
Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease


Abstract
:1. Introduction
2. Chemical Induced Colitis
2.1. Oxazolone Colitis
2.2. TNBS-Induced Colitis
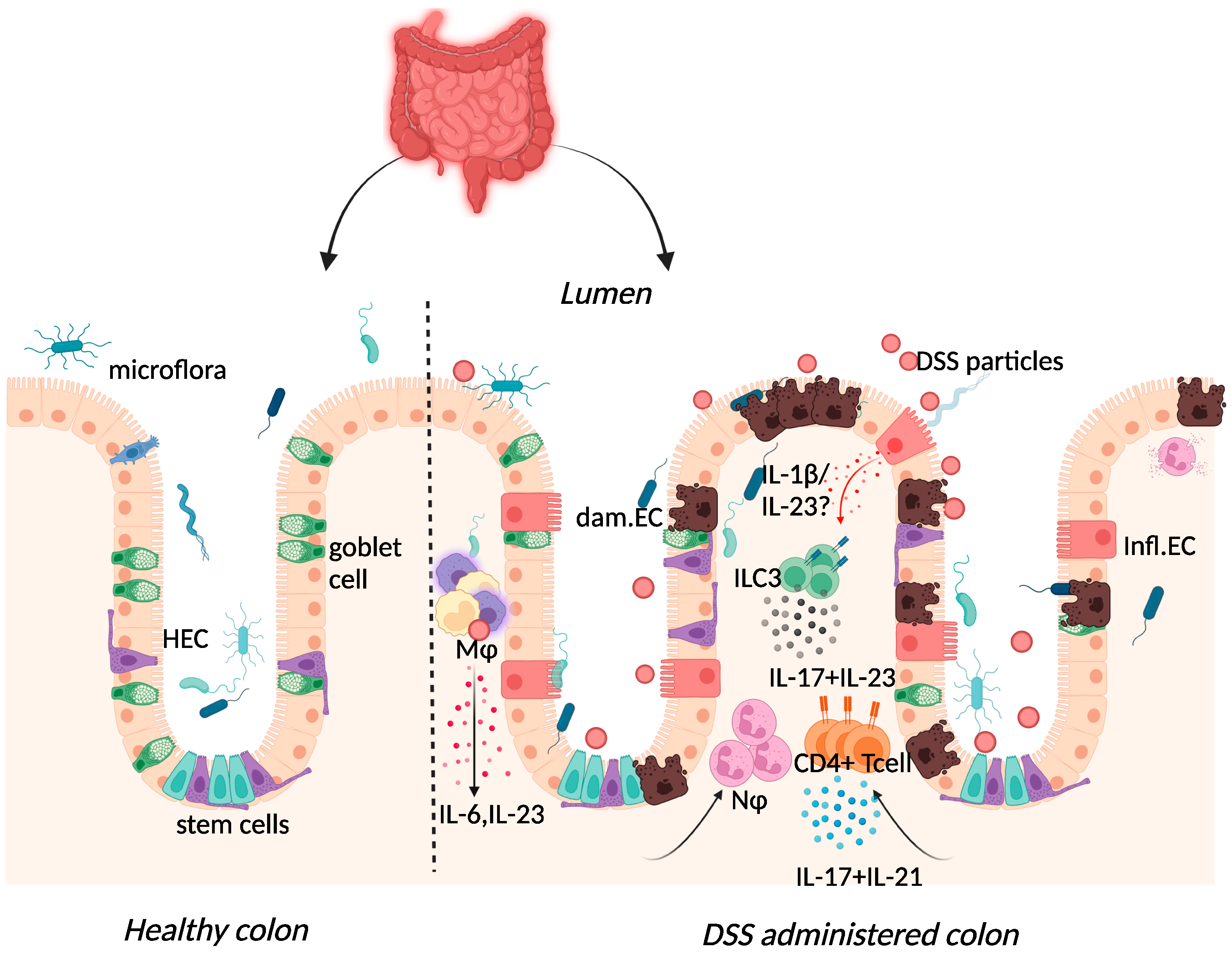
2.3. Dextran-Sulphate-Sodium-Induced Colitis
3. Spontaneous Colitis
3.1. Iκκ-γ (NEMO) Deficiency Colitis
3.2. Interleukin-10 (IL-10) Deficiency Colitis
4. Immune Cell Induced Colitis
T-Cell Adoptive Transfer Model
5. Conclusions
- Mice and humans differ in their immune responses due to different immune system development, immune activation, and immune responses to similar antigens [154].
- Mouse experiments tend to not recapitulate the genetic and environmental diversity inherent in human populations, although these can be closely modelled through diet, microbiome, and environmental manipulations.
- To determine the importance of certain genes in disease pathology, researchers tend to depend on transgenic knockout mice or antibody depletion, whereas human disease risk is rarely associated with the complete loss of function of a single gene or protein.
- Most pre-clinical murine experiments fail to account for variability in response to a therapeutic intervention that may appear in human trials due to genetic polymorphisms. This may be addressed through the use of outbred mice but may have ethical and economic implications, due to the likely need to use a large sample size to achieve statistical significance between treatment groups [155].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IBD | Inflammatory bowel disease |
| CD | Crohn’s disease |
| UC | Ulcerative colitis |
| TNBS | Trinitrobenzene sulfonic acid |
| DSS | Dextran sulphate sodium |
| OC | Oxazolone colitis |
| NKT | Natural killer T cell |
| IL- | Interleukin |
| TGF | Transforming growth factor |
| Infl.EC | Inflamed epithelial cells |
| HEC | Healthy epithelial cells |
| Dam.EC | Damaged epithelial cell |
| CD | Cluster of differentiation |
| ILC | Innate lymphoid cell |
| NOD2 | Nucleotide-binding and oligomerisation domain-containing 2 |
| SCID | Severe combined immunodeficiency |
| NF-κB | Nuclear factor kappa B |
| NEMO | NF-kB essential modulator (NEMO) |
| Iκκ-γ | I-kappa-B kinase gamma |
| RAG | Recombination-activating gene |
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Kappelman, M.D.; Rifas–Shiman, S.L.; Porter, C.Q.; Ollendorf, D.A.; Sandler, R.S.; Galanko, J.A.; Finkelstein, J.A. Direct Health Care Costs of Crohn’s Disease and Ulcerative Colitis in US Children and Adults. Gastroenterology 2008, 135, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone Colitis: A Murine Model of T Helper Cell Type 2 Colitis Treatable with Antibodies to Interleukin 4. J. Exp. Med. 1998, 188, 1929–1939. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; Van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stüber, E.; Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995, 182, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Powrie, F.; Leach, M.W.; Mauze, S.; Caddie, L.B.; Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 1993, 5, 1461–1471. [Google Scholar] [CrossRef]
- Heller, F.; Fuss, I.J.; E Nieuwenhuis, E.; Blumberg, R.S.; Strober, W. Oxazolone Colitis, a Th2 Colitis Model Resembling Ulcerative Colitis, Is Mediated by IL-13-Producing NK-T Cells. Immunity 2002, 17, 629–638. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, J.; Nakaguchi, T.; Gregersen, H. Biomechanical changes in oxazolone-induced colitis in BALB/C mice. J. Biomech. 2009, 42, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Kasaian, M.T.; Page, K.M.; Fish, S.; Brennan, A.; Cook, T.A.; Moreira, K.; Zhang, M.; Jesson, M.; Marquette, K.; Agostinelli, R.; et al. Therapeutic activity of an interleukin-4/interleukin-13 dual antagonist on oxazolone-induced colitis in mice. Immunology 2014, 143, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, Q.; Luo, W.J. Oxazolone-induced murine model of ulcerative colitis. Chin. J. Dig. Dis. 2004, 5, 165–168. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767.e1. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Strober, W. The role of IL-13 and NK T cells in experimental and human ulcerative colitis. Mucosal Immunol. 2008, 1 (Suppl. 1), S31–S33. [Google Scholar] [CrossRef]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef]
- Hoving, J.C.; Cutler, A.; Leeto, M.; Horsnell, W.G.C.; Dewals, B.G.; Nieuwenhuizen, N.; Brombacher, F. Interleukin 13-mediated colitis in the absence of IL-4Rα signalling. Gut 2017, 66, 2037–2039. [Google Scholar] [CrossRef]
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Iii, R.L.G.; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Rα2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186. [Google Scholar] [CrossRef]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Palamides, P.; Jodeleit, H.; Föhlinger, M.; Beigel, F.; Herbach, N.; Mueller, T.; Wolf, E.; Siebeck, M.; Gropp, R. A mouse model for ulcerative colitis based on NOD-scid IL2R gammanull mice reconstituted with peripheral blood mononuclear cells from affected individuals. Dis. Model. Mech. 2016, 9, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; Panes, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Rudziński, J.; Brandt, W.; Dupas, J.-L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, M.; Gapin, L. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2002, 2, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Hoving, J.C.; Kirstein, F.; Nieuwenhuizen, N.; Fick, L.C.; Hobeika, E.; Reth, M.; Brombacher, F. B Cells That Produce Immunoglobulin E Mediate Colitis in BALB/c Mice. Gastroenterology 2012, 142, 96–108. [Google Scholar] [CrossRef]
- Camelo, A.; Barlow, J.L.; Drynan, L.F.; Neill, D.R.; Ballantyne, S.J.; Wong, S.H.; Pannell, R.; Gao, W.; Wrigley, K.; Sprenkle, J.; et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J. Gastroenterol. 2012, 47, 1198–1211. [Google Scholar] [CrossRef]
- Forkel, M.; Van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 67–78. [Google Scholar] [CrossRef]
- De Salvo, C.; Buela, K.-A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 drives early IL-33–dependent expansion of group 2 innate lymphoid cells during Crohn’s disease–like ileitis. J. Clin. Investig. 2021, 131, e140624. [Google Scholar] [CrossRef]
- Nolte, T.; Zadeh-Khorasani, M.; Safarov, O.; Rueff, F.; Gülberg, V.; Herbach, N.; Wollenberg, A.; Mueller, T.; Siebeck, M.; Wolf, E.; et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rγnull mice engrafted with human peripheral blood mononuclear cells. Clin. Exp. Immunol. 2013, 172, 349–362. [Google Scholar] [CrossRef]
- Velde, A.A.T.; Verstege, M.I.; Hommes, D.W. Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm. Bowel Dis. 2006, 12, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Fichtner-Feigl, S.; Fuss, I.J.; Preiß, J.; Strober, W.; Kitani, A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF- B decoy oligonucleotides. J. Clin. Investig. 2005, 115, 3057–3071. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; De La Motte, C.; A Strong, S.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [PubMed]
- Dohi, T.; Fujihashi, K.; Rennert, P.D.; Iwatani, K.; Kiyono, H.; McGhee, J.R. Hapten-induced Colitis Is Associated with Colonic Patch Hypertrophy and T Helper Cell 2–Type Responses. J. Exp. Med. 1999, 189, 1169–1180. [Google Scholar] [CrossRef]
- Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014, 25, 415–421. [Google Scholar] [CrossRef]
- Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; Malefyt, R.D.W.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol. Rev. 2004, 202, 96–105. [Google Scholar] [CrossRef]
- Fuss, I.J.; Becker, C.; Yang, Z.; Groden, C.; Hornung, R.L.; Heller, F.; Neurath, M.F.; Strober, W.; Mannon, P.J. Both IL-12p70 and IL-23 are synthesized during active Crohnʼs disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 2006, 12, 9–15. [Google Scholar] [CrossRef]
- Mannon, P.J.; Fuss, I.J.; Mayer, L.; Elson, C.O.; Sandborn, W.J.; Present, D.; Dolin, B.; Goodman, N.; Groden, C.; Hornung, R.L.; et al. Anti–Interleukin-12 Antibody for Active Crohn’s Disease. N. Engl. J. Med. 2004, 351, 2069–2079. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef]
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohn’s Colitis 2022, 16, ii3–ii19. [Google Scholar] [CrossRef]
- Hugot, J.-P.; Laurent-Puig, P.; Gower, C.; Olson, J.M.; Lee, J.C.; Beaugerie, L.; Naom, I.; Dupas, J.-L.; Van Gossum, A.; Af, G.D.T.D.; et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 1996, 379, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Amendola, A.; Butera, A.; Sanchez, M.; Strober, W.; Boirivant, M. Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol. 2014, 7, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Kawakita, M.; Dussaillant, M.; Foligné, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 Is Required for Peyer’s Patches Homeostasis in Mice. PLoS ONE 2007, 2, e523. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-Dependent Regulation of Innate and Adaptive Immunity in the Intestinal Tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hsu, L.-C.; Liu, H.; Bankston, L.A.; Iimura, M.; Kagnoff, M.F.; Eckmann, L.; Karin, M. Nod2 Mutation in Crohn’s Disease Potentiates NF-κB Activity and IL-1ß Processing. Science 2005, 307, 734–738. [Google Scholar] [CrossRef]
- Watanabe, T.; Kitani, A.; Murray, P.J.; Wakatsuki, Y.; Fuss, I.J.; Strober, W. Nucleotide Binding Oligomerization Domain 2 Deficiency Leads to Dysregulated TLR2 Signaling and Induction of Antigen-Specific Colitis. Immunity 2006, 25, 473–485. [Google Scholar] [CrossRef]
- Yang, Z.; Fuss, I.J.; Watanabe, T.; Asano, N.; Davey, M.P.; Rosenbaum, J.T.; Strober, W.; Kitani, A. NOD2 Transgenic Mice Exhibit Enhanced MDP-Mediated Down-Regulation of TLR2 Responses and Resistance to Colitis Induction. Gastroenterology 2007, 133, 1510–1521. [Google Scholar] [CrossRef]
- Butera, A.; Di Paola, M.; Pavarini, L.; Strati, F.; Pindo, M.; Sanchez, M.; Cavalieri, D.; Boirivant, M.; De Filippo, C. Nod2 Deficiency in mice is Associated with Microbiota Variation Favouring the Expansion of mucosal CD4+ LAP+ Regulatory Cells. Sci. Rep. 2018, 8, 14241. [Google Scholar] [CrossRef]
- Cohen, L.J.; Cho, J.H.; Gevers, D.; Chu, H. Genetic Factors and the Intestinal Microbiome Guide Development of Microbe-Based Therapies for Inflammatory Bowel Diseases. Gastroenterology 2019, 156, 2174–2189. [Google Scholar] [CrossRef]
- Hall, A.B.; Tolonen, A.; Xavier, R.J. Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet. 2017, 18, 690–699. [Google Scholar] [CrossRef]
- Hu, S.; Vila, A.V.; Gacesa, R.; Collij, V.; Stevens, C.; Fu, J.M.; Wong, I.; E Talkowski, M.; A Rivas, M.; Imhann, F.; et al. Whole exome sequencing analyses reveal gene–microbiota interactions in the context of IBD. Gut 2021, 70, 285–296. [Google Scholar] [CrossRef]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef]
- Abad, C.; Martinez, C.; Juarranz, M.G.; Arranz, A.; Leceta, J.; Delgado, M.; Gomariz, R.P. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology 2003, 124, 961–971. [Google Scholar] [CrossRef]
- Laroui, H.; Ingersoll, S.A.; Liu, H.C.; Baker, M.T.; Ayyadurai, S.; Charania, M.A.; Laroui, F.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Dextran Sodium Sulfate (DSS) Induces Colitis in Mice by Forming Nano-Lipocomplexes with Medium-Chain-Length Fatty Acids in the Colon. PLoS ONE 2012, 7, e32084. [Google Scholar] [CrossRef]
- Kitajima, S.; Takuma, S.; Morimoto, M. Tissue Distribution of Dextran Sulfate Sodium(DSS) in the Acute Phase of Murine DSS-Induced Colitis. J. Vet. Med. Sci. 1999, 61, 67–70. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Perše, M.; Cerar, A. Dextran Sodium Sulphate Colitis Mouse Model: Traps and Tricks. J. Biomed. Biotechnol. 2012, 2012, 718617. [Google Scholar] [CrossRef]
- Kim, S.H.; Kwon, D.; Son, S.W.; Bin Jeong, T.; Lee, S.; Kwak, J.-H.; Cho, J.-Y.; Hwang, D.Y.; Seo, M.-S.; Kim, K.S.; et al. Inflammatory responses of C57BL/6NKorl mice to dextran sulfate sodium-induced colitis: Comparison between three C57BL/6 N sub-strains. Lab. Anim. Res. 2021, 37, 8. [Google Scholar] [CrossRef]
- A Dieleman, L.; Palmen, M.J.H.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.M.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef]
- Stevceva, L.; Pavli, P.; Husband, A.; Ramsay, A.; Doe, W. Dextran sulphate sodium-induced colitis is ameliorated in interleukin 4 deficient mice. Genes Immun. 2001, 2, 309–316. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: Correlation between symptoms and inflammation. Am. J. Physiol. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. [Google Scholar] [CrossRef]
- Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305. [Google Scholar] [CrossRef]
- Tanaka, M.; Riddell, R.H.; Saito, H.; Soma, Y.; Hidaka, H.; Kudo, H. Morphologic Criteria Applicable to Biopsy Specimens for Effective Distinction of Inflammatory Bowel Disease from Other Forms of Colitis and of Crohn’s Disease from Ulcerative Colitis. Scand. J. Gastroenterol. 1999, 34, 55–67. [Google Scholar] [CrossRef]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Gheita, T.A.; El Gazzar, I.I.; El-Fishawy, H.S.; Aboul-Ezz, M.A.; Kenawy, S.A. Involvement of IL-23 in enteropathic arthritis patients with inflammatory bowel disease: Preliminary results. Clin. Rheumatol. 2014, 33, 713–717. [Google Scholar] [CrossRef]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Agents Actions 2014, 63, 943–950. [Google Scholar] [CrossRef]
- Lucaciu, L.A.; Ilieș, M.; Vesa, C.; Seicean, R.; Din, S.; Iuga, C.A.; Seicean, A. Serum Interleukin (IL)-23 and IL-17 Profile in Inflammatory Bowel Disease (IBD) Patients Could Differentiate between Severe and Non-Severe Disease. J. Pers. Med. 2021, 11, 1130. [Google Scholar] [CrossRef]
- Mirsattari, D.; Seyyedmajidi, M.; Zojaji, H.; Haghazali, M.; Orimi, P.G.; Shoushtarizadeh, T.; Almasi, S. The relation between the level of interleukin-23 with duration and severity of ulcerative colitis. Gastroenterol. Hepatol. Bed Bench 2012, 5, 49–53. [Google Scholar]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef]
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and Adaptive Interleukin-22 Protects Mice from Inflammatory Bowel Disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Kobayashi, T.; Okamoto, S.; Hisamatsu, T.; Kamada, N.; Chinen, H.; Saito, R.; Kitazume, M.T.; Nakazawa, A.; Sugita, A.; Koganei, K.; et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 2008, 57, 1682–1689. [Google Scholar] [CrossRef]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Cárcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef]
- Caruso, R.; Fina, D.; Peluso, I.; Stolfi, C.; Fantini, M.C.; Gioia, V.; Caprioli, F.; Blanco, G.D.V.; Paoluzi, O.A.; MacDonald, T.T.; et al. A Functional Role for Interleukin-21 in Promoting the Synthesis of the T-Cell Chemoattractant, MIP-3α, by Gut Epithelial Cells. Gastroenterology 2007, 132, 166–175. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, M.H.; Ju, A.S.; Rhee, K.-J. Th17 Responses Are Not Induced in Dextran Sodium Sulfate Model of Acute Colitis. Immune Netw. 2011, 11, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Andoh, A.; Araki, Y.; Bamba, T.; Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 2004, 110, 55–62. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Kita, M.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Iwakura, Y.; Okanoue, T.; et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem. Biophys. Res. Commun. 2008, 377, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Kakuta, S.; Shimizu, K.; Kadoki, M.; Kamiya, T.; Shimazu, T.; Kubo, S.; Saijo, S.; Ishigame, H.; Nakae, S.; et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat. Immunol. 2018, 19, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Cao, P.; Su, W.; Zhan, N.; Dong, W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF-κB via the upregulation of CARD3 expression. J. Pathol. 2020, 250, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; E Sands, B.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients with Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef]
- Glatt, S.; Helmer, E.; Haier, B.; Strimenopoulou, F.; Price, G.; Vajjah, P.; Harari, O.A.; Lambert, J.; Shaw, S. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br. J. Clin. Pharmacol. 2017, 83, 991–1001. [Google Scholar] [CrossRef]
- Gordon, K.B.; Foley, P.; Krueger, J.G.; Pinter, A.; Reich, K.; Vender, R.; Vanvoorden, V.; Madden, C.; White, K.; Cioffi, C.; et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): A multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet 2021, 397, 475–486. [Google Scholar] [CrossRef]
- Reimund, J.M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Baumann, R.; Poindron, P.; Duclos, B. Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut 1996, 39, 684–689. [Google Scholar] [CrossRef]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Ma¨nnel, D.N.; Andus, T.; Scho¨lmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997, 107, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Diao, L.-H.; Gong, Y.; Liu, X.; Li, Y. NEMO differentially regulates TCR and TNF-α induced NF-κB pathways and has an inhibitory role in TCR-induced NF-κB activation. Cell. Signal. 2012, 24, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Hanauer, S.B.; Van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A Short-Term Study of Chimeric Monoclonal Antibody cA2 to Tumor Necrosis Factor α for Crohn’s Disease. N. Engl. J. Med. 1997, 337, 1029–1036. [Google Scholar] [CrossRef]
- Kopylov, U.; Seidman, E. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2016, 9, 513–526. [Google Scholar] [CrossRef]
- Dahmus, J.; Rosario, M.; Clarke, K. Risk of Lymphoma Associated with Anti-TNF Therapy in Patients with Inflammatory Bowel Disease: Implications for Therapy. Clin. Exp. Gastroenterol. 2020, 13, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Schierova, D.; Roubalova, R.; Kolar, M.; Stehlikova, Z.; Rob, F.; Jackova, Z.; Coufal, S.; Thon, T.; Mihula, M.; Modrak, M.; et al. Fecal Microbiome Changes and Specific Anti-Bacterial Response in Patients with IBD during Anti-TNF Therapy. Cells 2021, 10, 3188. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Polykratis, A.; Welz, P.-S.; van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2015, 65, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Fellermann, K.; Stange, D.E.; Schaeffeler, E.; Schmalzl, H.; Wehkamp, J.; Bevins, C.L.; Reinisch, W.; Teml, A.; Schwab, M.; Lichter, P.; et al. A Chromosome 8 Gene-Cluster Polymorphism with Low Human Beta-Defensin 2 Gene Copy Number Predisposes to Crohn Disease of the Colon. Am. J. Hum. Genet. 2006, 79, 439–448. [Google Scholar] [CrossRef]
- Vlantis, K.; Wullaert, A.; Polykratis, A.; Kondylis, V.; Dannappel, M.; Schwarzer, R.; Welz, P.; Corona, T.; Walczak, H.; Weih, F.; et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. [Google Scholar] [CrossRef]
- Kohn, L.L.; Braun, M.; Cordoro, K.M.; McCalmont, T.H.; Shah, S.D.; Frieden, I.J.; Mathur, A.N. Skin and Mucosal Manifestations in NEMO Syndrome: A Case Series and Literature Review. Pediatr. Dermatol. 2022, 39, 84–90. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Heyman, M.B.; Kirschner, B.S.; Gold, B.D.; Ferry, G.; Baldassano, R.; Cohen, S.A.; Winter, H.S.; Fain, P.; King, C.; Smith, T.; et al. Children with early-onset inflammatory bowel disease (IBD): Analysis of a pediatric IBD consortium registry. J. Pediatr. 2005, 146, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterol. Res. 2017, 10, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Davidson, N.; Kühn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Investig. 1996, 98, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef]
- Spencer, S.D.; Di Marco, F.; Hooley, J.; Pitts-Meek, S.; Bauer, M.; Ryan, A.M.; Sordat, B.; Gibbs, V.C.; Aguet, M. The Orphan Receptor CRF2-4 Is an Essential Subunit of the Interleukin 10 Receptor. J. Exp. Med. 1998, 187, 571–578. [Google Scholar] [CrossRef]
- Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.-H.; Vercauteren, K.; Jude, K.M.; Xiong, A.; Moraga, I.; Horton, T.M.; Glenn, J.S.; de Jong, Y.P.; et al. The IFN-λ-IFN-λR1-IL-10Rβ Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity. Immunity 2017, 46, 379–392. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell–mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef]
- Eyerich, K.; DiMartino, V.; Cavani, A. IL-17 and IL-22 in immunity: Driving protection and pathology. Eur. J. Immunol. 2017, 47, 607–614. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, J.; Ouyang, X.; Liu, W.; Li, H.; Yang, J.; Bromberg, J.; Chen, S.; Mayer, L.; Unkeless, J.C.; et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur. J. Immunol. 2008, 38, 1807–1813. [Google Scholar] [CrossRef]
- Gunasekera, D.C.; Ma, J.; Vacharathit, V.; Shah, P.; Ramakrishnan, A.; Uprety, P.; Shen, Z.; Sheh, A.; Brayton, C.F.; Whary, M.T.; et al. The development of colitis in Il10−/− mice is dependent on IL-22. Mucosal Immunol. 2020, 13, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.-F.; Lémann, M.; Cassagnou, M.; Bouhnik, Y.; Duclos, B.; Dupas, J.-L.; Notteghem, B.; Mary, J.-Y. A Controlled Trial Comparing Ciprofloxacin With Mesalazine for The Treatment of Active Crohn’s Disease. Am. J. Gastroenterol. 1999, 94, 674–678. [Google Scholar] [CrossRef]
- Sutherland, L.; Singleton, J.; Sessions, J.; Hanauer, S.; Krawitt, E.; Rankin, G.; Summers, R.; Mekhjian, H.; Greenberger, N.; Kelly, M. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 1991, 32, 1071–1075. [Google Scholar] [CrossRef]
- Madsen, K.L.; Doyle, J.S.; Tavernini, M.M.; Jewell, L.D.; Rennie, R.P.; Fedorak, R.N. Antibiotic therapy attenuates colitis in interleukin 10 gene–deficient mice. Gastroenterology 2000, 118, 1094–1105. [Google Scholar] [CrossRef]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Burich, A.; Hershberg, R.; Waggie, K.; Zeng, W.; Brabb, T.; Westrich, G.; Viney, J.L.; Maggio-Price, L. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am. J. Physiol. Liver Physiol. 2001, 281, G764–G778. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Ward, J.M.; Gorelick, P.L.; Caspar, P.; Hieny, S.; Cheever, A.; Jankovic, D.; Sher, A. Helicobacter hepaticus Triggers Colitis in Specific-Pathogen-Free Interleukin-10 (IL-10)-Deficient Mice through an IL-12- and Gamma Interferon-Dependent Mechanism. Infect. Immun. 1998, 66, 5157–5166. [Google Scholar] [CrossRef]
- Balish, E.; Warner, T. Enterococcus faecalis Induces Inflammatory Bowel Disease in Interleukin-10 Knockout Mice. Am. J. Pathol. 2002, 160, 2253–2257. [Google Scholar] [CrossRef]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef]
- Madsen, K.L.; Doyle, J.S.; Jewell, L.D.; Tavernini, M.M.; Fedorak, R.N. Lactobacillus species prevents colitis in interleukin 10 gene–deficient mice. Gastroenterology 1999, 116, 1107–1114. [Google Scholar] [CrossRef]
- Madsen, K.; Cornish, A.; Soper, P.; McKaigney, C.; Jijon, H.; Yachimec, C.; Doyle, J.; Jewell, L.; De Simone, C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001, 121, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, Y.; Fu, W.; Cox, A.D.; Lee, D.; Reddivari, L. Intermittent antibiotic treatment accelerated the development of colitis in IL-10 knockout mice. Biomed. Pharmacother. 2022, 146, 112486. [Google Scholar] [CrossRef] [PubMed]
- van Deventer, S.; Elson, C.; Fedorak, R. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology 1997, 113, 383–389. [Google Scholar] [CrossRef]
- Fedorak, R.N.; Gangl, A.; Elson, C.O.; Rutgeerts, P.; Schreiber, S.; Wild, G.; Hanauer, S.B.; Kilian, A.; Cohard, M.; LeBeaut, A.; et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. Gastroenterology 2000, 119, 1473–1482. [Google Scholar] [CrossRef]
- Huhn, R.D.; Radwanski, E.; Gallo, J.; Affrime, M.B.; Sabo, R.; Gonyo, G.; Monge, A.; Cutler, D.L. Pharmacodynamics of subcutaneous recombinant human interleukin-10 in healthy volunteers*. Clin. Pharmacol. Ther. 1997, 62, 171–180. [Google Scholar] [CrossRef]
- Schreiber, S.; Fedorak, R.N.; Nielsen, O.H.; Wild, G.; Williams, C.; Nikolaus, S.; Jacyna, M.; Lashner, B.A.; Gangl, A.; Rutgeerts, P.; et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Gastroenterology 2000, 119, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Verma, A.; Moss, S.; Sorokin, P.; Blend, M.; Bradlow, B.; Chachlani, N.; Cutler, D.; Sabo, R.; Nelson, M.; et al. Interleukin 10-induced thrombocytopenia in normal healthy adult volunteers: Evidence for decreased platelet production. Br. J. Haematol. 2000, 111, 104–111. [Google Scholar] [CrossRef]
- Tilg, H.; Van Montfrans, C.; Ende, A.V.D.; Kaser, A.; Van Deventer, S.J.H.; Schreiber, S.; Gregor, M.; Ludwiczek, O.; Rutgeerts, P.; Gasche, C.; et al. Treatment of Crohn’s disease with recombinant human interleukin 10 induces the proinflammatory cytokine interferon gamma. Gut 2002, 50, 191–195. [Google Scholar] [CrossRef]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of Murine Colitis by Lactococcus lactis Secreting Interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef]
- Zurita-Turk, M.; Souza, B.M.; De Castro, C.P.; Pereira, V.B.; Da Cunha, V.P.; Preisser, T.M.; De Faria, A.M.C.; Machado, D.C.C.; Miyoshi, A. Attenuation of intestinal inflammation in IL-10 deficient mice by a plasmid carrying Lactococcus lactis strain. BMC Biotechnol. 2020, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Fay, N.C.; Muthusamy, B.-P.; Nyugen, L.P.; Desai, R.C.; Taverner, A.; Mackay, J.; Seung, M.; Hunter, T.; Liu, K.; Chandalia, A.; et al. A Novel Fusion of IL-10 Engineered to Traffic across Intestinal Epithelium to Treat Colitis. J. Immunol. 2020, 205, 3191–3204. [Google Scholar] [CrossRef] [PubMed]
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A Phase I Trial With Transgenic Bacteria Expressing Interleukin-10 in Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2006, 4, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Kucharzik, T.; Stoll, R.; Lügering, N.; Domschke, W. Circulating antiinflammatory cytokine IL-10 in patients with inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 100, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.-F.; Rutgeerts, P.; Malchow, H.; Jacyna, M.; Nielsen, O.H.; Rask-Madsen, J.; Van Deventer, S.; Ferguson, A.; Desreumaux, P.; Forbes, A.; et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut 2001, 49, 42–46. [Google Scholar] [CrossRef]
- Aranda, R.; Sydora, B.C.; McAllister, P.L.; Binder, S.W.; Yang, H.Y.; Targan, S.R.; Kronenberg, M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J. Immunol. 1997, 158, 3464–3473. [Google Scholar]
- Feng, T.; Wang, L.; Schoeb, T.R.; Elson, C.O.; Cong, Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010, 207, 1321–1332. [Google Scholar] [CrossRef]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef]
- Kjellev, S.; Lundsgaard, D.; Poulsen, S.S.; Markholst, H. Reconstitution of Scid mice with CD4+CD25− T cells leads to rapid colitis: An improved model for pharmacologic testing. Int. Immunopharmacol. 2006, 6, 1341–1354. [Google Scholar] [CrossRef]
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Thl responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell–mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Brasseit, J.; Chung, C.K.C.K.; Noti, M.; Zysset, D.; Hoheisel-Dickgreber, N.; Genitsch, V.; Corazza, N.; Mueller, C. Divergent Roles of Interferon-γ and Innate Lymphoid Cells in Innate and Adaptive Immune Cell-Mediated Intestinal Inflammation. Front. Immunol. 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [PubMed]
- Poholek, C.; Dulson, S.J.; Zajac, A.J.; Harrington, L.E. IL-21 Controls ILC3 Cytokine Production and Promotes a Protective Phenotype in a Mouse Model of Colitis. ImmunoHorizons 2019, 3, 194–202. [Google Scholar] [CrossRef]
- Kole, A.; He, J.; Rivollier, A.; Silveira, D.D.; Kitamura, K.; Maloy, K.J.; Kelsall, B.L. Type I IFNs Regulate Effector and Regulatory T Cell Accumulation and Anti-Inflammatory Cytokine Production during T Cell–Mediated Colitis. J. Immunol. 2013, 191, 2771–2779. [Google Scholar] [CrossRef]
- Fort, M.M.; Leach, M.W.; Rennick, D.M. A role for NK cells as regulators of CD4+ T cells in a transfer model of colitis. J. Immunol. 1998, 161, 3256–3261. [Google Scholar]
- A Keilbaugh, S.; E Shin, M.; Banchereau, R.F.; McVay, L.D.; Boyko, N.; Artis, D.; Cebra, J.J.; Wu, G.D. Activation of RegIII/and interferon expression in the intestinal tract of SCID mice: An innate response to bacterial colonisation of the gut. Gut 2005, 54, 623–629. [Google Scholar] [CrossRef]
- Kjellev, S.; Haase, C.; Lundsgaard, D.; Ursø, B.; Tornehave, D.; Markholst, H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. Eur. J. Immunol. 2007, 37, 1397–1406. [Google Scholar] [CrossRef]
- Yusung, S.; McGovern, D.; Lin, L.; Hommes, D.; Lagishetty, V.; Braun, J. NK cells are biologic and biochemical targets of 6-mercaptopurine in Crohn’s disease patients. Clin. Immunol. 2017, 175, 82–90. [Google Scholar] [CrossRef]
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 167, 1016–1033. [Google Scholar] [CrossRef] [PubMed]
- Karo, J.M.; Schatz, D.G.; Sun, J.C. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell 2014, 159, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Chia, R.; Achilli, F.; Festing, M.F.W.; Fisher, E.M.C. The origins and uses of mouse outbred stocks. Nat. Genet. 2005, 37, 1181–1186. [Google Scholar] [CrossRef]
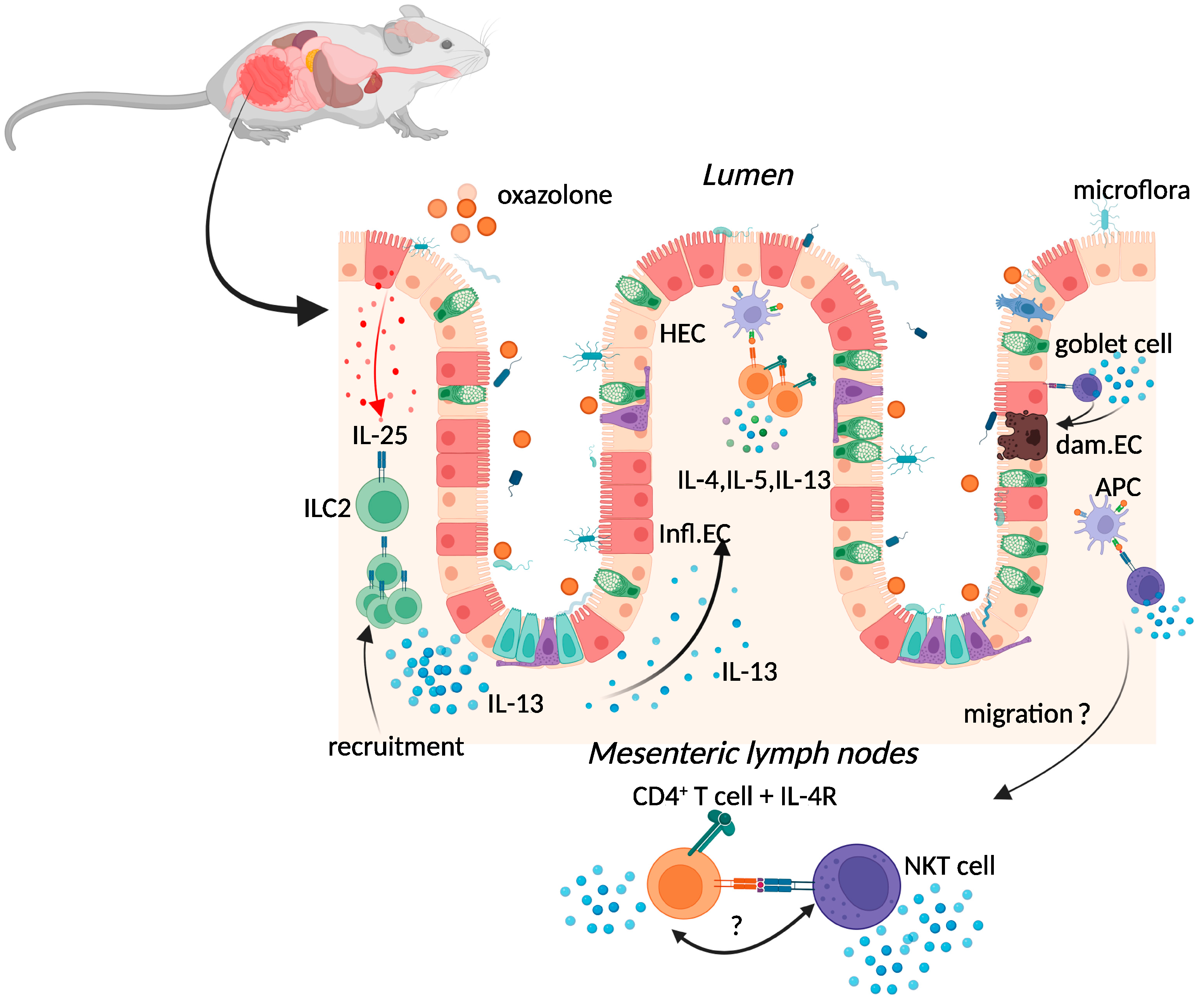


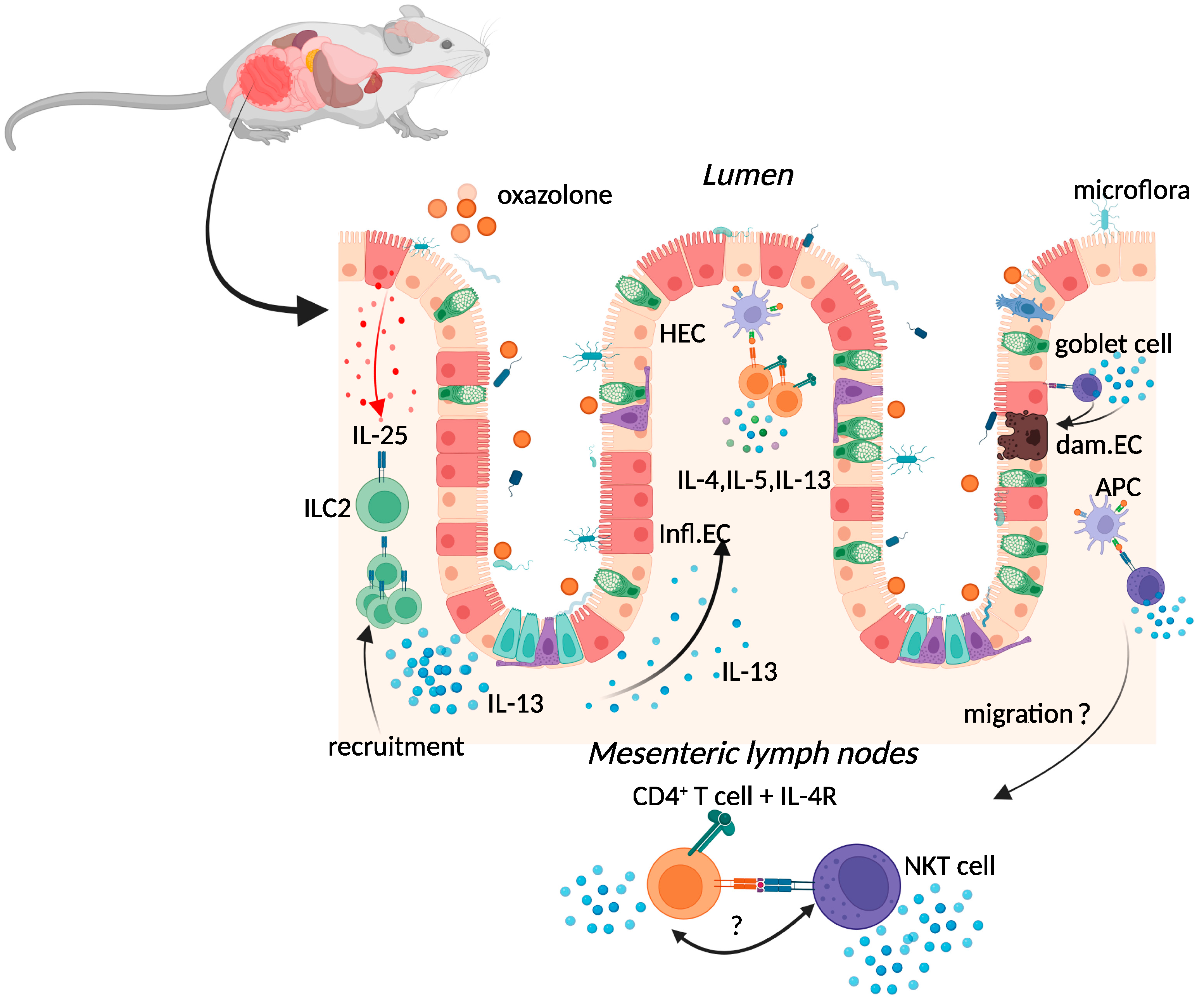{kind=link}
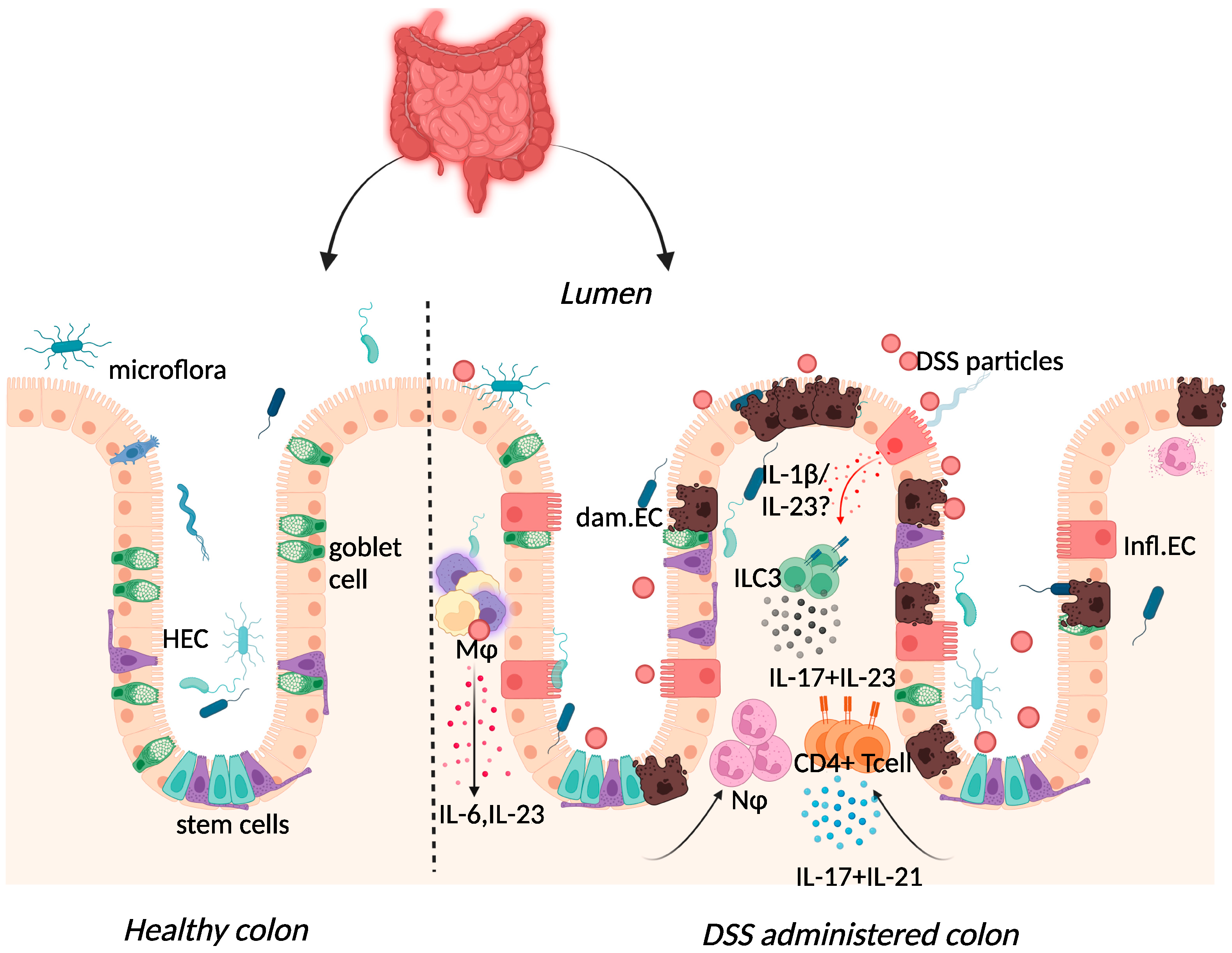{kind=link}
{kind=link}
| Induction Method | Examples | Disease Modelled | Advantages | Disadvantages |
|---|---|---|---|---|
| Chemical Induced Colitis | Oxazolone colitis | UC |
|
|
| Trinitrobenzene sulfonic acid (TNBS) colitis | CD |
|
| |
| Dextran Sulfate Sodium (DSS) | UC and CD |
|
| |
| Spontaneous colitis | Iκκ-γ (NEMO) Deficiency Colitis | NEMO-deficiency |
|
|
| IL-10 Deficiency Colitis | Childhood IBD |
|
| |
| Immune cell Induced Colitis | CD4+ T cell transfer | CD |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsandegwaza, B.; Horsnell, W.; Smith, K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. Int. J. Mol. Sci. 2022, 23, 9344. https://doi.org/10.3390/ijms23169344
Katsandegwaza B, Horsnell W, Smith K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. International Journal of Molecular Sciences. 2022; 23(16):9344. https://doi.org/10.3390/ijms23169344
Chicago/Turabian StyleKatsandegwaza, Brunette, William Horsnell, and Katherine Smith. 2022. "Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease" International Journal of Molecular Sciences 23, no. 16: 9344. https://doi.org/10.3390/ijms23169344
APA StyleKatsandegwaza, B., Horsnell, W., & Smith, K. (2022). Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. International Journal of Molecular Sciences, 23(16), 9344. https://doi.org/10.3390/ijms23169344