| GaGGPPSL-3 | XP_017622622 | 5E8H | 59.9 | C25 | C25 |
| GaGGPPSL-4 | XP_017649086 | 3AQ0 | 71.8 | ≥C30 | NA |
| GaGGPPSL-5 | XP_017640526 | 3AQ0 | 74.7 | ≥C30 | ≥C30 |
| TcGGPPSL-1 | KAB8318528 | 2FOR | 49.8 | C15 | C15 |
| TcGGPPSL-2 | KAB8317749 | 3AQ0 | 39.4 | C20 | C10 |
| TcGGPPSL-3 | KAB8318557 | 5E8H | 50.9 | C20 | C20 |
| TcGGPPSL-4 | KAB8315220 | 5E8H | 51.0 | C20/C25 | C25 |
| SaGGPPSL-1 | WP_059416577 | 2FOR | 36.1 | C15 | C15 |
| SaGGPPSL-2 | WP_059424690 | 3Q2Q | 43.5 | C20 | NA |
| SaGGPPSL-3 | WP_078945674 | 3OYR | 35.2 | C20/C25 | C15 |
| Protein ID | Organism | PTS-Pre Predicted | Experimentally Determined |
|---|---|---|---|
| WP_084743876 | Tolypothrix campylonemoides * | C20/C25 | C25 |
| RCJ15756 | Nostoc sp. ATCC 43529 * | C20/C25 | C25 |
| WP_100897628 | Nostoc flagelliforme * | C20/C25 | C25 |
| XP_009395333 | Musa acuminata | C20/C25 | C20 |
| XP_025814700 | Panicum hallii | C25 | C20 |
| XP_021311468 | Sorghum bicolor | C25 | C20 |
| RCV24163 | Setaria italica | C25 | C20 |
| XP_009406992 | Musa acuminata | C25 | C20 |
| XP_009386740 | Musa acuminata | C25 | C25 |
| RWW69357 | Ensete ventricosum | C25 | C25 |
| PIA55534 | Aquilegia coerulea | C25 | C20 |
| XP_023881877 | Quercus suber | C25 | NA |
| PIA55630 | Aquilegia coerulea | C25 | C25 |
| XP_017622622 | Gossypium arboreum | C25 | C25 |
| OMO95617 | Corchorus olitorius | C25 | C25 |
| XP_021296398 | Herrania umbratica | C25 | C25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ma, Y.; Chen, Q.; Yang, M.; Feng, D.; Zhou, F.; Wang, G.; Wang, C. Functional Prediction of trans-Prenyltransferases Reveals the Distribution of GFPPSs in Species beyond the Brassicaceae Clade. Int. J. Mol. Sci. 2022, 23, 9471. https://doi.org/10.3390/ijms23169471
Zhang J, Ma Y, Chen Q, Yang M, Feng D, Zhou F, Wang G, Wang C. Functional Prediction of trans-Prenyltransferases Reveals the Distribution of GFPPSs in Species beyond the Brassicaceae Clade. International Journal of Molecular Sciences. 2022; 23(16):9471. https://doi.org/10.3390/ijms23169471
Chicago/Turabian StyleZhang, Jing, Yihua Ma, Qingwen Chen, Mingxia Yang, Deyu Feng, Fei Zhou, Guodong Wang, and Chengyuan Wang. 2022. "Functional Prediction of trans-Prenyltransferases Reveals the Distribution of GFPPSs in Species beyond the Brassicaceae Clade" International Journal of Molecular Sciences 23, no. 16: 9471. https://doi.org/10.3390/ijms23169471
APA StyleZhang, J., Ma, Y., Chen, Q., Yang, M., Feng, D., Zhou, F., Wang, G., & Wang, C. (2022). Functional Prediction of trans-Prenyltransferases Reveals the Distribution of GFPPSs in Species beyond the Brassicaceae Clade. International Journal of Molecular Sciences, 23(16), 9471. https://doi.org/10.3390/ijms23169471