
Functional Diversity of Microbial Communities in the Soybean (Glycine max L.) Rhizosphere from Free State, South Africa



Abstract
:1. Introduction
2. Results
2.1. Physicochemical Analysis of Soybean Soil
2.2. Sequence Processes
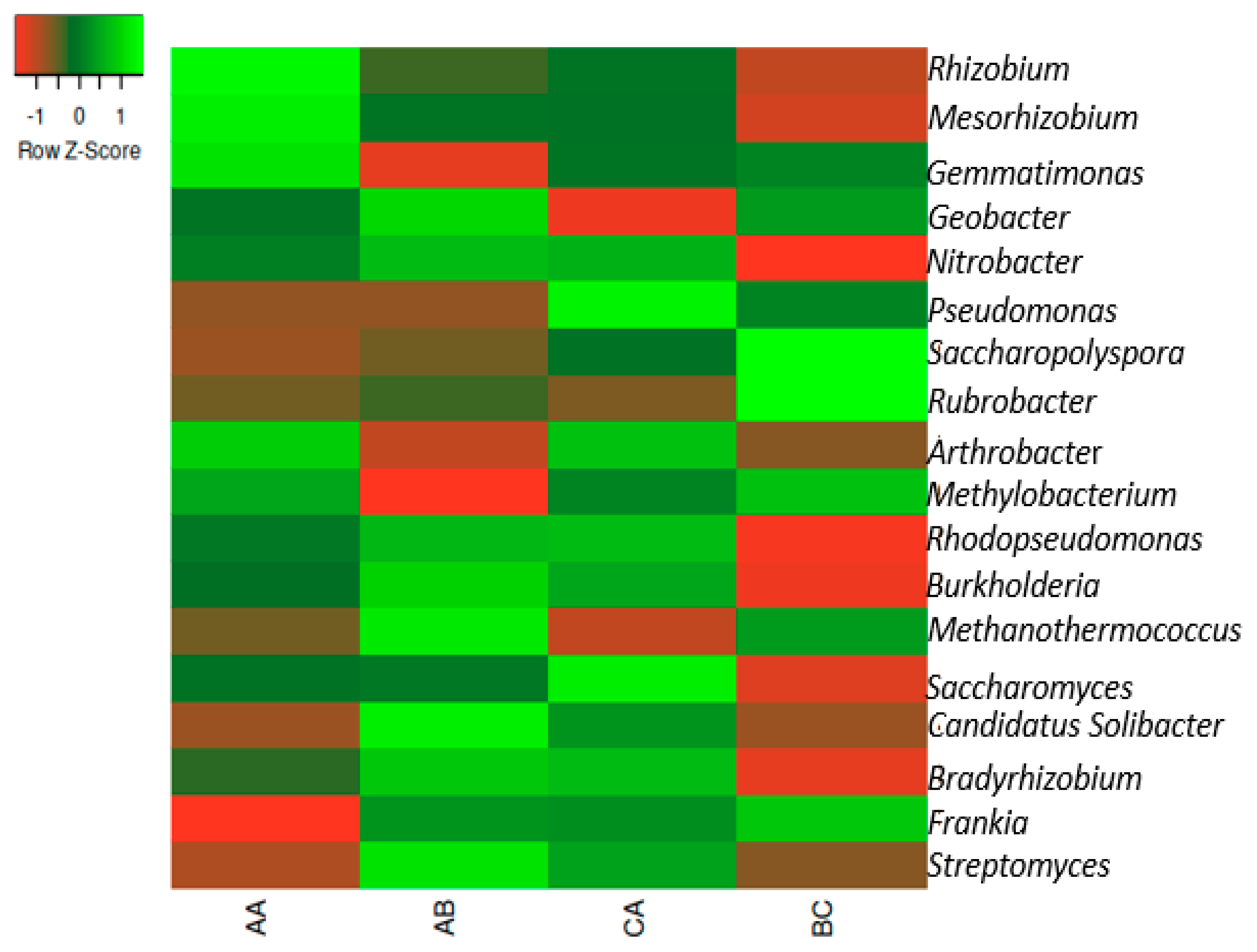
2.3. Taxonomy of the Microbiome in Both the Rhizosphere and Bulk Sample
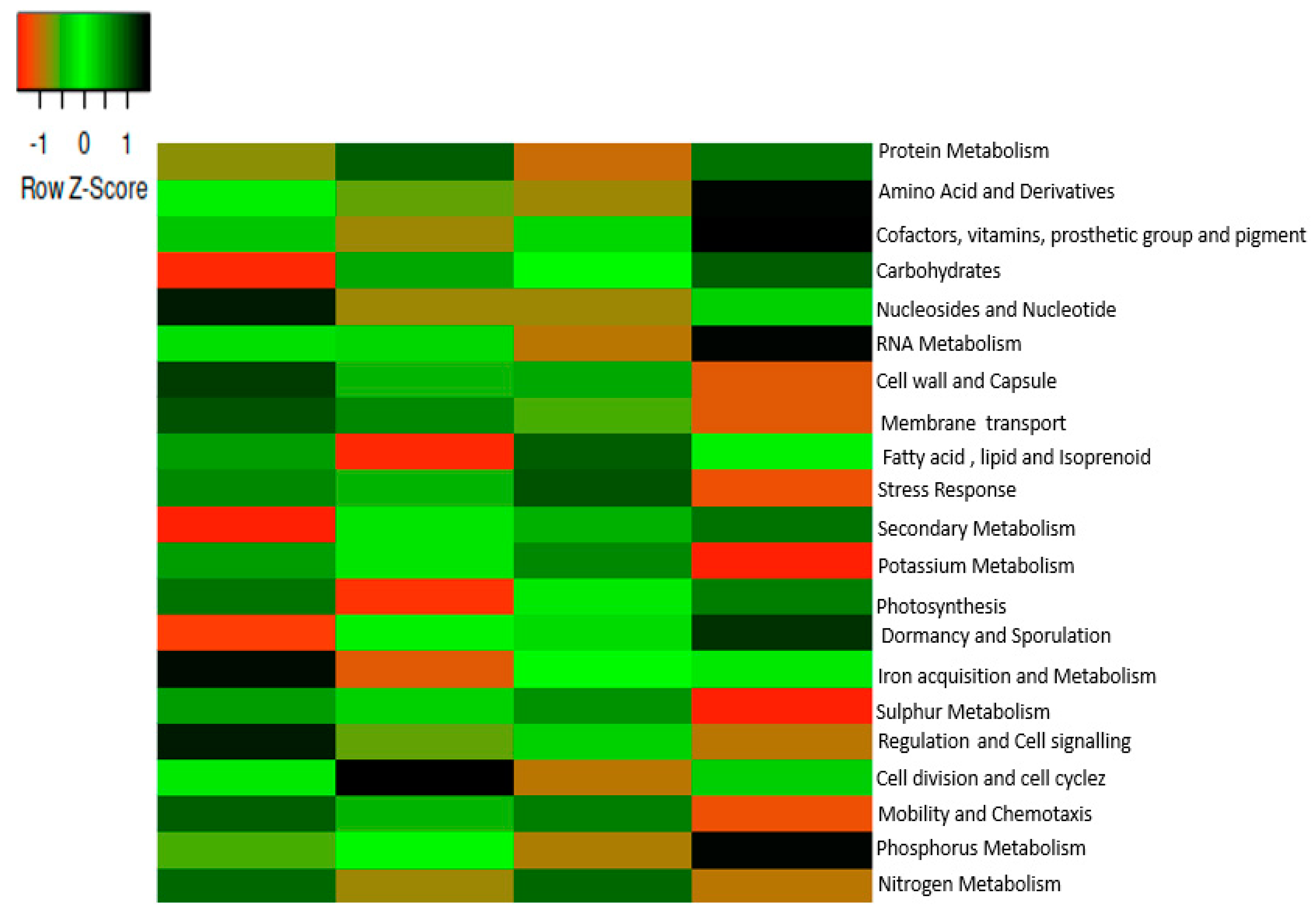
2.4. Functions of Microbiomes in the Soil
2.5. Microbiome Indices of Function in Soybean Soil Samples
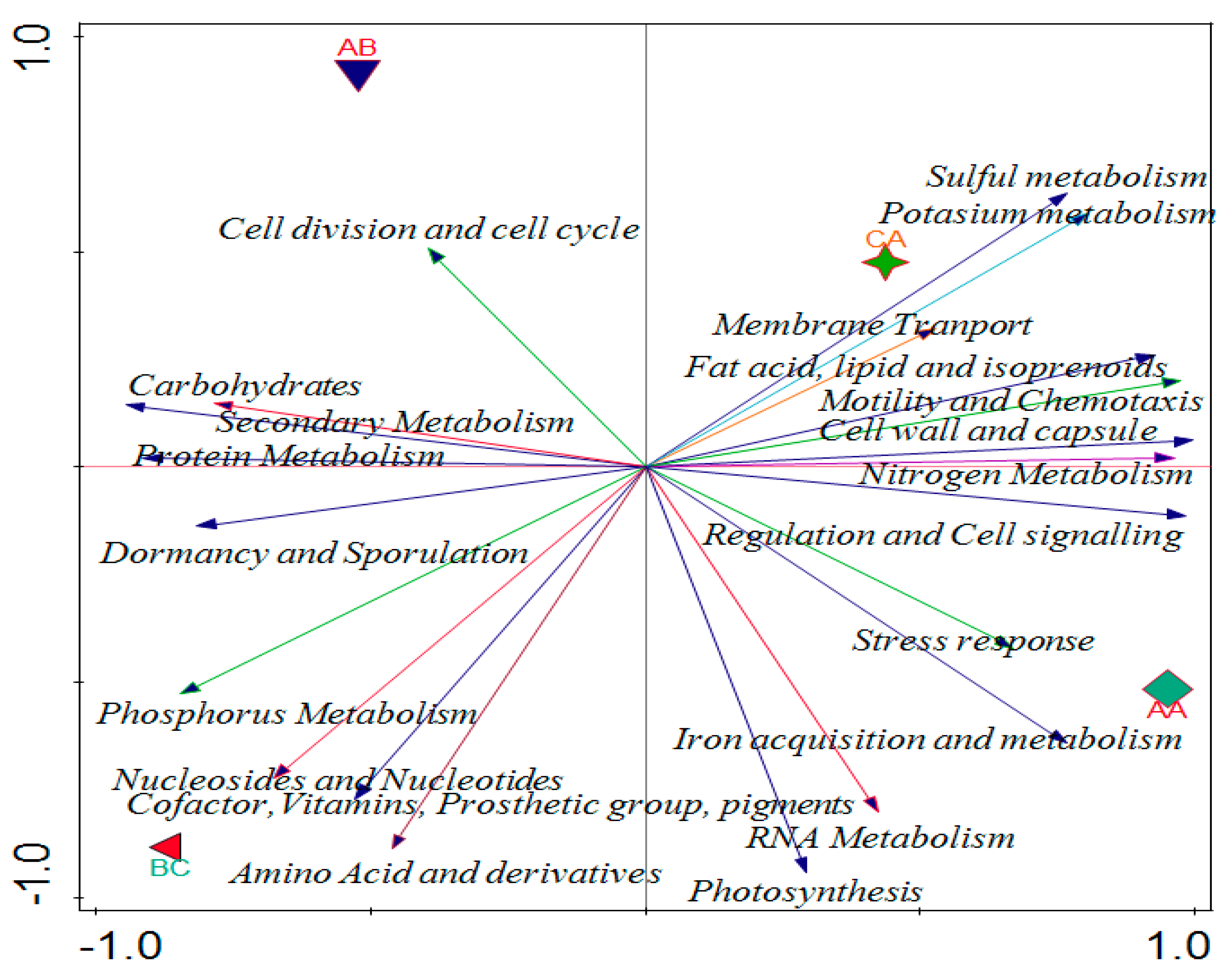
2.6. Effects of Soil Properties on the Functional Categories of the Microbiome in the Soil Samples
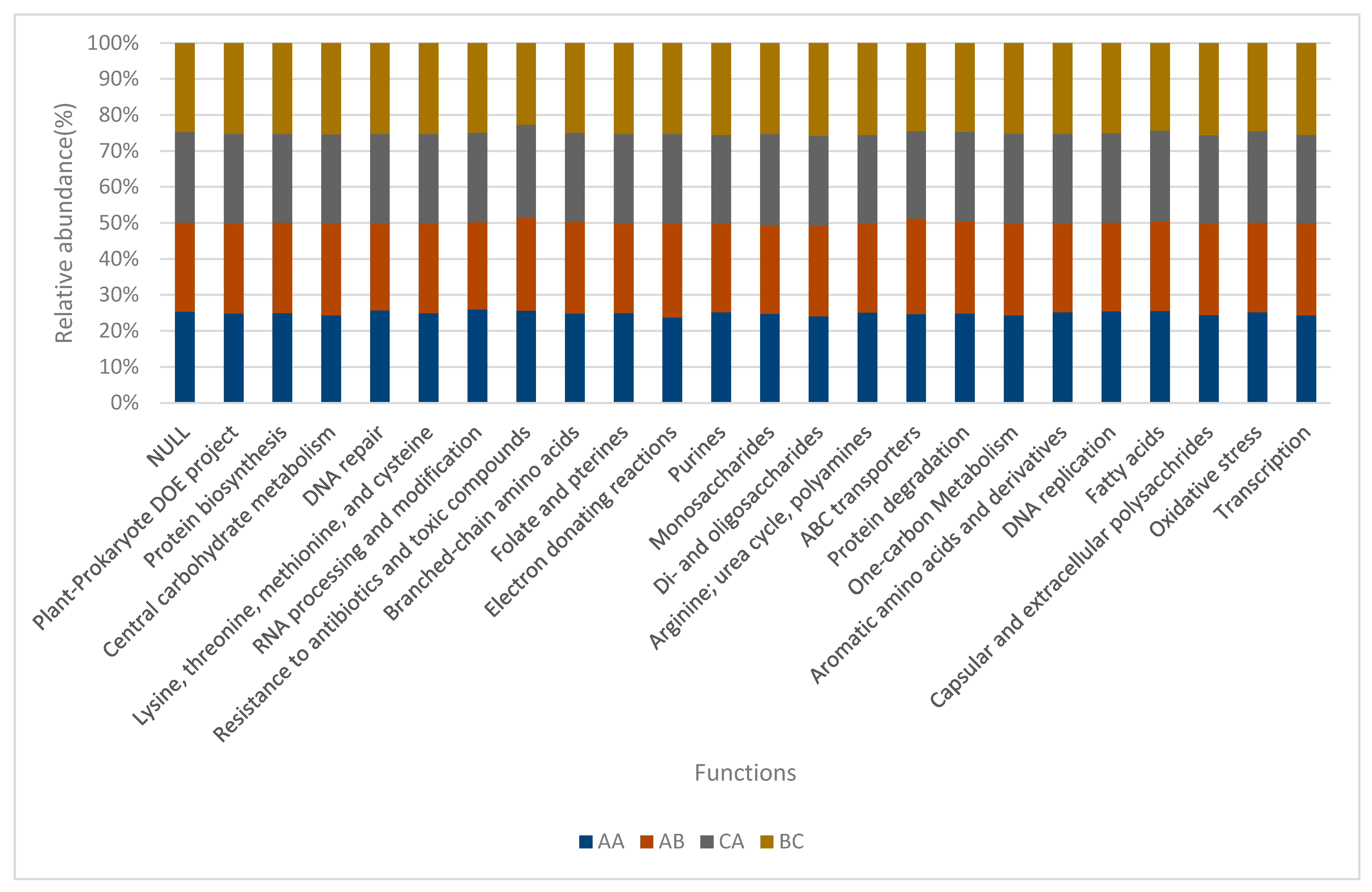
2.7. Pathways Revealing the Functions of Microbiomes Living in the Soil Samples
3. Discussion
4. Material and Method
4.1. Soil Sample Collection
4.2. Physicochemical Analysis of the Soil Samples
4.3. Extraction of DNA and Sample Sequencing
4.4. Metagenomics Data Annotation and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakei, M.D.; Venkataramana, P.B.; Ndakidemi, P.A. Soybean-nodulating rhizobia: Ecology, characterization, diversity, and growth promoting functions. Front. Sustain. Food Syst. 2022, 6, 824444. [Google Scholar] [CrossRef]
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean—Worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Fernandez-Gnecco, G.; Smalla, K.; Maccario, L.; Sørensen, S.J.; Barbieri, P.; Consolo, V.F.; Covacevich, F.; Babin, D. Microbial community analysis of soils under different soybean cropping regimes in the Argentinean south-eastern Humid Pampas. FEMS Microbiol. Ecol. 2021, 97, fiab007. [Google Scholar] [CrossRef] [PubMed]
- Santos, M. Soybean varieties in sub-Saharan Africa. Afr. J. Food Agric. Nutr. Dev. 2019, 19, 15136–15139. [Google Scholar] [CrossRef]
- Sugiyama, A.; Ueda, Y.; Zushi, T.; Takase, H.; Yazaki, K. Changes in the bacterial community of soybean rhizospheres during growth in the field. PLoS ONE 2014, 9, e100709. [Google Scholar]
- Trivedi, P.; Batista, B.D.; Bazany, K.E.; Singh, B.K. Plant–microbiome interactions under a changing world: Responses, consequences and perspectives. New Phytol. 2022, 234, 1951–1959. [Google Scholar] [CrossRef]
- Singh, B.K.; Trivedi, P.; Egidi, E.; Macdonald, C.A.; Delgado-Baquerizo, M. Crop microbiome and sustainable agriculture. Nat. Rev. Microbiol. 2020, 18, 601–602. [Google Scholar] [CrossRef]
- Sugiyama, A. The soybean rhizosphere: Metabolites, microbes, and beyond—A review. J. Adv. Res. 2019, 19, 67–73. [Google Scholar] [CrossRef]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; Van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Singh, R.P.; Meng, C.; Ma, S.; Jing, C.; Li, Y.; Zhang, C. Nodule and root zone microbiota of salt-tolerant wild soybean in coastal sand and saline-alkali soil. Front. Microbiol. 2020, 11, 2178. [Google Scholar] [CrossRef]
- Chukwuneme, C.F.; Ayangbenro, A.S.; Babalola, O.O.; Raphael Kutu, F. Functional diversity of microbial communities in two contrasting maize rhizosphere soils. Rhizosphere 2021, 17, 100282. [Google Scholar] [CrossRef]
- Odelade, K.A.; Babalola, O.O. Bacteria, fungi and archaea domains in rhizospheric soil and their effects in enhancing agricultural productivity. Int. J. Envion. Res. Public Health 2019, 16, 3873. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Lagos, L.; Maruyama, F.; Nannipieri, P.; Mora, M.; Ogram, A.; Jorquera, M. Current overview on the study of bacteria in the rhizosphere by modern molecular techniques: A mini–review. J. Soil Sci. Plant Nutr. 2015, 15, 504–523. [Google Scholar]
- LeBlanc, N.; Kinkel, L.L.; Kistler, H.C. Soil fungal communities respond to grassland plant community richness and soil edaphics. Microb. Ecol. 2015, 70, 188–195. [Google Scholar] [CrossRef]
- Spence, C.; Alff, E.; Johnson, C.; Ramos, C.; Donofrio, N.; Sundaresan, V.; Bais, H. Natural rice rhizospheric microbes suppress rice blast infections. BMC Plant Biol. 2014, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Meena, R.S.; Vijayakumar, V.; Yadav, G.S.; Mitran, T. Response and interaction of Bradyrhizobium japonicum and arbuscular mycorrhizal fungi in the soybean rhizosphere. Plant Growth Regul. 2018, 84, 207–223. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Mendes, L.W.; Braga, L.P.P.; Navarrete, A.A.; Souza, D.G.D.; Silva, G.G.Z.; Tsai, S.M. Using metagenomics to connect microbial community biodiversity and functions. Curr. Issues Mol. Biol. 2017, 24, 103–118. [Google Scholar] [CrossRef]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 2019, 19, 1–19. [Google Scholar] [CrossRef]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.J.N. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Kong, W.; Wu, Y.; Wang, J. Effect of monoculture soybean on soil microbial community in the Northeast China. Plant Soil 2010, 330, 423–433. [Google Scholar] [CrossRef]
- Kumar, A.; Somasundaram, J.; Biswas, A.; Sinha, N.K.; Mishra, V.; Chaudhary, R.; Mohanty, M.; Hati, K.; Saha, R.; Patra, A. Short-term effect of conservation agriculture practices on soil quality of a vertisol in central India. Appl. Biol. Res. 2017, 19, 26–34. [Google Scholar] [CrossRef]
- Dubey, A.; Malla, M.A.; Kumar, A. Taxonomical and functional bacterial community profiling in disease-resistant and disease-susceptible soybean cultivars. Braz. J. Microbiol. 2022, 1–16. [Google Scholar] [CrossRef]
- Prabha, R.; Singh, D.P.; Gupta, S.; Gupta, V.K.; El-Enshasy, H.A.; Verma, M.K. Rhizosphere metagenomics of Paspalum scrobiculatum L.(kodo millet) reveals rhizobiome multifunctionalities. Microorganisms 2019, 7, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianoulis, T.A.; Raes, J.; Patel, P.V.; Bjornson, R.; Korbel, J.O.; Letunic, I.; Yamada, T.; Paccanaro, A.; Jensen, L.J.; Snyder, M. Quantifying environmental adaptation of metabolic pathways in metagenomics. Proc. Natl. Acad. Sci. USA 2009, 106, 1374–1379. [Google Scholar] [CrossRef] [Green Version]
- Rigobelo, E.C.; Baron, N.C. Endophytic fungi: A tool for plant growth promotion and sustainable agriculture. Mycology 2022, 13, 39–55. [Google Scholar]
- Ohyama, T.; Ohtake, N.; Sueyoshi, K.; Ono, Y.; Tsutsumi, K.; Ueno, M.; Tanabata, S.; Sato, T.; Takahashi, Y. Amino acid metabolism and transport in soybean plants. Amino Acid-New Insights Roles Plant Anima 2017, 171–196. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Sun, S.; Ji, J.; Wu, H.; Meng, F.; Zhang, M.; Zheng, X.; Wu, C.; Zhang, Z. Comparison of the rhizosphere bacterial communities of Zigongdongdou soybean and a high-methionine transgenic line of this cultivar. PLoS ONE 2014, 9, e103343. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, R.P.; Succurro, A.; Kopriva, S. Nitrogen Substrate Utilization in Three Rhizosphere Bacterial Strains Investigated Using Proteomics. Front. Microbiol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Nason, A.; McElroy, W.D. Modes of action of the essential mineral elements. Plant Physiol. 1963, 3, 451–536. [Google Scholar]
- Fatima, Z.; Zia, M.; Chaudhary, M.F. Interactive effect of Rhizobium strains and P on soybean yield, nitrogen fixation and soil fertility. Pak. J. Bot. 2007, 39, 255. [Google Scholar]
- Yin, H.; Zhang, X.; Li, X.; He, Z.; Liang, Y.; Guo, X.; Hu, Q.; Xiao, Y.; Cong, J.; Ma, L. Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans. BMC Microbiol. 2014, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Niu, J.; Liang, Y.; Liu, X.; Yin, H. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 2016, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Ohtsu, I. L-Cysteine metabolism and fermentation in microorganisms. Amino Acid Ferment. 2016, 129–151. [Google Scholar] [CrossRef]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar] [CrossRef]
- Klimek, B.; Chodak, M.; Jaźwa, M.; Niklińska, M. Functional diversity of soil microbial communities in boreal and temperate Scots pine forests. Eur. J. For. Res. 2016, 135, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.M.; Wang, M.K.; Zhuang, S.Y.; Chiang, P.N. Chemical and physical properties of rhizosphere and bulk soils of three tea plants cultivated in Ultisols. Geoderma 2006, 136, 378–387. [Google Scholar] [CrossRef]
- Sáez-Plaza, P.; Navas, M.J.; Wybraniec, S.; Michałowski, T.; Asuero, A.G. An overview of the Kjeldahl method of nitrogen determination. Part II. Sample preparation, working scale, instrumental finish, and quality control. Crit. Rev. Anal. Chem. 2013, 43, 224–272. [Google Scholar] [CrossRef]
- Bray, R.H.; Kurtz, L.T. Determination of total, organic, and available forms of phosphorus in soils. Soil Sci. 1945, 59, 39–46. [Google Scholar] [CrossRef]
- Kachurina, O.; Zhang, H.; Raun, W.; Krenzer, E. Simultaneous determination of soil aluminum, ammonium-and nitrate-nitrogen using 1M potassium chloride extraction. Commun. Soil Sci. Plant Anal. 2000, 31, 893–903. [Google Scholar] [CrossRef]
- Wright, A.F.; Bailey, J.S. Organic carbon, total carbon, and total nitrogen determinations in soils of variable calcium carbonate contents using a Leco CN-2000 dry combustion analyzer. Commun. Soil Sci. Plant Anal. 2001, 32, 3243–3258. [Google Scholar] [CrossRef]
- Hoogsteen, M.J.; Lantinga, E.A.; Bakker, E.J.; Groot, J.C.; Tittonell, P.A. Estimating soil organic carbon through loss on ignition: Effects of ignition conditions and structural water loss. Eur. J. Soil Sci. 2015, 66, 320–328. [Google Scholar] [CrossRef]
- Walkley, A.; Black, I.A. An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Khomtchouk, B.B.; Hennessy, J.R.; Wahlestedt, C. shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE 2017, 12, e0176334. [Google Scholar] [CrossRef] [Green Version]
- Ter Braak, C.J.; Smilauer, P. Canoco Reference Manual and User’s Guide: Software for Ordination, Version 5.0; Microcomputer Power: Ithaca, NY, USA, 2012. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | AA | AB | CA | BC |
|---|---|---|---|---|
| Sand (%) | 88.00 ± 1.53 a | 86.00 ± 2.00 a,b | 88.30 ± 0.58 a | 84.00 ± 1.00 b |
| Silt (%) | 2.00 ± 1.00 a | 4.00 ± 1.00 a | 2.00 ± 1.00 a | 2.00 ± 1.00 a |
| Clay (%) | 10.00 ± 2.00 b | 10.00 ± 1.00 b | 10.00 ± 1.00 b | 14.00 ± 2.00 a |
| pH | 6.94 ± 0.04 a | 6.80 ± 0.03 b | 6.58 ± 0.03 c | 6.63 ± 0.01 c |
| S (mg/kg) | 543 ± 3.00 b | 563 ± 0.01 a | 501 ± 1.00 c | 496.3 ± 0.58 d |
| Org C (%) | 0.36 ± 0.03 b | 0.24 ± 0.02 d | 0.30 ± 0.02 c | 0.63 ± 0.01 a |
| Org M (%) | 1.62 ± 0.02 b | 1.44 ± 0.01 c | 1.40 ± 0.02 c | 2.66 ± 0.01 a |
| P (mg/kg) | 46.68 ± 0.01 b | 41.57 ± 0.03 c | 48.59 ± 0.01 a | 7.40 ± 0.01 d |
| K (mg/kg) | 81.46 ± 0.02 d | 93.14 ± 0.02 c | 97.48 ± 0.02 b | 106.58 ± 0.03 a |
| Na (cmol(+)/kg | 10.23 ± 0.03 a | 9.74 ± 0.04 b | 8.49 ± 0.00 c | 8.52 ± 0.03 c |
| N-NO3− (mg/kg) | 2.29 ± 0.03 b | 3.48 ± 0.01 a | 2.22 ± 0.01 c | 2.01 ± 0.01 d |
| N-NH4+ (mg/kg) | 9.83 ± 0.01 c | 12.64 ± 0.03 a | 10.33 ± 0.01 b | 5.90 ± 0.01 d |
| Total C (%) | 0.38 ± 0.005 b | 0.26 ± 0.005 d | 0.32 ± 0.00 c | 0.64 ± 0.00 a |
| Total N (%) | 0.04 ± 0.002 b | 0.03 ± 0.003 d | 0.03 ± 0.00 c | 0.06 ± 0.00 a |
| Diversity | Sample AA | Sample AB | Sample CA | Sample BC |
|---|---|---|---|---|
| Shannon | 1.851 | 1.947 | 1.815 | 1.763 |
| Simpson | 0.6712 | 0.7152 | 0.6652 | 0.6408 |
| Evenness | 0.1027 | 0.113 | 0.08902 | 0.09257 |
| Name | Explain% | Contribution% | Pseudo-F | p-Value |
|---|---|---|---|---|
| Total N | 30.8 | 30.8 | 0.90 | 0.91 |
| P | 64.3 | 64.3 | 13.2 | 0.718 |
| K | 4.9 | 4.9 | <0.1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajiboye, T.T.; Ayangbenro, A.S.; Babalola, O.O. Functional Diversity of Microbial Communities in the Soybean (Glycine max L.) Rhizosphere from Free State, South Africa. Int. J. Mol. Sci. 2022, 23, 9422. https://doi.org/10.3390/ijms23169422
Ajiboye TT, Ayangbenro AS, Babalola OO. Functional Diversity of Microbial Communities in the Soybean (Glycine max L.) Rhizosphere from Free State, South Africa. International Journal of Molecular Sciences. 2022; 23(16):9422. https://doi.org/10.3390/ijms23169422
Chicago/Turabian StyleAjiboye, Titilope Tinu, Ayansina Segun Ayangbenro, and Olubukola Oluranti Babalola. 2022. "Functional Diversity of Microbial Communities in the Soybean (Glycine max L.) Rhizosphere from Free State, South Africa" International Journal of Molecular Sciences 23, no. 16: 9422. https://doi.org/10.3390/ijms23169422
APA StyleAjiboye, T. T., Ayangbenro, A. S., & Babalola, O. O. (2022). Functional Diversity of Microbial Communities in the Soybean (Glycine max L.) Rhizosphere from Free State, South Africa. International Journal of Molecular Sciences, 23(16), 9422. https://doi.org/10.3390/ijms23169422