
Neural Networks Reveal the Impact of the Vibrational Dynamics in the Prediction of the Long-Time Mobility of Molecular Glassformers

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- the test of the approach of Ref. [14] by developing Neural Network (NN) architectures to predict the propensity in a molecular liquid, a melt of fully-flexible short chains. Melts of fully-flexible chains are known to have structural relaxation independent of the chain length [23] and thus are of large interest in polymer science to investigate long-time dynamics with considerable saving of computational effort [24]. Even if the results address conventional linear structural polymers, they are anticipated to provide at least useful hints concerning more complex polymer systems, such as bio-polymers and proteins.
- As further advance, in order to improve the prediction of propensity in glassy systems with ML techniques, we consider as an additional source of information the picosecond vibrational dynamics of the particles wiggling within the transient cage formed by their neighbors (augmented with further information on local structure drawn by Voronoi tessellation and potential energy per particle). Despite the huge difference of time-scales, strong correlations between the vibrational dynamics and the long time structural relaxation, rearranging the cage with considerable spatial distribution of mobility—so-called dynamical heterogeneity (DH)—are well known, see a recent short review [25]. As a matter of fact, the vibrational dynamics inside the cage was one of the first predictors of the long time propensity [19]. We show that this novel scheme improves the accuracy of the prediction, which is found to be significantly temperature-independent.
2. Results and Discussion
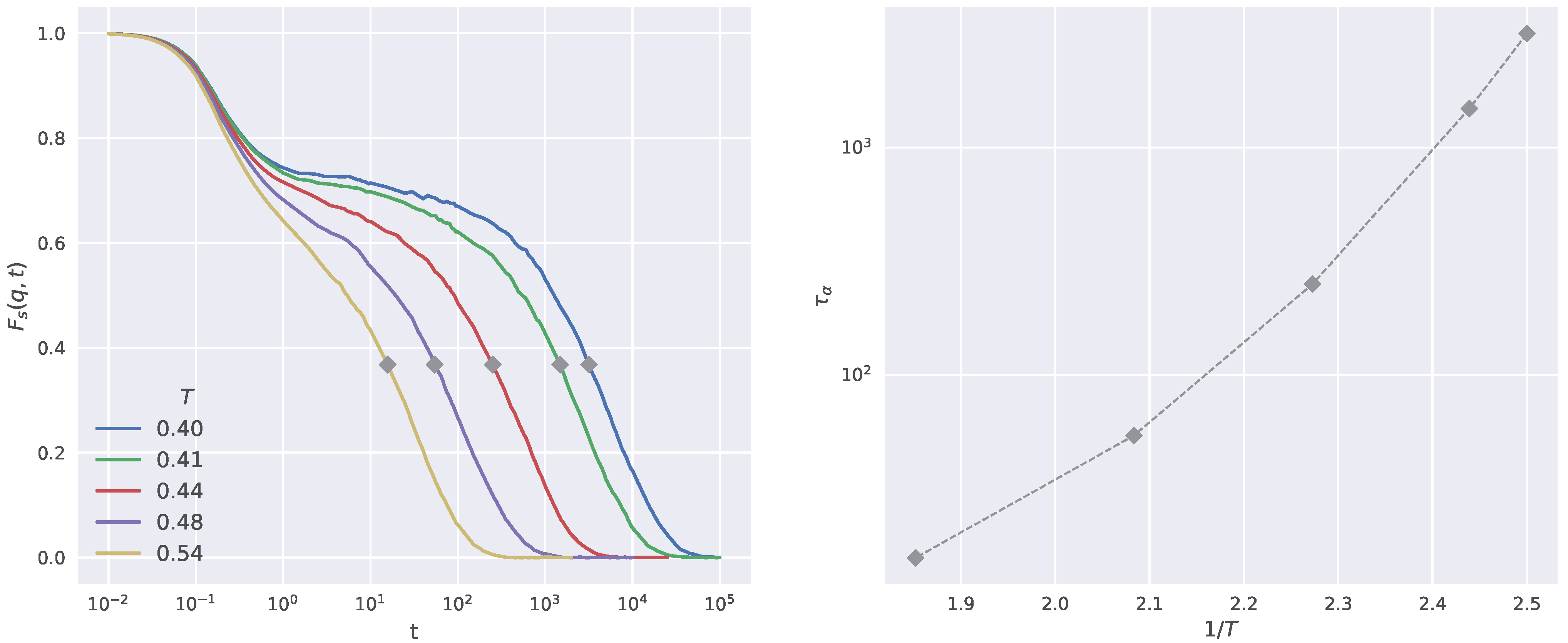
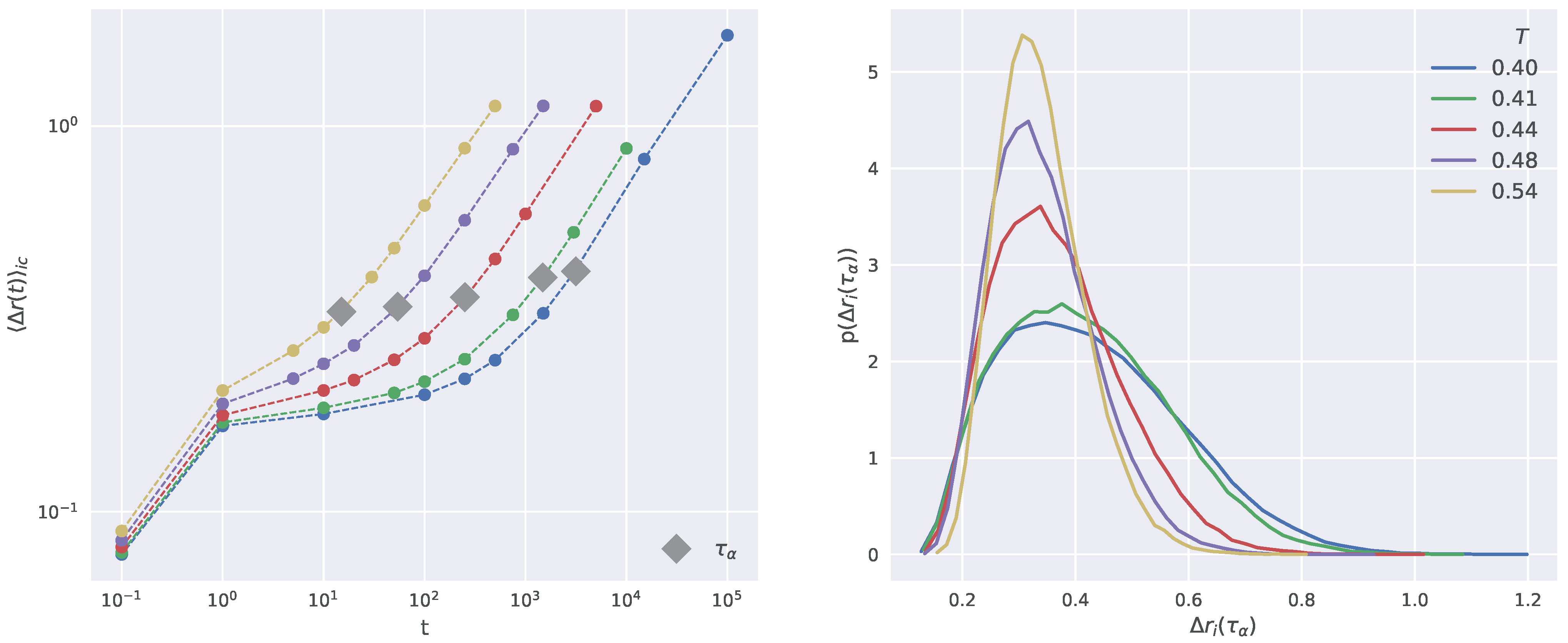
2.1. Structural Relaxation and Propensity
2.2. Local Descriptors
2.2.1. Structural Descriptors
2.2.2. Short-Time Dynamics Descriptor
2.3. Neural Network
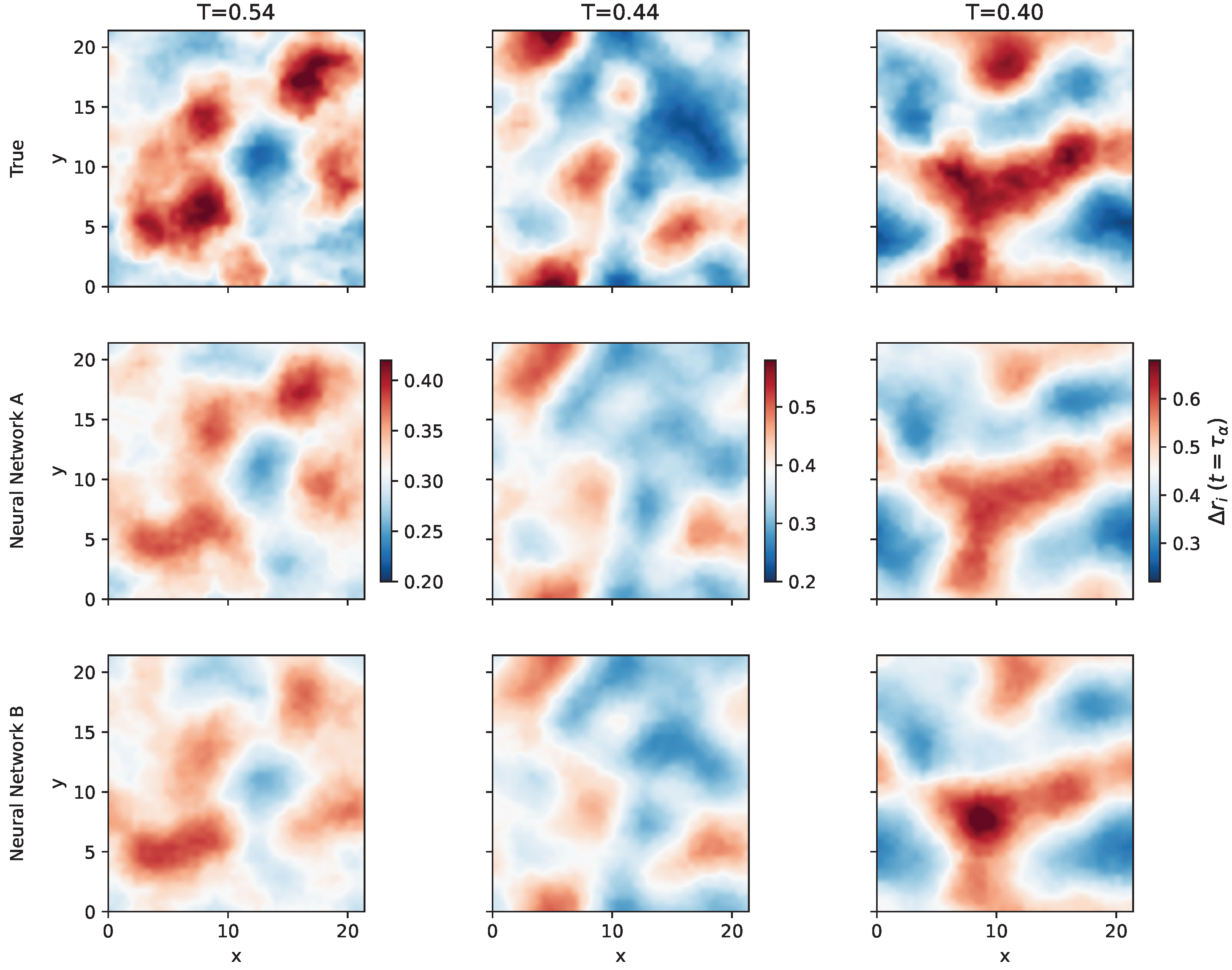
2.4. Predicting the Propensity
- the sole consideration of the particle position at a given time provides poor account of long-time large particle displacement in connected systems, especially at high temperatures;
- the information encoded in the local DW counteracts the drawback.
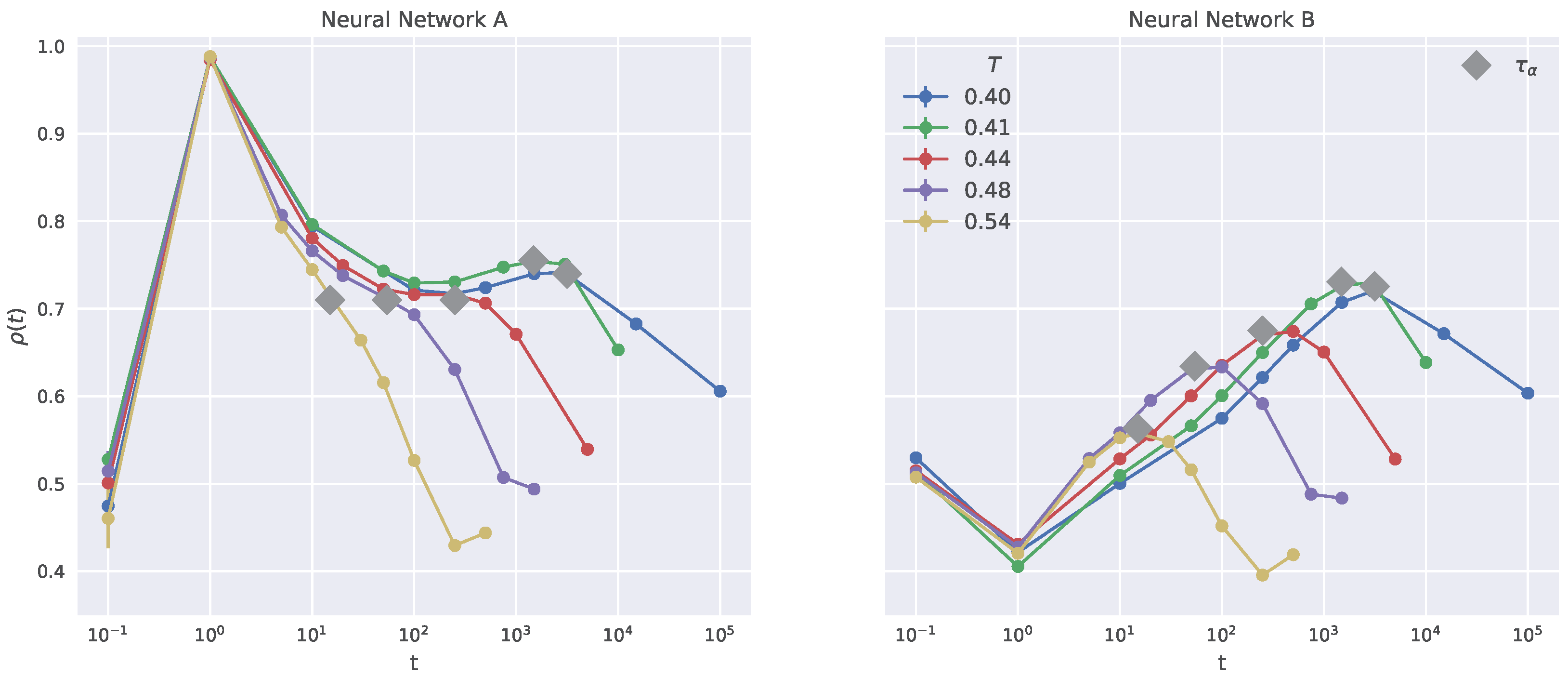
2.5. Dynamical Heterogeneity
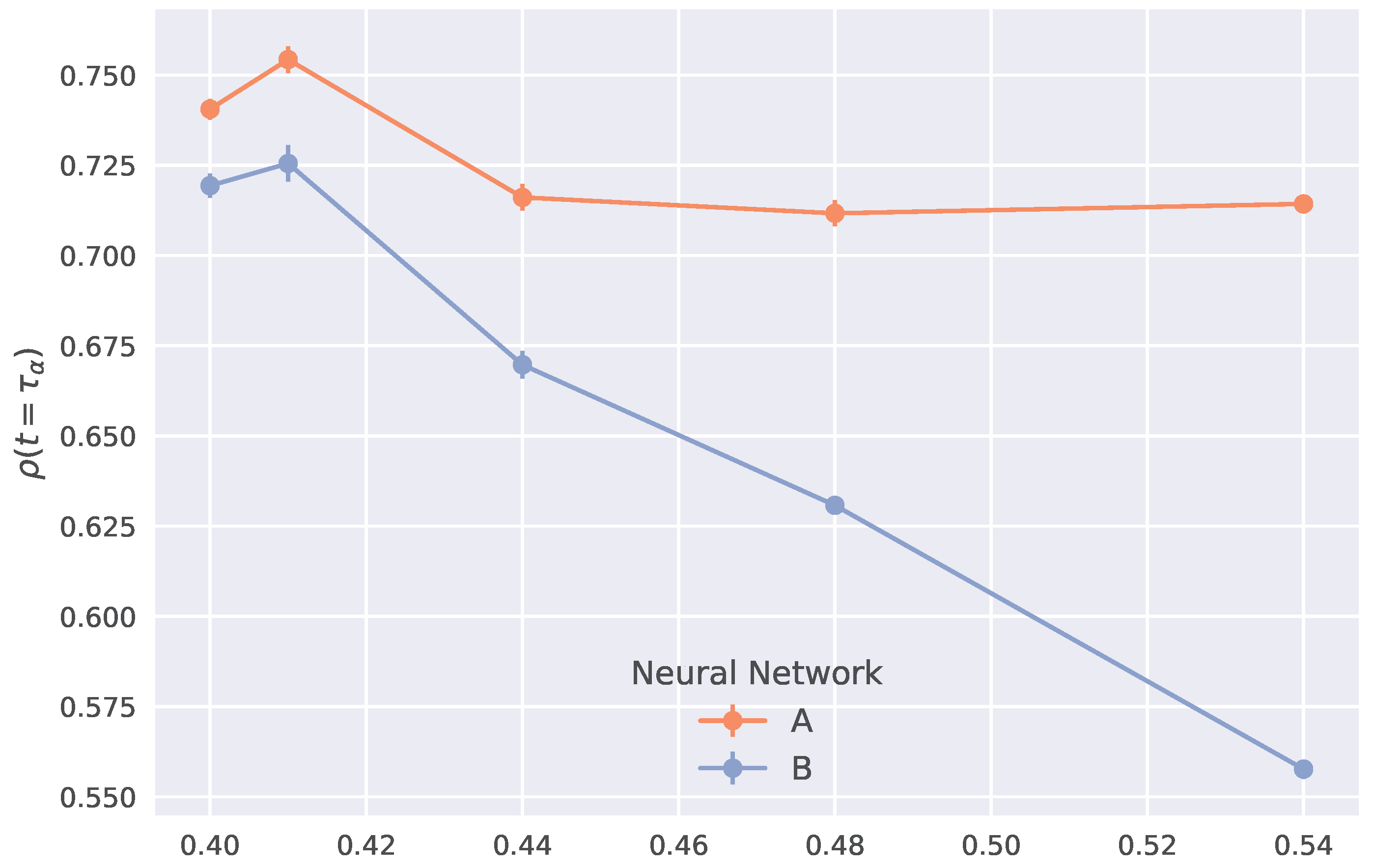
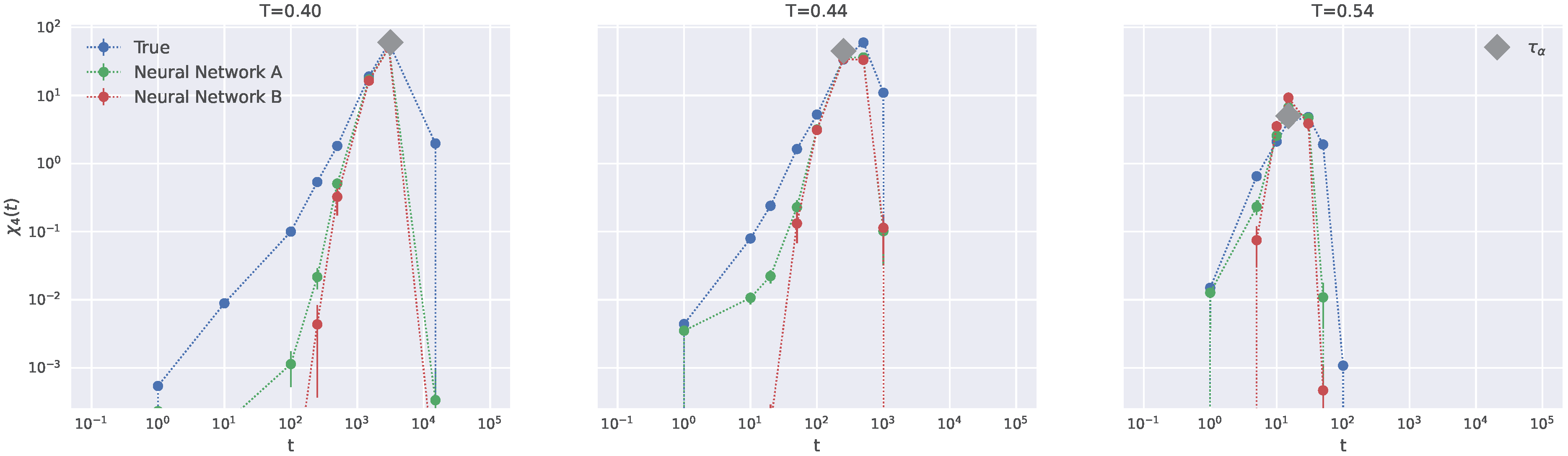
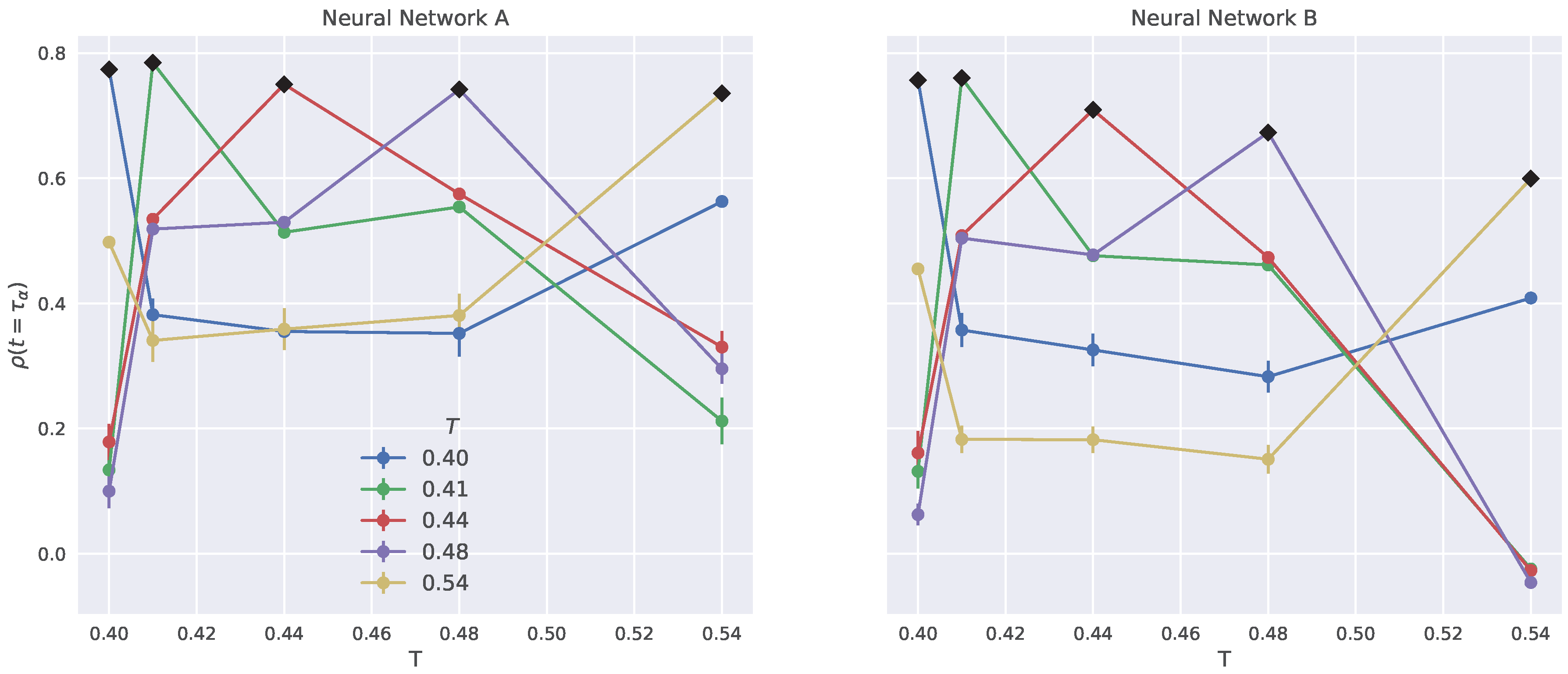
2.6. Generalization to Other Temperatures
3. Methods and Materials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DH | Dynamical Heterogeneity |
| DW | Debye–Waller |
| GNN | Graph Neural Network |
| ICE | Isoconfigurational Ensemble |
| ISF | Intermediate Scattering Function |
| ML | Machine Learning |
| NN | Neural Network |
References
- Angell, C.A. Formation of glasses from liquids and biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Debenedetti, P.G.; Stillinger, F.H. Supercooled liquids and the glass transition. Nature 2001, 410, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hocky, G.M.; Coslovich, D.; Ikeda, A.; Reichman, D.R. Correlation of Local Order with Particle Mobility in Supercooled Liquids Is Highly System Dependent. Phys. Rev. Lett. 2014, 113, 157801. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, P.; Tarjus, G. Decorrelation of the static and dynamic length scales in hard-sphere glass formers. Phys. Rev. E 2013, 87, 042305. [Google Scholar] [CrossRef]
- Berthier, L.; Biroli, G.; Bouchaud, J.P.; Tarjus, G. Can the glass transition be explained without a growing static length scale? J. Chem. Phys. 2019, 150, 094501. [Google Scholar] [CrossRef]
- Karmakar, S.; Dasgupta, C.; Sastry, S. Length scales in glass-forming liquids and related systems: A review. Rep. Prog. Phys. 2015, 79, 016601. [Google Scholar] [CrossRef]
- Kob, W.; Roldán-Vargas, S.; Berthier, L. Non-monotonic temperature evolution of dynamic correlations in glass-forming liquids. Nat. Phys. 2012, 8, 164–167. [Google Scholar] [CrossRef]
- Royall, C.P.; Williams, S.R. The role of local structure in dynamical arrest. Phys. Rep. 2015, 560, 1–75. [Google Scholar] [CrossRef]
- Tripodo, A.; Giuntoli, A.; Malvaldi, M.; Leporini, D. Mutual information does not detect growing correlations in the propensity of a model molecular liquid. Soft Matter 2019, 15, 6784–6790. [Google Scholar] [CrossRef]
- Tripodo, A.; Puosi, F.; Malvaldi, M.; Leporini, D. Vibrational scaling of the heterogeneous dynamics detected by mutual information. Eur. Phys. J. E 2019, 42, 1–10. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Cubuk, E.D.; Schoenholz, S.S.; Rieser, J.M.; Malone, B.D.; Rottler, J.; Durian, D.J.; Kaxiras, E.; Liu, A.J. Identifying Structural Flow Defects in Disordered Solids Using Machine-Learning Methods. Phys. Rev. Lett. 2015, 114, 108001. [Google Scholar] [CrossRef]
- Alkemade, R.M.; Boattini, E.; Filion, L.; Smallenburg, F. Comparing machine learning techniques for predicting glassy dynamics. J. Chem. Phys. 2022, 156, 204503. [Google Scholar] [CrossRef]
- Boattini, E.; Smallenburg, F.; Filion, L. Averaging Local Structure to Predict the Dynamic Propensity in Supercooled Liquids. Phys. Rev. Lett. 2021, 127, 088007. [Google Scholar] [CrossRef]
- Boattini, E.; Marín-Aguilar, S.; Mitra, S.; Foffi, G.; Smallenburg, F.; Filion, L. Autonomously revealing hidden local structures in supercooled liquids. Nat. Commun. 2020, 11, 5479. [Google Scholar] [CrossRef]
- Paret, J.; Jack, R.L.; Coslovich, D. Assessing the structural heterogeneity of supercooled liquids through community inference. J. Chem. Phys. 2020, 152, 144502. [Google Scholar] [CrossRef]
- Schoenholz, S.S.; Cubuk, E.D.; Sussman, D.M.; Kaxiras, E.; Liu, A.J. A structural approach to relaxation in glassy liquids. Nat. Phys. 2016, 12, 469–471. [Google Scholar] [CrossRef]
- Landes, F.m.c.P.; Biroli, G.; Dauchot, O.; Liu, A.J.; Reichman, D.R. Attractive versus truncated repulsive supercooled liquids: The dynamics is encoded in the pair correlation function. Phys. Rev. E 2020, 101, 010602. [Google Scholar] [CrossRef]
- Widmer-Cooper, A.; Harrowell, P. Predicting the Long-Time Dynamic Heterogeneity in a Supercooled Liquid on the Basis of Short-Time Heterogeneities. Phys. Rev. Lett. 2006, 96, 185701. [Google Scholar] [CrossRef]
- Widmer-Cooper, A.; Harrowell, P.; Fynewever, H. How Reproducible Are Dynamic Heterogeneities in a Supercooled Liquid? Phys. Rev. Lett. 2004, 93, 135701. [Google Scholar] [CrossRef]
- Balbuena, C.; Gianetti, M.M.; Soulé, E.R. Looking at the dynamical heterogeneity in a supercooled polymer system through isoconfigurational ensemble. J. Chem. Phys. 2018, 149, 094506. [Google Scholar] [CrossRef]
- Bapst, V.; Keck, T.; Grabska-Barwińska, A.; Donner, C.; Cubuk, E.D.; Schoenholz, S.S.; Obika, A.; Nelson, A.W.R.; Back, T.; Hassabis, D.; et al. Unveiling the predictive power of static structure in glassy systems. Nat. Phys. 2020, 16, 448–454. [Google Scholar] [CrossRef]
- Larini, L.; Ottochian, A.; De Michele, C.; Leporini, D. Universal scaling between structural relaxation and vibrational dynamics in glass-forming liquids and polymers. Nat. Phys. 2008, 4, 42–45. [Google Scholar] [CrossRef]
- Baschnagel, J.; Varnik, F. Computer simulations of supercooled polymer melts in the bulk and in confined geometry. J. Phys. Condens. Matter 2005, 17, R851–R953. [Google Scholar] [CrossRef]
- Puosi, F.; Tripodo, A.; Leporini, D. Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. Int. J. Mol. Sci. 2019, 20, 5708. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids, 3rd ed.; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. [Google Scholar] [CrossRef]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Kröger, M. Simple models for complex nonequilibrium fluids. Phys. Rep. 2004, 390, 453–551. [Google Scholar] [CrossRef]
- Candelier, R.; Widmer-Cooper, A.; Kummerfeld, J.K.; Dauchot, O.; Biroli, G.; Harrowell, P.; Reichman, D.R. Spatiotemporal Hierarchy of Relaxation Events, Dynamical Heterogeneities, and Structural Reorganization in a Supercooled Liquid. Phys. Rev. Lett. 2010, 105, 135702. [Google Scholar] [CrossRef]
- Pastore, R.; Pesce, G.; Sasso, A.; Pica Ciamarra, M. Cage Size and Jump Precursors in Glass-Forming Liquids: Experiment and Simulations. J. Phys. Chem. Lett. 2017, 8, 1562–1568. [Google Scholar] [CrossRef]
- Géron, A. Hands-On Machine Learning with Scikit-Learn and TensorFlow: Concepts, Tools, and Techniques to Build Intelligent Systems, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2017. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. 2015. Available online: tensorflow.org (accessed on 30 October 2021).
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar] [CrossRef]
- Schnell, B.; Meyer, H.; Fond, C.; Wittmer, J.P.; Baschnagel, J. Simulated glass-forming polymer melts: Glass transition temperature and elastic constants of the glassy state. Eur. Phys. J. E 2011, 34, 97. [Google Scholar] [CrossRef] [PubMed]
- Keys, A.S.; Abate, A.R.; Glotzer, S.C.; Durian, D.J. Measurement of growing dynamical length scales and prediction of the jamming transition in a granular material. Nat. Phys. 2007, 3, 260–264. [Google Scholar] [CrossRef]
- Berthier, L.; Jack, R.L. Structure and dynamics of glass formers: Predictability at large length scales. Phys. Rev. E 2007, 76, 041509. [Google Scholar] [CrossRef] [PubMed]
- Berthier, L. Dynamic heterogeneity in amorphous materials. Physics 2011, 42. [Google Scholar] [CrossRef]
- Available online: http://lammps.sandia.gov (accessed on 26 February 2020).
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1988. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodo, A.; Cordella, G.; Puosi, F.; Malvaldi, M.; Leporini, D. Neural Networks Reveal the Impact of the Vibrational Dynamics in the Prediction of the Long-Time Mobility of Molecular Glassformers. Int. J. Mol. Sci. 2022, 23, 9322. https://doi.org/10.3390/ijms23169322
Tripodo A, Cordella G, Puosi F, Malvaldi M, Leporini D. Neural Networks Reveal the Impact of the Vibrational Dynamics in the Prediction of the Long-Time Mobility of Molecular Glassformers. International Journal of Molecular Sciences. 2022; 23(16):9322. https://doi.org/10.3390/ijms23169322
Chicago/Turabian StyleTripodo, Antonio, Gianfranco Cordella, Francesco Puosi, Marco Malvaldi, and Dino Leporini. 2022. "Neural Networks Reveal the Impact of the Vibrational Dynamics in the Prediction of the Long-Time Mobility of Molecular Glassformers" International Journal of Molecular Sciences 23, no. 16: 9322. https://doi.org/10.3390/ijms23169322
APA StyleTripodo, A., Cordella, G., Puosi, F., Malvaldi, M., & Leporini, D. (2022). Neural Networks Reveal the Impact of the Vibrational Dynamics in the Prediction of the Long-Time Mobility of Molecular Glassformers. International Journal of Molecular Sciences, 23(16), 9322. https://doi.org/10.3390/ijms23169322