
Role of HMGB1 in Cutaneous Melanoma: State of the Art

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
1.1. Cutaneous Melanoma
1.1.1. Cutaneous Melanoma Clinical Types and Pathogenesis
1.1.2. Molecular Categories and Therapy
1.2. HMGB1
1.2.1. Structure and Localization
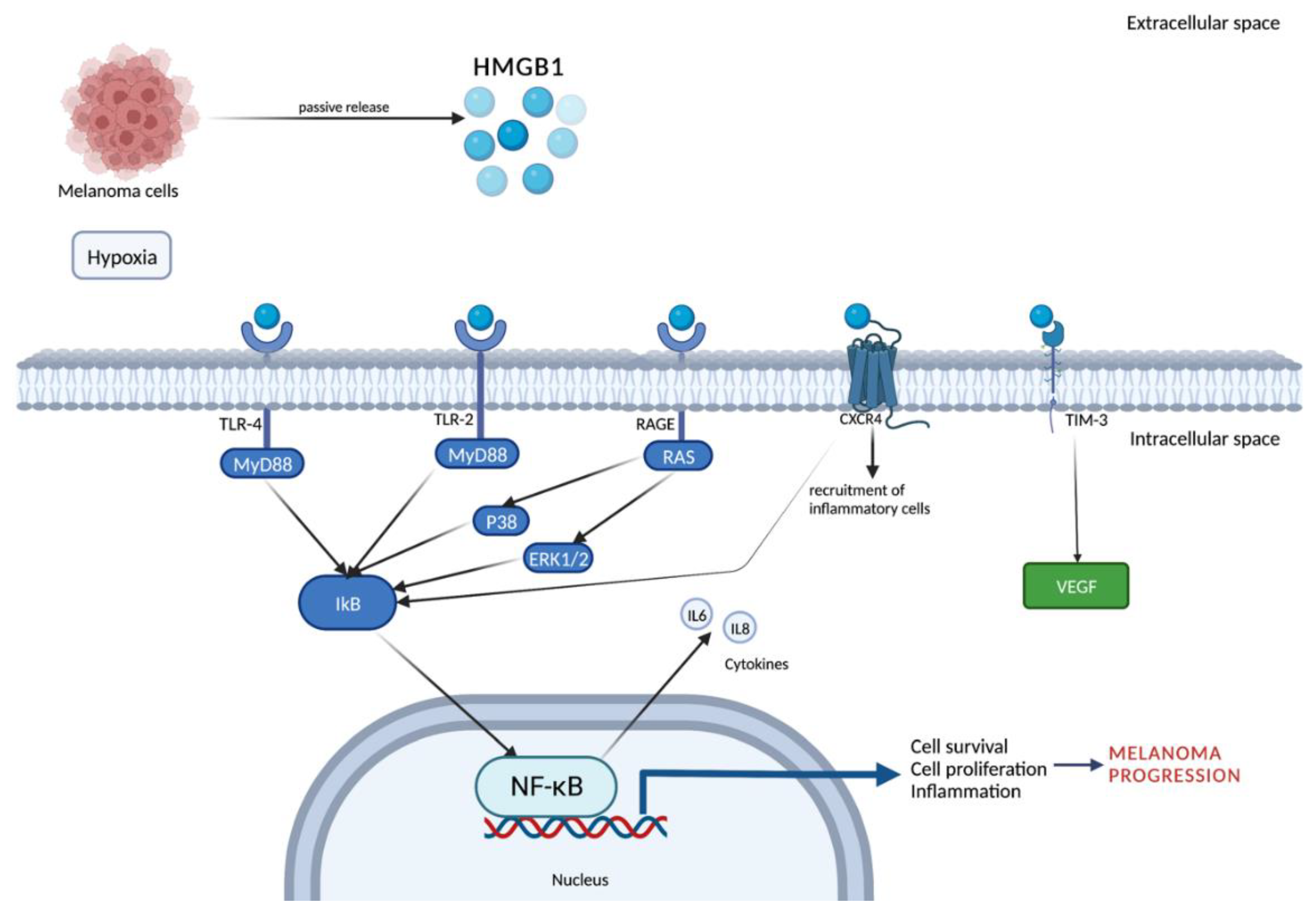
1.2.2. Mechanism of Release and Receptors
1.2.3. Function
2. Discussion
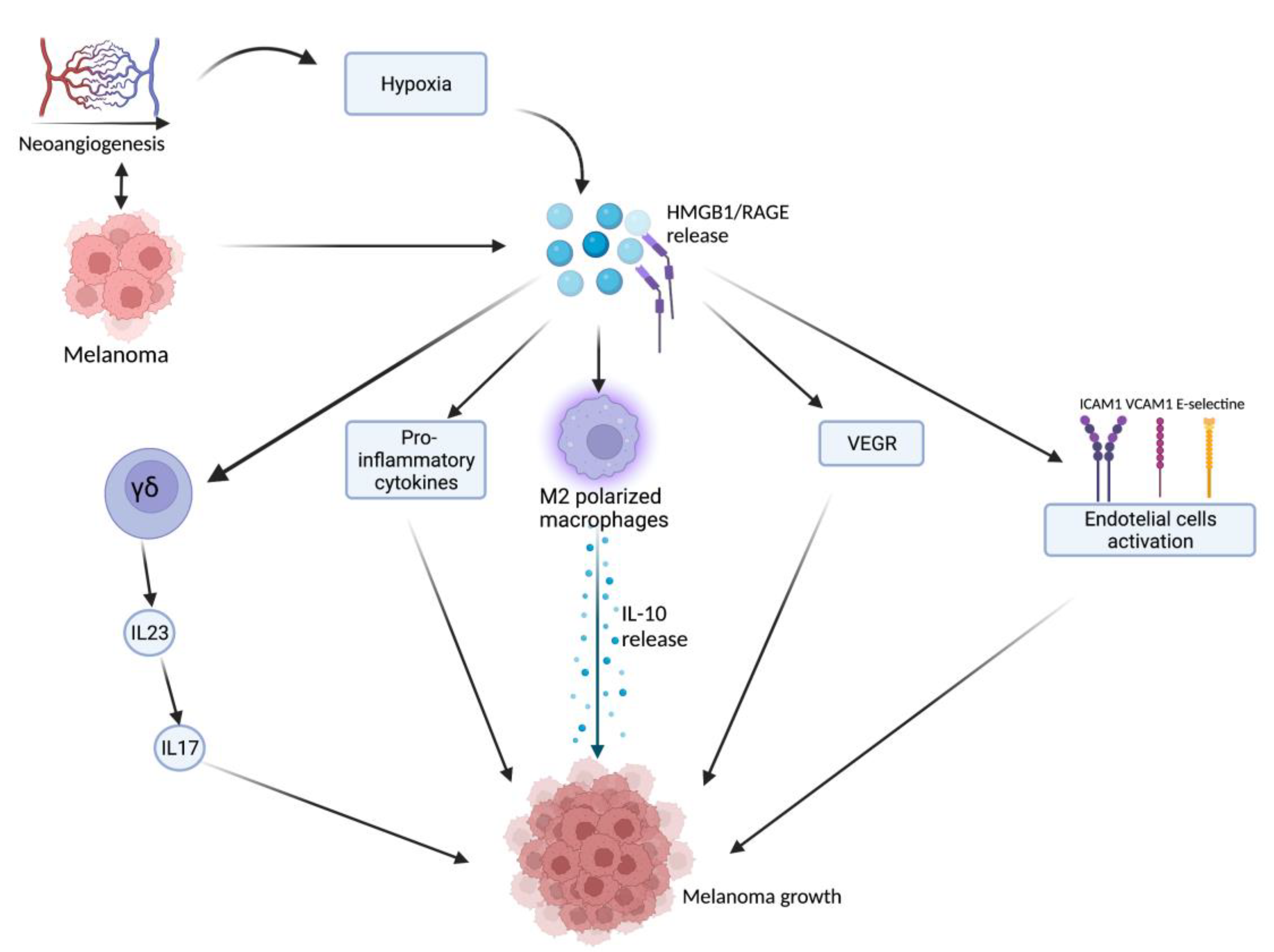
2.1. HMGB1-Related Melanoma Growth
2.2. HMGB1 and UVB
2.3. HMGB1 as a Marker
2.4. Melanoma Metastases and HMGB-1-Based Possible Future Therapies
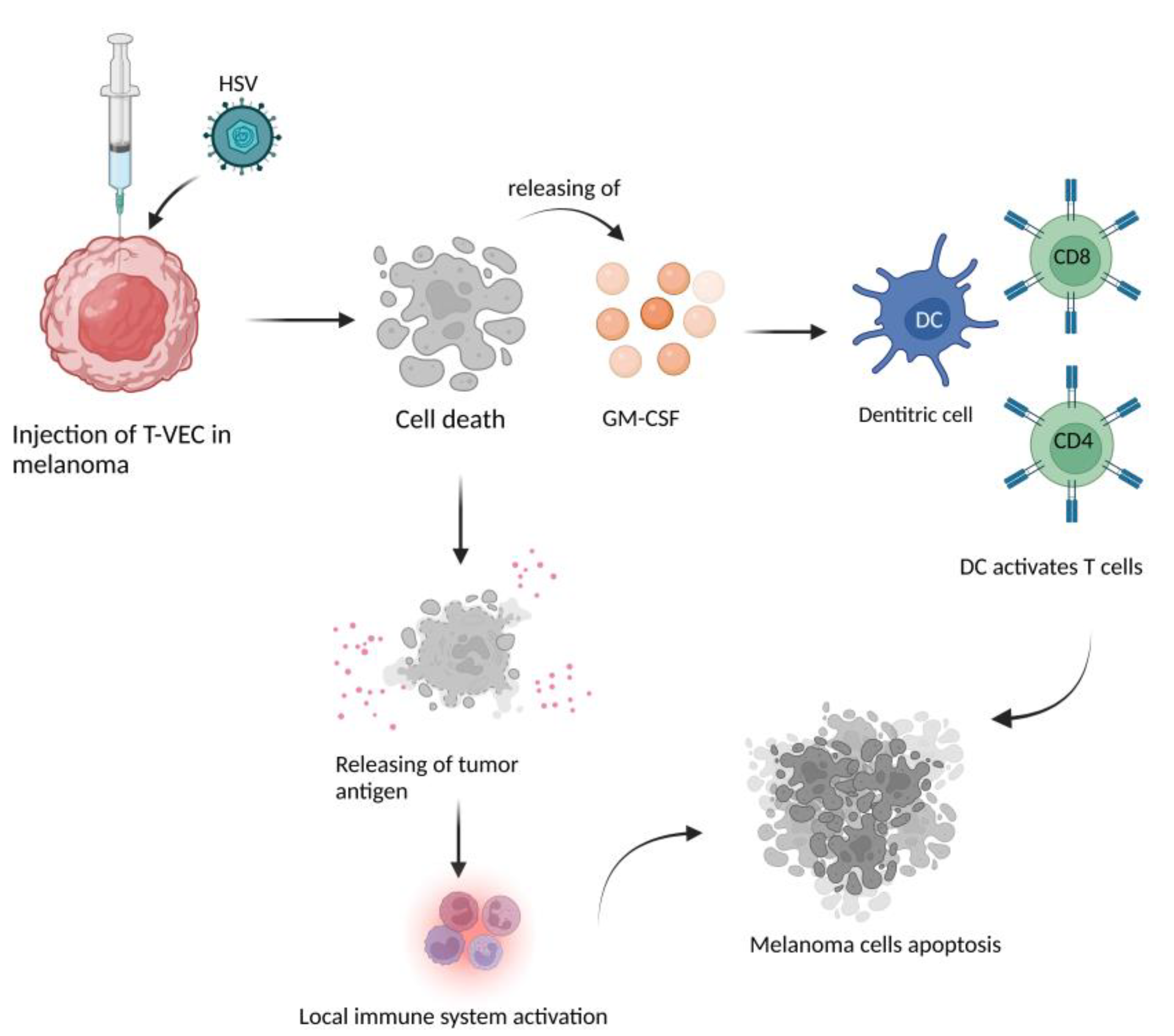
2.5. HMGB1 and Immunological Cell Death
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhanyamraju, P.K.; Rotimi, S.; Bhattacharya, P.; Patel, T.N. Revisiting the Melanomagenic Pathways and Current Therapeutic Approaches. Mol. Biol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Olaoba, O.T.; Kadasah, S.; Vetter, S.W.; Leclerc, E. RAGE Signaling in Melanoma Tumors. Int. J. Mol. Sci. 2020, 21, 8989. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; et al. European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Incorvaia, L.; Badalamenti, G.; Rinaldi, G.; Iovanna, J.L.; Olive, D.; Swayden, M.; Terruso, L.; Vincenzi, B.; Fulfaro, F.; Bazan, V.; et al. Can the Plasma PD-1 Levels Predict the Presence and Efficiency of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma? Ther. Adv. Med. Oncol. 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef] [PubMed]
- Tímár, J.; Ladányi, A. Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. Int. J. Mol. Sci. 2022, 23, 5384. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; et al. European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 2: Treatment—Update 2022. Eur. J. Cancer 2022, 170, 256–284. [Google Scholar] [CrossRef]
- Huynh, S.; Mortier, L.; Dutriaux, C.; Maubec, E.; Boileau, M.; Dereure, O.; Leccia, M.-T.; Arnault, J.-P.; Brunet-Possenti, F.; Aubin, F.; et al. Combined Therapy with Anti-PD1 and BRAF and/or MEK Inhibitor for Advanced Melanoma: A Multicenter Cohort Study. Cancers 2020, 12, 1666. [Google Scholar] [CrossRef]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef]
- Deng, M.; Scott, M.J.; Fan, J.; Billiar, T.R. Location Is the Key to Function: HMGB1 in Sepsis and Trauma-induced Inflammation. J. Leukoc. Biol. 2019, 106, 161–169. [Google Scholar] [CrossRef]
- Xue, J.; Suarez, J.S.; Minaai, M.; Li, S.; Gaudino, G.; Pass, H.I.; Carbone, M.; Yang, H. HMGB1 as a Therapeutic Target in Disease. J. Cell. Physiol. 2021, 236, 3406–3419. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Allegra, A.; Paladin, F.; Calapai, F.; Musolino, C.; Gangemi, S. Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 9039. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Murakami, M.; Tsuda, T.; Kameda, K.; Utsunomiya, R.; Masuda, K.; Shiraishi, K.; Dai, X.; Tohyama, M.; Nakaoka, H.; et al. Reduced-HMGB1 Suppresses Poly(I:C)-Induced Inflammation in Keratinocytes. J. Dermatol. Sci. 2018, 90, 154–165. [Google Scholar] [CrossRef]
- Xu, T.; Jiang, L.; Wang, Z. The Progression of HMGB1-Induced Autophagy in Cancer Biology. OncoTargets Ther. 2018, 12, 365–377. [Google Scholar] [CrossRef]
- Feng, X. MiR-548b Suppresses Melanoma Cell Growth, Migration, and Invasion by Negatively Regulating Its Target Gene HMGB1. Cancer Biother. Radiopharm. 2021, 36, 189–201. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.; Yan, Z.; et al. HMGB1 in Health and Disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Meier, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour Hypoxia Promotes Melanoma Growth and Metastasis via High Mobility Group Box-1 and M2-like Macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef]
- Cannavò, S.P.; Tonacci, A.; Bertino, L.; Casciaro, M.; Borgia, F.; Gangemi, S. The Role of Oxidative Stress in the Biology of Melanoma: A Systematic Review. Pathol. Res. Pract. 2019, 215, 21–28. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Siveen, K.S.; Kuttan, G. Role of Macrophages in Tumour Progression. Immunol. Lett. 2009, 123, 97–102. [Google Scholar] [CrossRef]
- Simões, R.L.; De-Brito, N.M.; Cunha-Costa, H.; Morandi, V.; Fierro, I.M.; Roitt, I.M.; Barja-Fidalgo, C. Lipoxin A4 Selectively Programs the Profile of M2 Tumor-Associated Macrophages Which Favour Control of Tumor Progression. Int. J. Cancer 2017, 140, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-Associated Macrophages and the Related Myeloid-Derived Suppressor Cells as a Paradigm of the Diversity of Macrophage Activation. Hum. Immunol. 2009, 70, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, J.; Zhu, H.; Li, P.; Zou, Z.; Xiao, Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 Axis Promotes Tumor Growth in Murine Models of Melanoma. Mediat. Inflamm. 2013, 2013, 713859. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.-H. Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-ΚB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef]
- Syed, D.N.; Aljohani, A.; Waseem, D.; Mukhtar, H. Ousting RAGE in Melanoma: A Viable Therapeutic Target? Semin. Cancer Biol. 2018, 49, 20–28. [Google Scholar] [CrossRef]
- Logsdon, C.; Fuentes, M.; Huang, E.; Arumugam, T. RAGE and RAGE Ligands in Cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Zhang, K.; Anumanthan, G.; Scheaffer, S.; Cornelius, L.A. HMGB1/RAGE Mediates UVB-Induced Secretory Inflammatory Response and Resistance to Apoptosis in Human Melanocytes. J. Investig. Dermatol. 2019, 139, 202–212. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Wondrak, G.T. TLR4 in Skin Cancer: From Molecular Mechanisms to Clinical Interventions. Mol. Carcinog. 2019, 58, 1086–1093. [Google Scholar] [CrossRef]
- Li, Q.; Li, J.; Wen, T.; Zeng, W.; Peng, C.; Yan, S.; Tan, J.; Yang, K.; Liu, S.; Guo, A.; et al. Overexpression of HMGB1 in Melanoma Predicts Patient Survival and Suppression of HMGB1 Induces Cell Cycle Arrest and Senescence in Association with P21 (Waf1/Cip1) up-Regulation via a P53-Independent, Sp1-Dependent Pathway. Oncotarget 2014, 5, 6387–6403. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.-L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune Profile and Mitotic Index of Metastatic Melanoma Lesions Enhance Clinical Staging in Predicting Patient Survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y. HMGB1 in Inflammation and Cancer. J. Hematol. Oncol. 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Meng, H.; Dong, X.; Li, X.; Shi, Z.; Li, H.; Zhang, L.; Yang, Y.; Liu, R.; Pei, C.; et al. IRGM Promotes Melanoma Cell Survival through Autophagy and Is a Promising Prognostic Biomarker for Clinical Application. Mol. Ther. Oncolytics 2021, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, S.; Chen, L.; Ito, T.; Tong, Y.; Onodera, T.; Sasaki, Y.; Nakamura, S.; Mauri, P.; Sanada, Y.; Igaki, H.; et al. Extracellular Release of HMGB1 as an Early Potential Biomarker for the Therapeutic Response in a Xenograft Model of Boron Neutron Capture Therapy. Biology 2022, 11, 420. [Google Scholar] [CrossRef]
- Bouwhuis, M.G.; Suciu, S.; Kruit, W.; Salès, F.; Stoitchkov, K.; Patel, P.; Cocquyt, V.; Thomas, J.; Liénard, D.; Eggermont, A.M.M.; et al. Prognostic Value of Serial Blood S100B Determinations in Stage IIB–III Melanoma Patients: A Corollary Study to EORTC Trial 18952. Eur. J. Cancer 2011, 47, 361–368. [Google Scholar] [CrossRef]
- Karonidis, A.; Mantzourani, M.; Gogas, H.; Tsoutsos, D. Serum S100B Levels Correlate with Stage, N Status, Mitotic Rate and Disease Outcome in Melanoma Patients Independent to LDH. J. BUON 2017, 22, 1296–1302. [Google Scholar] [PubMed]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T.K. S100B and LDH as Early Prognostic Markers for Response and Overall Survival in Melanoma Patients Treated with Anti-PD-1 or Combined Anti-PD-1 plus Anti-CTLA-4 Antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High Mobility Group Box 1 (HMGB1): A Pivotal Regulator of Hematopoietic Malignancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef]
- Liu, Z.; Falo, L.D.; You, Z. Knockdown of HMGB1 in Tumor Cells Attenuates Their Ability to Induce Regulatory T Cells and Uncovers Naturally Acquired CD8 T Cell-Dependent Antitumor Immunity. J. Immunol. 2011, 187, 118–125. [Google Scholar] [CrossRef]
- Yokomizo, K.; Waki, K.; Ozawa, M.; Yamamoto, K.; Ogasawara, S.; Yano, H.; Yamada, A. Knockout of High-Mobility Group Box 1 in B16F10 Melanoma Cells Induced Host Immunity-Mediated Suppression of in Vivo Tumor Growth. Med. Oncol. 2022, 39, 58. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-J.; Park, I.H.; Rhee, W.J.; Kim, H.S.; Shin, J.-S. HMGB1 Modulates the Balance between Senescence and Apoptosis in Response to Genotoxic Stress. FASEB J. 2019, 33, 10942–10953. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiang, N.; Lin, M.; Huang, J.; Zhang, J.; Cheng, B.; Ji, C. miR-26a Sensitizes Melanoma Cells to Dabrafenib Via Targeting HMGB1-Dependent Autophagy Pathways. Drug Des. Dev. Ther. 2019, 13, 3717–3726. [Google Scholar] [CrossRef]
- Maia, V.S.C.; Berzaghi, R.; Arruda, D.C.; Machado, F.C.; Loureiro, L.L.; Melo, P.M.S.; Morais, A.S.; Budu, A.; Travassos, L.R. PLP2-Derived Peptide Rb4 Triggers PARP-1-Mediated Necrotic Death in Murine Melanoma Cells. Sci. Rep. 2022, 12, 2890. [Google Scholar] [CrossRef]
- Hiramoto, K.; Yamate, Y.; Goto, K.; Ohnishi, S.; Morita, A.; Yoshikawa, N.; Kawanishi, S. Glycyrrhizin Ameliorates Melanoma Cell Extravasation into Mouse Lungs by Regulating Signal Transduction through HMGB1 and Its Receptors. J. Clin. Biochem. Nutr. 2021, 69, 20–125. [Google Scholar] [CrossRef]
- Li, P.; Ren, K.; Liang, Y.Y.; Liu, J.K.; Liang, Z.W.; Zhang, Y.F. Aloin Promotes Cell Apoptosis by Targeting HMGB1-TLR4-ERK Axis in Human Melanoma Cells. EXCLI J. 2020, 19, 641–651. [Google Scholar]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Troitskaya, O.S.; Novak, D.D.; Richter, V.A.; Koval, O.A. Immunogenic Cell Death in Cancer Therapy. Acta Nat. 2022, 14, 40–53. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting Immunogenic Cell Death in Cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-W.; Wang, S.-T.; Chang, S.-H.; Chuang, K.-C.; Wang, H.-Y.; Kao, J.-K.; Liang, S.-M.; Wu, C.-Y.; Kao, S.-H.; Chen, Y.-J.; et al. Imiquimod Exerts Antitumor Effects by Inducing Immunogenic Cell Death and Is Enhanced by the Glycolytic Inhibitor 2-Deoxyglucose. J. Investig. Dermatol. 2020, 140, 1771–1783.e6. [Google Scholar] [CrossRef] [PubMed]
- Giglio, P.; Gagliardi, M.; Tumino, N.; Antunes, F.; Smaili, S.; Cotella, D.; Santoro, C.; Bernardini, R.; Mattei, M.; Piacentini, M.; et al. PKR and GCN2 Stress Kinases Promote an ER Stress-Independent EIF2α Phosphorylation Responsible for Calreticulin Exposure in Melanoma Cells. OncoImmunology 2018, 7, e1466765. [Google Scholar] [CrossRef] [PubMed]
- Pasquereau-Kotula, E.; Habault, J.; Kroemer, G.; Poyet, J.-L. The Anticancer Peptide RT53 Induces Immunogenic Cell Death. PLoS ONE 2018, 13, e0201220. [Google Scholar] [CrossRef] [PubMed]
- Borgia, F.; Giuffrida, R.; Caradonna, E.; Vaccaro, M.; Guarneri, F.; Cannavò, S. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting Immunogenic Cancer Cell Death by Photodynamic Therapy: Past, Present and Future. J. ImmunoTher. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef]
- Konda, P.; Roque Iii, J.A.; Lifshits, L.M.; Alcos, A.; Azzam, E.; Shi, G.; Cameron, C.G.; McFarland, S.A.; Gujar, S. Photodynamic Therapy of Melanoma with New, Structurally Similar, NIR-Absorbing Ruthenium (II) Complexes Promotes Tumor Growth Control via Distinct Hallmarks of Immunogenic Cell Death. Am. J. Cancer Res. 2022, 12, 210–228. [Google Scholar]
- Morais, J.A.V.; Almeida, L.R.; Rodrigues, M.C.; Azevedo, R.B.; Muehlmann, L.A. The Induction of Immunogenic Cell Death by Photodynamic Therapy in B16F10 Cells in Vitro Is Effected by the Concentration of the Photosensitizer. Photodiagn. Photodyn. Ther. 2021, 35, 102392. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Podolska, M.J.; Shan, X.; Janko, C.; Boukherroub, R.; Gaipl, U.S.; Szunerits, S.; Frey, B.; Muñoz, L.E. Graphene-Induced Hyperthermia (GIHT) Combined with Radiotherapy Fosters Immunogenic Cell Death. Front. Oncol. 2021, 11, 664615. [Google Scholar] [CrossRef]
- Del Rodríguez-Salazar, M.C.; Franco-Molina, M.A.; Mendoza-Gamboa, E.; Martínez-Torres, A.C.; Zapata-Benavides, P.; López-González, J.S.; Coronado-Cerda, E.E.; Alcocer-González, J.M.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. The Novel Immunomodulator IMMUNEPOTENT CRP Combined with Chemotherapy Agent Increased the Rate of Immunogenic Cell Death and Prevented Melanoma Growth. Oncol. Lett. 2017, 14, 844–852. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dharmadhikari, N.; Mehnert, J.M.; Kaufman, H.L. Oncolytic Virus Immunotherapy for Melanoma. Curr. Treat. Options Oncol. 2015, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic Virus Therapy in Cancer: A Current Review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A Phase I Study of OncoVEXGM-CSF, a Second-Generation Oncolytic Herpes Simplex Virus Expressing Granulocyte Macrophage Colony-Stimulating Factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bines, S.D. OPTIM Trial: A Phase III Trial of an Oncolytic Herpes Virus Encoding GM-CSF for Unresectable Stage III or IV Melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab Versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Study of RP1 Monotherapy and RP1 in Combination with Nivolumab (IGNYTE). Available online: https://clinicaltrials.gov/ct2/show/NCT03767348 (accessed on 20 September 2017).
- Watanabe, D.; Goshima, F.; Mori, I.; Tamada, Y.; Matsumoto, Y.; Nishiyama, Y. Oncolytic Virotherapy for Malignant Melanoma with Herpes Simplex Virus Type 1 Mutant HF10. J. Dermatol. Sci. 2008, 50, 185–196. [Google Scholar] [CrossRef]
- Au, G.; Lindberg, A.; Barry, R.; Shafren, D. Oncolysis of Vascular Malignant Human Melanoma Tumors by Coxsackievirus A21. Int. J. Oncol. 2005, 26, 1471–1476. [Google Scholar] [CrossRef]
- Shao, X.; Wang, X.; Guo, X.; Jiang, K.; Ye, T.; Chen, J.; Fang, J.; Gu, L.; Wang, S.; Zhang, G.; et al. STAT3 Contributes to Oncolytic Newcastle Disease Virus-Induced Immunogenic Cell Death in Melanoma Cells. Front. Oncol. 2019, 9, 436. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Topic | Author, Reference | Study Characteristics |
|---|---|---|
| Metastases | Yokomizo, K et al. [41] | In a mouse model, knockout of HMGB1 in tumor cells converted tumors from scarcely immunogenic phenotypes to inflammation-prone-ones, inhibiting in vivo tumor progression. |
| Metastases | Lee, Y-Y et al. [42] | HMGB1 plays a role in the senescence or apoptosis of cancer cells. Highly metastatic tumor cells preferentially enter senescence and adopt survival mechanisms, while apoptosis predominates in weakly metastatic tumor cells. |
| Possible future therapies | Yu, Y et al. [44] | miR-26a is involved in the upregulation of dabrafenib efficacy via an HMGB1-dependent autophagy pathway in melanoma. The treatment with a miR-26a mimic and HMGB1 shRNA increased the efficacy of dabrafenib in melanoma cells. |
| Possible future therapies | Maia, V.S.C. et al. [45] | Peptide Rb4 acts directly on tumor cells inducing the expression of HMGB1, which trigger the immunoprotective effect in vivo against melanoma cells. |
| Possible future therapies | Hiramoto, K et al. [46] | Glycirrizin inhibits HMGB1/RAGE pathway and reduces NF-κB expression, phosphorylation and nuclear translocation, which altogether induces cellular invasion. |
| Possible future therapies | Li, P et al. [47] | HMGB1 facilitates ALO-mediated apoptosis by binding to its receptor, TLR4, and activation of ERK signal pathways. |
| Topic | Author, Reference | Study Characteristics |
|---|---|---|
| ICD | Huang, SW et al. [52] | Exposure of HMGB1 and calreticulin in a mouse model of B16F10 melanoma, following direct injection in situ or vaccinating mice with IMQ, reduced tumor growth, demonstrating the role of HMGB1 in ICD. |
| ICD | Giglio, P et al. [53] | Mitoxantrone and doxorubicin could induce cell death through the release of high levels of HMGB1 in melanoma cell lines. |
| ICD | Konda, P et al. [57] | In a mouse model, PDT treatment with ML19B01 and ML19B02 induced dying melanoma cells contained hallmarks of the ICD, including HMGB1. |
| ICD | Chesney, J et al. [68] | T-VEC plus Ipi showed an almost doubled response rate and a higher degree of regression in visceral metastases with no unexpected increase in toxicity. The ICD was confirmed by the release of HMGB1. |
| ICD | IGNYTE study [69] | The expression of GALV-GP-R- improved the oncolytic ability in several tumor cell lines in vitro and mouse xenograft models. The increased immunogenic cell death in vitro was confirmed by the release of HMGB1 and ATP and by high levels of CRT on the cell surface. |
| ICD | Shao, X et al. [72] | HMGB1, HSP70, HSP90, CRT exposure and the secretion of ATP in melanoma cells were detected in melanoma cells line after treating with oncolytc NDV/FMW, demonstrating the role of these molecules in the ICD. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li Pomi, F.; Borgia, F.; Custurone, P.; Vaccaro, M.; Pioggia, G.; Gangemi, S. Role of HMGB1 in Cutaneous Melanoma: State of the Art. Int. J. Mol. Sci. 2022, 23, 9327. https://doi.org/10.3390/ijms23169327
Li Pomi F, Borgia F, Custurone P, Vaccaro M, Pioggia G, Gangemi S. Role of HMGB1 in Cutaneous Melanoma: State of the Art. International Journal of Molecular Sciences. 2022; 23(16):9327. https://doi.org/10.3390/ijms23169327
Chicago/Turabian StyleLi Pomi, Federica, Francesco Borgia, Paolo Custurone, Mario Vaccaro, Giovanni Pioggia, and Sebastiano Gangemi. 2022. "Role of HMGB1 in Cutaneous Melanoma: State of the Art" International Journal of Molecular Sciences 23, no. 16: 9327. https://doi.org/10.3390/ijms23169327
APA StyleLi Pomi, F., Borgia, F., Custurone, P., Vaccaro, M., Pioggia, G., & Gangemi, S. (2022). Role of HMGB1 in Cutaneous Melanoma: State of the Art. International Journal of Molecular Sciences, 23(16), 9327. https://doi.org/10.3390/ijms23169327