
Nonsteroidal Mineralocorticoid Receptor Antagonism by Finerenone—Translational Aspects and Clinical Perspectives across Multiple Organ Systems



Abstract
:1. Introduction
2. The Effects of Finerenone’s Mechanism of Action
2.1. Sodium Retention
2.2. Oxidative Stress—ROS Generation
2.2.1. Kidney ROS
2.2.2. Cardiac ROS
2.2.3. Vascular ROS
2.3. Inflammation
2.3.1. Renal Inflammation
2.3.2. Cardiac Inflammation
2.4. Fibrosis
2.4.1. Renal Fibrosis
2.4.2. Cardiac and Vascular Fibrosis
2.5. Hypertrophy/Remodeling
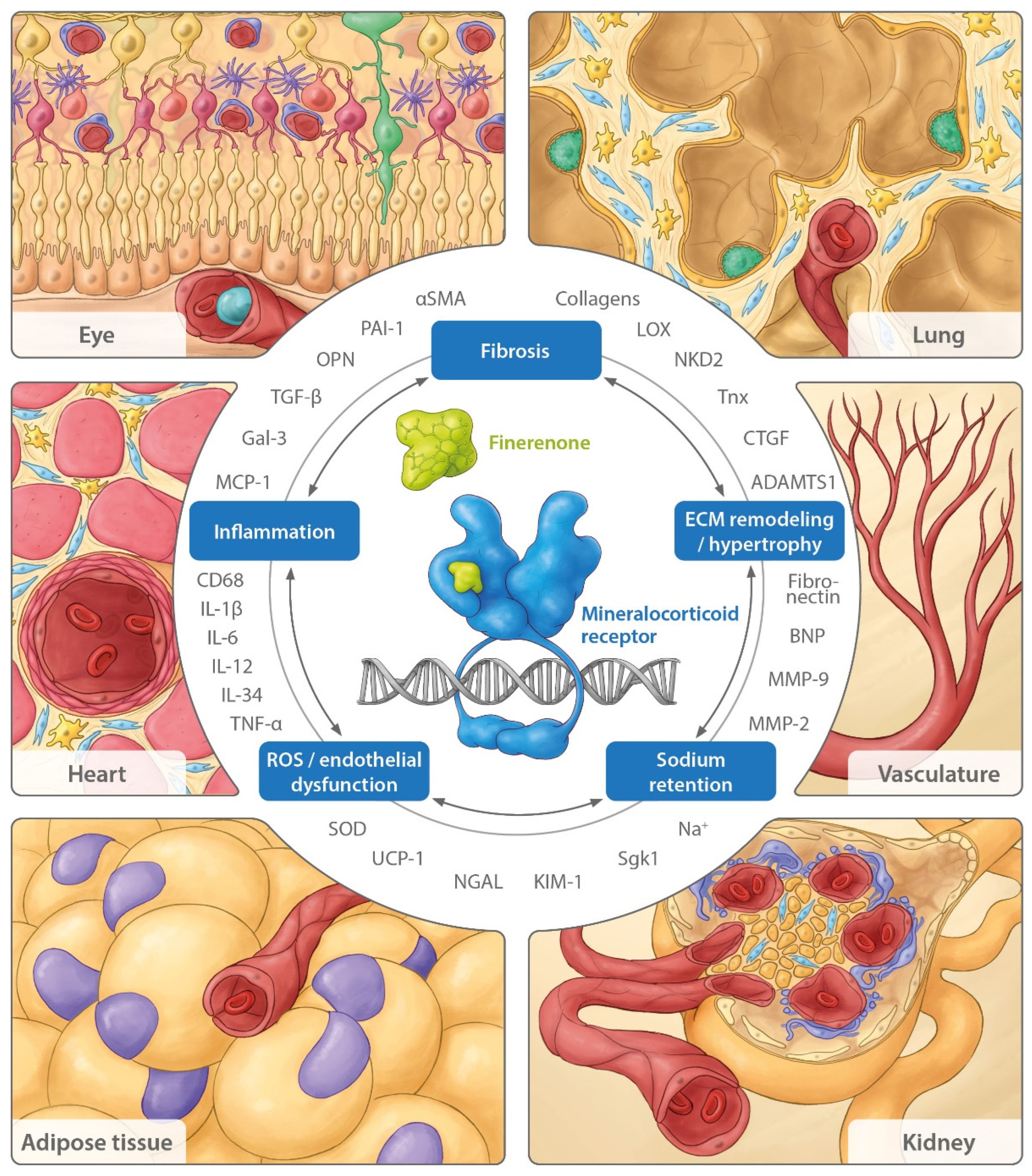
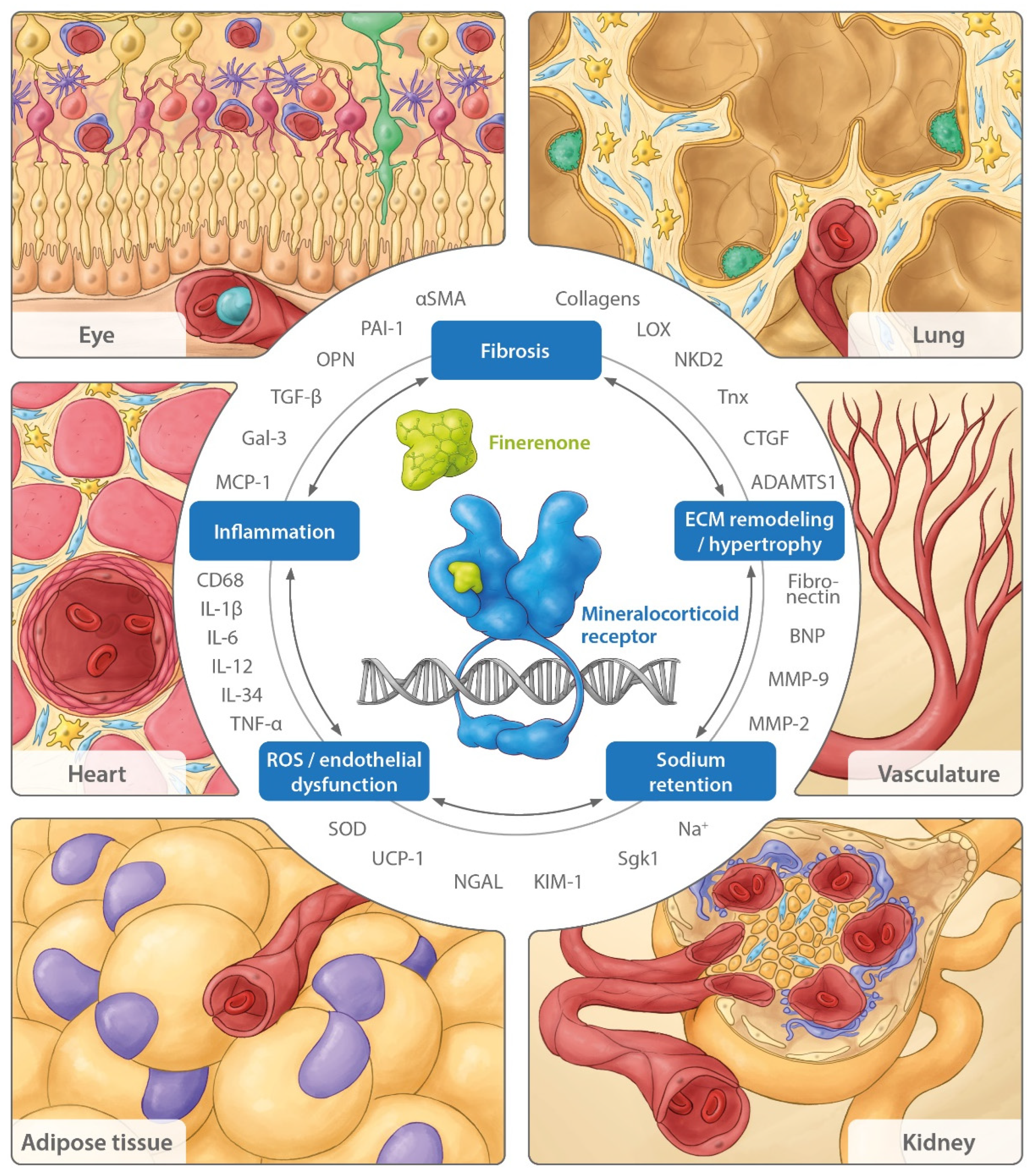
3. Beyond the Cardiovascular and Renal Systems: Consequences of MR Overactivation in Other Organs
3.1. Lung
3.2. Adipose Tissue
3.3. Eye
4. Finerenone in Clinical Studies: Mechanistic Explanations and Future Prospects
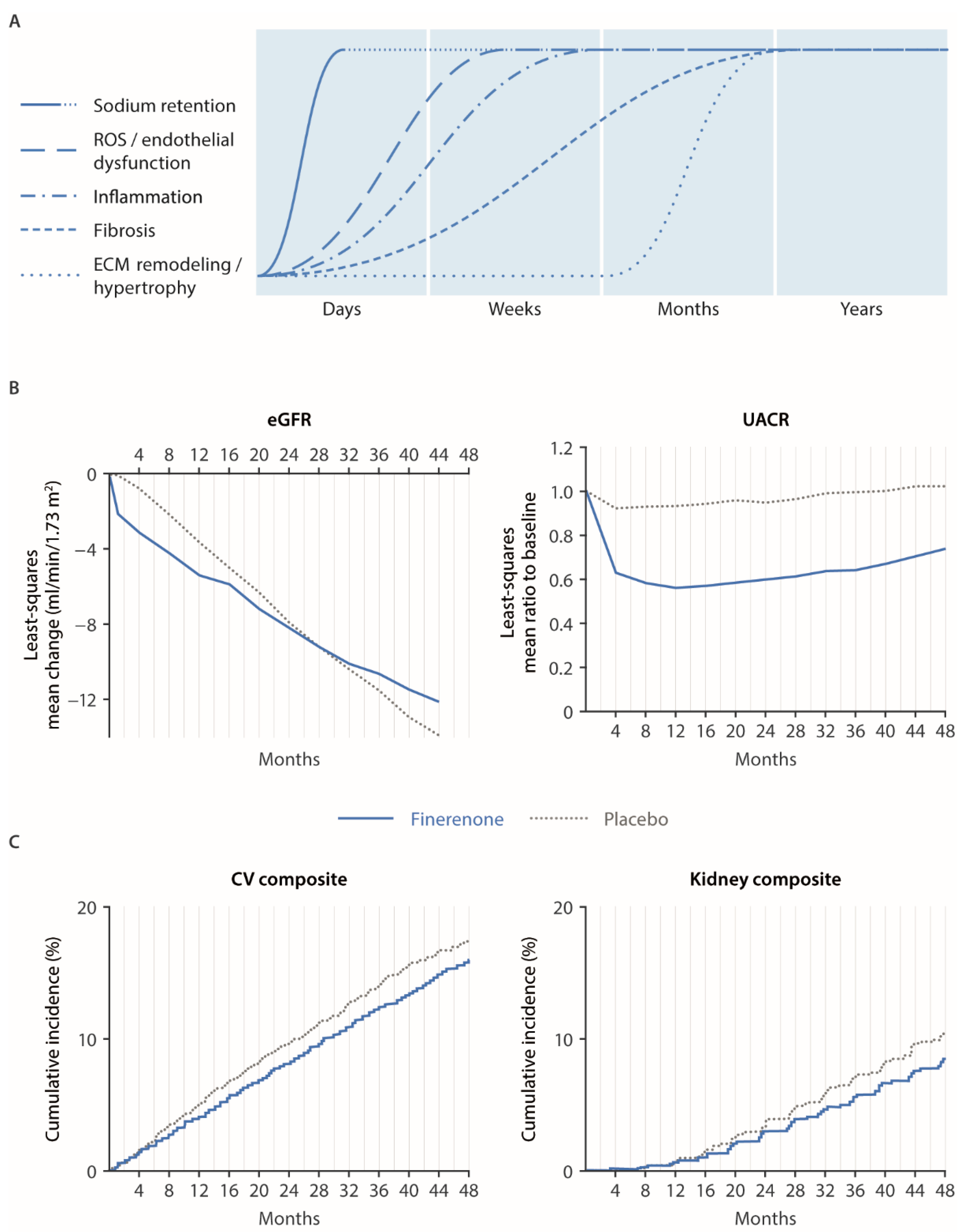
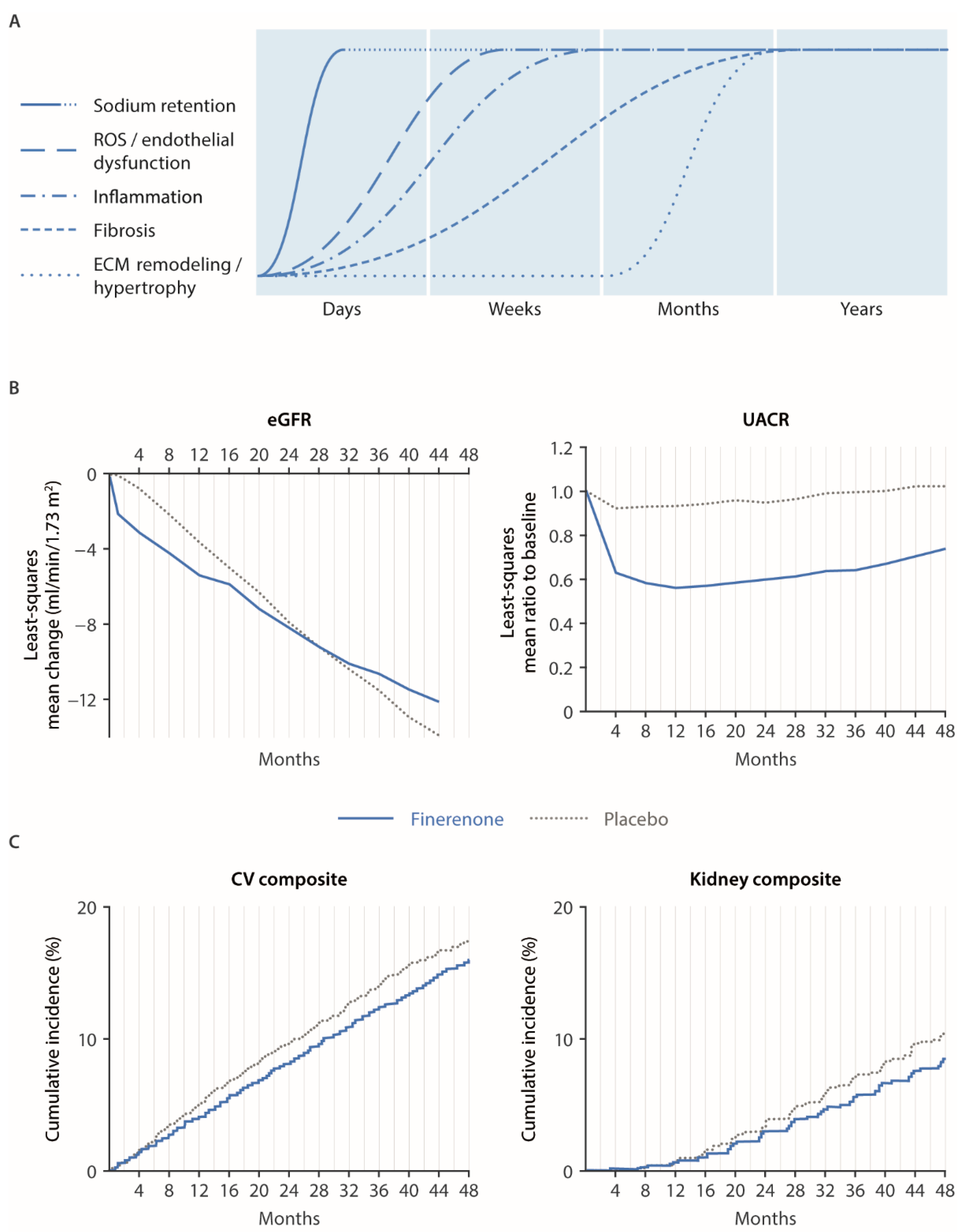
4.1. Renal and Cardiovascular Outcomes in FIGARO/FIDELIO and Mechanistic Explanations
4.2. “Systemic” MR Antagonism vs. “Local” (i.e., Proximal Tubule-Specific) SGLT-2 Inhibition and Potential for Combination Therapy
4.3. Potential Future Clinical Indications for Finerenone and Ongoing Clinical Studies
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Jaisser, F.; Farman, N. Emerging roles of the mineralocorticoid receptor in pathology: Toward new paradigms in clinical pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Selzer, C.; Schuster, P.; Lenski, M.; Adam, O.; Schäfers, H.J.; Böhm, M.; Laufs, U. The mineralocorticoid receptor promotes fibrotic remodeling in atrial fibrillation. J. Biol. Chem. 2014, 289, 6656–6668. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J.; Jaisser, F.; Toto, R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension 2015, 65, 257–263. [Google Scholar] [CrossRef]
- Kolkhof, P.; Joseph, A.; Kintscher, U. Nonsteroidal mineralocorticoid receptor antagonism for cardiovascular and renal disorders—New perspectives for combination therapy. Pharmacol. Res. 2021, 172, 105859. [Google Scholar] [CrossRef] [PubMed]
- Viengchareun, S.; Le Menuet, D.; Martinerie, L.; Munier, M.; Pascual-Le Tallec, L.; Lombès, M. The mineralocorticoid receptor: Insights into its molecular and (patho)physiological biology. Nucl. Recept. Signal. 2007, 5, e012. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Jaisser, F.; Anders, H.J. The mineralocorticoid receptor in chronic kidney disease. Br. J. Pharmacol. 2022, 179, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Jaisser, F. Vascular mineralocorticoid receptor activation and disease. Exp. Eye Res. 2019, 188, 107796. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Bonnard, B.; Jaisser, F. Roles of mineralocorticoid receptors in cardiovascular and cardiorenal diseases. Annu. Rev. Physiol. 2022, 84, 585–610. [Google Scholar] [CrossRef]
- Hawkins, U.A.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.M.; Gomez-Sanchez, C.E. The ubiquitous mineralocorticoid receptor: Clinical implications. Curr. Hypertens. Rep. 2012, 14, 573–580. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- Nagase, M.; Ayuzawa, N.; Kawarazaki, W.; Ishizawa, K.; Ueda, K.; Yoshida, S.; Fujita, T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 2012, 59, 500–506. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Kolkhof, P.; Lima-Posada, I.; Joachim, A.; Rossignol, P.; Jaisser, F. Differentiation between emerging non-steroidal and established steroidal mineralocorticoid receptor antagonists: Head-to-head comparisons of pharmacological and clinical characteristics. Expert Opin. Investig. Drugs 2021, 30, 1141–1157. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Jaisser, F. Pathophysiologic mechanisms in diabetic kidney disease: A focus on current and future therapeutic targets. Diabetes Obes. Metab. 2020, 22 (Suppl. S1), 16–31. [Google Scholar] [CrossRef]
- Bauersachs, J.; López-Andrés, N. Mineralocorticoid receptor in cardiovascular diseases–clinical trials and mechanistic insights. Br. J. Pharmacol. 2022, 179, 3119–3134. [Google Scholar] [CrossRef]
- Lalevée, N.; Rebsamen, M.C.; Barrère-Lemaire, S.; Perrier, E.; Nargeot, J.; Bénitah, J.P.; Rossier, M.F. Aldosterone increases T-type calcium channel expression and in vitro beating. Cardiovasc. Res. 2005, 67, 216–224. [Google Scholar] [CrossRef]
- Rossier, M.F. The cardiac mineralocorticoid receptor (MR): A therapeutic target against ventricular arrhythmias. Front. Endocrinol. (Lausanne) 2021, 28, 694758. [Google Scholar] [CrossRef]
- Dudenbostel, T.; Calhoun, D.A. Use of aldosterone antagonists for treatment of uncontrolled resistant hypertension. Am. J. Hypertens. 2017, 30, 103–109. [Google Scholar] [CrossRef]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Kolkhof, P.; Jaisser, F.; Kim, S.Y.; Filippatos, G.; Nowack, C.; Pitt, B. Steroidal and novel non-steroidal mineralocorticoid receptor antagonists in heart failure and cardiorenal diseases: Comparison at bench and bedside. Handb. Exp. Pharmacol. 2017, 243, 271–305. [Google Scholar]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gomez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Andre-Gregoire, G.; Nguyen Dinh Cat, A.; Lechner, S.M.; Cau, J.; Prince, S.; Kolkhof, P.; Loirand, G.; Sauzeau, V.; Hauet, T.; et al. Benefit of mineralocorticoid receptor antagonism in AKI: Role of vascular smooth muscle Rac1. J. Am. Soc. Nephrol. 2017, 28, 1216–1226. [Google Scholar] [CrossRef]
- Lattenist, L.; Lechner, S.M.; Messaoudi, S.; Le Mercier, A.; El Moghrabi, S.; Prince, S.; Bobadilla, N.A.; Kolkhof, P.; Jaisser, F.; Barrera-Chimal, J. Nonsteroidal mineralocorticoid receptor antagonist finerenone protects against acute kidney injury-mediated chronic kidney disease: Role of oxidative stress. Hypertension 2017, 69, 870–878. [Google Scholar] [CrossRef]
- Buonafine, M.; Bonnard, B.; Jaisser, F. Mineralocorticoid receptor and cardiovascular disease. Am. J. Hypertens. 2018, 31, 1165–1174. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef]
- Fuller, P.J.; Young, M.J. Mechanisms of mineralocorticoid action. Hypertension 2005, 46, 1227–1235. [Google Scholar] [CrossRef]
- Lentini, S.; Kimmeskamp-Kirschbaum, N.; Wensing, G.; Heinig, R. BAY 94-8862 exerts a potent natriuretic effect in healthy male subjects pre-treated with fludrocortisone: Findings from a proof-of-concept study. Circulation 2012, 126, A10732. [Google Scholar]
- Soi, V.; Yee, J. Sodium homeostasis in chronic kidney disease. Adv. Chronic Kidney Dis. 2017, 24, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.T.; Schrier, R.W. Edematous disorders: Pathophysiology of renal sodium and water retention and treatment with diuretics. Curr. Opin. Nephrol. Hypertens. 1993, 2, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H. Treatment of disorders of sodium balance in chronic kidney disease. Adv. Chronic Kidney Dis. 2017, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, D.; Nishimoto, M.; Ayuzawa, N.; Kawarazaki, W.; Fujii, W.; Oba, S.; Shibata, S.; Marumo, T.; Fujita, T. Activation of Rac1-mineralocorticoid receptor pathway contributes to renal injury in salt-loaded db/db mice. Hypertension 2021, 78, 82–93. [Google Scholar] [CrossRef]
- Schatz, V.; Neubert, P.; Schröder, A.; Binger, K.; Gebhard, M.; Müller, D.N.; Luft, F.C.; Titze, J.; Jantsch, J. Elementary immunology: Na+ as a regulator of immunity. Pediatr. Nephrol. 2017, 32, 201–210. [Google Scholar] [CrossRef]
- Sahinoz, M.; Tintara, S.; Deger, S.M.; Alsouqi, A.; Crescenzi, R.L.; Mambungu, C.; Vincz, A.; Mason, O.; Prigmore, H.L.; Guide, A.; et al. Tissue sodium stores in peritoneal dialysis and hemodialysis patients determined by 23-sodium magnetic resonance imaging. Nephrol. Dial. Transpl. 2020, 36, 1307–1317. [Google Scholar] [CrossRef]
- Iyer, A.; Chan, V.; Brown, L. The DOCA-salt hypertensive rat as a model of cardiovascular oxidative and inflammatory stress. Curr. Cardiol. Rev. 2010, 6, 291–297. [Google Scholar] [CrossRef]
- Kitiyakara, C.; Chabrashvili, T.; Chen, Y.; Blau, J.; Karber, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J. Am. Soc. Nephrol. 2003, 14, 2775–2782. [Google Scholar] [CrossRef]
- Rubattu, S.; Mennuni, S.; Testa, M.; Mennuni, M.; Pierelli, G.; Pagliaro, B.; Gabriele, E.; Coluccia, R.; Autore, C.; Volpe, M. Pathogenesis of chronic cardiorenal syndrome: Is there a role for oxidative stress? Int. J. Mol. Sci. 2013, 14, 23011–23032. [Google Scholar] [CrossRef]
- Zannad, F.; Rossignol, P. Cardiorenal syndrome revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Araujo, M.; Wilcox, C.S. Oxidative stress in hypertension: Role of the kidney. Antioxid. Redox Signal. 2014, 20, 74–101. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Yao, L.; Nagai, Y.; Miyata, K.; Yoshizumi, M.; Kagami, S.; Kondo, S.; Kiyomoto, H.; Shokoji, T.; Kimura, S.; et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension 2004, 43, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Prince, S.; Fadel, F.; El Moghrabi, S.; Warnock, D.G.; Kolkhof, P.; Jaisser, F. Sulfenic acid modification of endothelin B receptor is responsible for the benefit of a nonsteroidal mineralocorticoid receptor antagonist in renal ischemia. J. Am. Soc. Nephrol. 2016, 27, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, I.; Sohara, E.; Ito, E.; Okado, T.; Rai, T.; Uchida, S. Fibronectin glomerulopathy. Clin. Kidney J. 2013, 6, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Liangos, O.; Perianayagam, M.C.; Vaidya, V.S.; Han, W.K.; Wald, R.; Tighiouart, H.; MacKinnon, R.W.; Li, L.; Balakrishnan, V.S.; Pereira, B.J.; et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J. Am. Soc. Nephrol. 2007, 18, 904–912. [Google Scholar] [CrossRef]
- Kolkhof, P.; Delbeck, M.; Kretschmer, A.; Steinke, W.; Hartmann, E.; Barfacker, L.; Eitner, F.; Albrecht-Kupper, B.; Schafer, S. Finerenone, a novel selective nonsteroidal mineralocorticoid receptor antagonist protects from rat cardiorenal injury. J. Cardiovasc. Pharmacol. 2014, 64, 69–78. [Google Scholar] [CrossRef]
- Upadhyay, A.; Larson, M.G.; Guo, C.Y.; Vasan, R.S.; Lipinska, I.; O’Donnell, C.J.; Kathiresan, S.; Meigs, J.B.; Keaney, J.F., Jr.; Rong, J.; et al. Inflammation, kidney function and albuminuria in the Framingham offspring cohort. Nephrol. Dial. Transplant. 2011, 26, 920–926. [Google Scholar] [CrossRef]
- Murea, M.; Register, T.C.; Divers, J.; Bowden, D.W.; Carr, J.J.; Hightower, C.R.; Xu, J.; Smith, S.C.; Hruska, K.A.; Langefeld, C.D.; et al. Relationships between serum MCP-1 and subclinical kidney disease: African American-Diabetes Heart Study. BMC Nephrol. 2012, 13, 148. [Google Scholar] [CrossRef]
- Tesch, G.H. MCP-1/CCL2: A new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2008, 294, F697–F701. [Google Scholar] [CrossRef]
- Titan, S.M.; Vieira, J.M., Jr.; Dominguez, W.V.; Moreira, S.R.; Pereira, A.B.; Barros, R.T.; Zatz, R. Urinary MCP-1 and RBP: Independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. J. Diabetes Complications. 2012, 26, 546–553. [Google Scholar] [CrossRef]
- Mansour, S.G.; Puthumana, J.; Coca, S.G.; Gentry, M.; Parikh, C.R. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: A systematic review. BMC Nephrol. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ortega, M.; Vega-Martin, E.; Martin-Ramos, M.; González-Blázquez, R.; Pulido-Olmo, H.; Ruiz-Hurtado, G.; Schulz, A.; Ruilope, L.M.; Kolkhof, P.; Somoza, B.; et al. Finerenone reduces intrinsic arterial stiffness in Munich Wistar Frömter rats, a genetic model of chronic kidney disease. Am. J. Nephrol. 2020, 51, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.W.; Kuo, K.L.; Hung, S.C.; Huang, P.H.; Chen, J.W.; Tarng, D.C. Progression of kidney disease in non-diabetic patients with coronary artery disease: Predictive role of circulating matrix metalloproteinase-2, -3, and -9. PLoS ONE 2013, 8, e70132. [Google Scholar] [CrossRef]
- Pulido-Olmo, H.; García-Prieto, C.F.; Álvarez-Llamas, G.; Barderas, M.G.; Vivanco, F.; Aranguez, I.; Somoza, B.; Segura, J.; Kreutz, R.; Fernández-Alfonso, M.S.; et al. Role of matrix metalloproteinase-9 in chronic kidney disease: A new biomarker of resistant albuminuria. Clin. Sci. 2016, 130, 525–538. [Google Scholar] [CrossRef]
- Bolignano, D.; Lacquaniti, A.; Coppolino, G.; Donato, V.; Campo, S.; Fazio, M.R.; Nicocia, G.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Lv, L.L.; Pan, M.M.; Cao, Y.H.; Liu, H.; Feng, Y.; Ni, H.F.; Liu, B.C. Are urinary tubular injury markers useful in chronic kidney disease? A systematic review and meta analysis. PLoS ONE 2016, 11, e0167334. [Google Scholar] [CrossRef]
- Droebner, K.; Pavkovic, M.; Grundmann, M.; Hartmann, E.; Goea, L.; Nordlohne, J.; Klar, J.; Eitner, F.; Kolkhof, P. Direct blood pressure-independent anti-fibrotic effects by the selective nonsteroidal mineralocorticoid receptor antagonist finerenone in progressive models of kidney fibrosis. Am. J. Nephrol. 2021, 52, 588–601. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Paton, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Abedini, A.; Wu, J.; Ma, Z.; Frederick, J.; Cernecka, H.; Kolkhof, P.; Susztak, K. Revealing the antifibrotic mechanism of finerenone in the DOCA-salt nephropathy rat model using single nuclei and bulk transcriptomics. J. Am. Soc. Nephrol. 2021, 32, 765. [Google Scholar]
- Steinbrenner, I.; Sekula, P.; Kotsis, F.; von Cube, M.; Cheng, Y.; Nadal, J.; Schmid, M.; Schneider, M.P.; Krane, V.; Nauck, M.; et al. Association of osteopontin with kidney function and kidney failure in chronic kidney disease patients: The GCKD study. Nephrol. Dial. Transpl. 2022, gfac173. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Estrela, G.R.; Lechner, S.M.; Giraud, S.; El Moghrabi, S.; Kaaki, S.; Kolkhof, P.; Hauet, T.; Jaisser, F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018, 93, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A.; Fogo, A.B. Plasminogen activator inhibitor-1 in chronic kidney disease: Evidence and mechanisms of action. J. Am. Soc. Nephrol. 2006, 17, 2999–3012. [Google Scholar] [CrossRef] [PubMed]
- Scurt, F.G.; Menne, J.; Brandt, S.; Bernhardt, A.; Mertens, P.R.; Haller, H.; Chatzikyrkou, C.; Ito, S.; Izzo, J.L.; Januszewicz, A.; et al. Systemic inflammation precedes microalbuminuria in diabetes. Kidney Int. Rep. 2019, 4, 1373–1386. [Google Scholar] [CrossRef]
- Kolkhof, P.; Hartmann, E.; Freyberger, A.; Pavkovic, M.; Mathar, I.; Sandner, P.; Droebner, K.; Joseph, A.; Hüser, J.; Eitner, F. Effects of finerenone combined with empagliflozin in a model of hypertension-induced end-organ damage. Am. J. Nephrol. 2021, 52, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Juul-Nielsen, C.; Shen, J.; Stenvinkel, P.; Scholze, A. Systematic review of the nuclear factor erythroid 2-related factor 2 (NRF2) system in human chronic kidney disease: Alterations, interventions and relation to morbidity. Nephrol. Dial. Transplant. 2022, 37, 904–916. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular superoxide dismutase protects against proteinuric kidney disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and progression of CKD: The CRIC study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef]
- Fan, Q.; Yan, X.; Zhang, H.; Lu, L.; Zhang, Q.; Wang, F.; Xi, R.; Hu, J.; Chen, Q.; Niu, W.; et al. IL-34 is associated with the presence and severity of renal dysfunction and coronary artery disease in patients with heart failure. Sci. Rep. 2016, 6, 39324. [Google Scholar] [CrossRef]
- Grune, J.; Beyhoff, N.; Smeir, E.; Chudek, R.; Blumrich, A.; Ban, Z.; Brix, S.; Betz, I.R.; Schupp, M.; Foryst-Ludwig, A.; et al. Selective mineralocorticoid receptor cofactor modulation as molecular basis for finerenone’s antifibrotic activity. Hypertension 2018, 71, 599–608. [Google Scholar] [CrossRef]
- Lachaux, M.; Barrera-Chimal, J.; Nicol, L.; Rémy-Jouet, I.; Renet, S.; Dumesnil, A.; Wecker, D.; Richard, V.; Kolkhof, P.; Jaisser, F.; et al. Short- and long-term administration of the non-steroidal mineralocorticoid receptor antagonist finerenone opposes metabolic syndrome-related cardio-renal dysfunction. Diabetes Obes. Metab. 2018, 20, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Jacobs, N.; Mahfoud, F.; Kolkhof, P.; Bohm, M.; Laufs, U. The non-steroidal mineralocorticoid receptor antagonist finerenone prevents cardiac fibrotic remodeling. Biochem. Pharmacol. 2019, 168, 173–183. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Niwano, K.; Watanabe, A.; Endoh, M.; Suguta, M.; Yokoyama, T.; Tada, H.; Toyama, T.; Adachi, H.; et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.L.; Katz, R.; Bellovich, K.A.; Bhat, Z.Y.; Brosius, F.C.; de Boer, I.H.; Gadegbeku, C.A.; Gipson, D.S.; Hawkins, J.J.; Himmelfarb, J.; et al. Soluble ST2 and galectin-3 and progression of CKD. Kidney Int. Rep. 2018, 4, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.B.; Cheung, C.L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Galectin-3 is independently associated with progression of nephropathy in type 2 diabetes mellitus. Diabetologia 2018, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Wang, Y.Y.; Jia, P.; Xiong, Q.; Hu, Y.; Chang, Y.; Lai, S.; Xu, Y.; Zhao, Z.; Song, J. Multi-level transcriptome sequencing identifies COL1A1 as a candidate marker in human heart failure progression. BMC Med. 2020, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- López, B.; Querejeta, R.; González, A.; Beaumont, J.; Larman, M.; Díez, J. Impact of treatment on myocardial lysyl oxidase expression and collagen cross-linking in patients with heart failure. Hypertension 2009, 53, 236–242. [Google Scholar] [CrossRef]
- Martínez-Martínez, E.; Buonafine, M.; Boukhalfa, I.; Ibarrola, J.; Fernández-Celis, A.; Kolkhof, P.; Rossignol, P.; Girerd, N.; Mulder, P.; López-Andrés, N.; et al. Aldosterone target NGAL (neutrophil gelatinase-associated lipocalin) is involved in cardiac remodeling after myocardial infarction through NFκB pathway. Hypertension 2017, 70, 1148–1156. [Google Scholar] [CrossRef]
- Parajuli, N.; Patel, V.B.; Wang, W.; Basu, R.; Oudit, G.Y. Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin. Sci. (Lond.) 2014, 127, 331–340. [Google Scholar] [CrossRef]
- Grune, J.; Benz, V.; Brix, S.; Salatzki, J.; Blumrich, A.; Hoft, B.; Klopfleisch, R.; Foryst-Ludwig, A.; Kolkhof, P.; Kintscher, U. Steroidal and nonsteroidal mineralocorticoid receptor antagonists cause differential cardiac gene expression in pressure overload-induced cardiac hypertrophy. J. Cardiovasc. Pharmacol. 2016, 67, 402–411. [Google Scholar] [CrossRef]
- Gonzalez-Blazquez, R.; Somoza, B.; Gil-Ortega, M.; Martin Ramos, M.; Ramiro-Cortijo, D.; Vega-Martin, E.; Schulz, A.; Ruilope, L.M.; Kolkhof, P.; Kreutz, R.; et al. Finerenone attenuates endothelial dysfunction and albuminuria in a chronic kidney disease model by a reduction in oxidative stress. Front. Pharmacol. 2018, 9, 1131. [Google Scholar] [CrossRef] [PubMed]
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The role of the mineralocorticoid receptor in inflammation: Focus on kidney and vasculature. Am. J. Nephrol. 2017, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Nikolic-Paterson, D.J.; Han, Y.; Ozols, E.; Ma, F.Y.; Young, M.J.; Tesch, G.H. Myeloid mineralocorticoid receptor activation contributes to progressive kidney disease. J. Am. Soc. Nephrol. 2014, 25, 2231–2240. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr. Nephrol. 2015, 30, 199–209. [Google Scholar] [CrossRef]
- van der Heijden, C.D.C.C.; Deinum, J.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc. Res. 2018, 114, 944–953. [Google Scholar] [CrossRef]
- Luettges, K.; Bode, M.; Diemer, J.N.; Schwanbeck, J.; Wirth, E.K.; Klopfleisch, R.; Kappert, K.; Thiele, A.; Ritter, D.; Foryst-Ludwig, A.; et al. Finerenone reduces renal RORgammat gammadelta T cells and protects against cardiorenal damage. Am. J. Nephrol. 2022, 53, 552–564. [Google Scholar] [CrossRef]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Rocha, L.; Amador-Martinez, I.; Perez-Villalva, R.; Gonzalez, R.; Cortes-Gonzalez, C.; Uribe, N.; Ramirez, V.; Berman, N.; Gamba, G.; et al. Delayed spironolactone administration prevents the transition from acute kidney injury to chronic kidney disease through improving renal inflammation. Nephrol. Dial. Transplant. 2019, 34, 794–801. [Google Scholar] [CrossRef]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef]
- Usher, M.G.; Duan, S.Z.; Ivaschenko, C.Y.; Frieler, R.A.; Berger, S.; Schutz, G.; Lumeng, C.N.; Mortensen, R.M. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 2010, 120, 3350–3364. [Google Scholar] [CrossRef]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage polarization in cardiac tissue repair following myocardial infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef]
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Aspects Med. 2019, 65, 16–36. [Google Scholar] [CrossRef]
- Thomas, T.P.; Grisanti, L.A. The dynamic interplay between cardiac inflammation and fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef]
- Lowe, J.; Kolkhof, P.; Haupt, M.J.; Peczkowski, K.K.; Rastogi, N.; Hauck, J.S.; Kadakia, F.K.; Zins, J.G.; Ciccone, P.C.; Smart, S.; et al. Mineralocorticoid receptor antagonism by finerenone is sufficient to improve function in preclinical muscular dystrophy. ESC Heart Fail. 2020, 7, 3983–3995. [Google Scholar] [CrossRef]
- Gueret, A.; Harouki, N.; Favre, J.; Galmiche, G.; Nicol, L.; Henry, J.P.; Besnier, M.; Thuillez, C.; Richard, V.; Kolkhof, P.; et al. Vascular smooth muscle mineralocorticoid receptor contributes to coronary and left ventricular dysfunction after myocardial infarction. Hypertension 2016, 67, 717–723. [Google Scholar] [CrossRef]
- Hill, J.A.; Karimi, M.; Kutschke, W.; Davisson, R.L.; Zimmerman, K.; Wang, Z.; Kerber, R.E.; Weiss, R.M. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation 2000, 101, 2863–2869. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.Y.; Frieler, R.A.; Zheng, X.J.; Zhang, W.C.; Sun, X.N.; Yang, Q.Z.; Ma, S.M.; Huang, B.; Berger, S.; et al. Myeloid mineralocorticoid receptor deficiency inhibits aortic constriction-induced cardiac hypertrophy in mice. PLoS ONE 2014, 9, e110950. [Google Scholar] [CrossRef]
- Dutzmann, J.; Musmann, R.J.; Haertle, M.; Daniel, J.M.; Sonnenschein, K.; Schafer, A.; Kolkhof, P.; Bauersachs, J.; Sedding, D.G. The novel mineralocorticoid receptor antagonist finerenone attenuates neointima formation after vascular injury. PLoS ONE 2017, 12, e0184888. [Google Scholar] [CrossRef]
- Kramer, G.; Mitteregger, D.; Marberger, M. Is benign prostatic hyperplasia (BPH) an immune inflammatory disease? Eur. Urol. 2007, 51, 1202–1216. [Google Scholar] [CrossRef]
- Saponaro, M.; Giacomini, I.; Morandin, G.; Cocetta, V.; Ragazzi, E.; Orso, G.; Carnevali, I.; Berretta, M.; Mancini, M.; Pagano, F.; et al. Serenoa repens and urtica dioica fixed combination: In-vitro validation of a therapy for benign prostatic hyperplasia (BPH). Int. J. Mol. Sci. 2020, 21, 9178. [Google Scholar] [CrossRef]
- Hirasawa, G.; Sasano, H.; Takahashi, K.; Fukushima, K.; Suzuki, T.; Hiwatashi, N.; Toyota, T.; Krozowski, Z.S.; Nagura, H. Colocalization of 11 beta-hydroxysteroid dehydrogenase type II and mineralocorticoid receptor in human epithelia. J. Clin. Endocrinol. Metab. 1997, 82, 3859–3863. [Google Scholar]
- Rosenbloom, J.; Macarak, E.; Piera-Velazquez, S.; Jimenez, S.A. Human fibrotic diseases: Current challenges in fibrosis research. Methods Mol. Biol. 2017, 1627, 1–23. [Google Scholar]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The bleomycin model of pulmonary fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar]
- Delbeck, M.; Joseph, A.; Pitt, B.; Kolkhof, P. Anti-fibrotic and anti-inflammatory effects of the selective nonsteroidal MR antagonist finerenone in preclinical pulmonary fibrosis. Eur. Heart J. 2021, 42, ehab724-2932. [Google Scholar] [CrossRef]
- Tu, L.; Thuillet, R.; Perrot, J.; Ottaviani, M.; Ponsardin, E.; Kolkhof, P.; Humbert, M.; Viengchareun, S.; Lombes, M.; Guignabert, C. Mineralocorticoid receptor antagonism by finerenone attenuates established pulmonary hypertension in rats. Hypertension 2022, in press. [CrossRef]
- Tsoutsou, P.G.; Gourgoulianis, K.I.; Petinaki, E.; Germenis, A.; Tsoutsou, A.G.; Mpaka, M.; Efremidou, S.; Molyvdas, P.A. Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respir. Med. 2006, 100, 938–945. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Zou, R.; Chen, H.; Liu, X.; Qiu, X.; Xiao, Y.; Cai, H.; Dai, J. Associations of serological biomarkers of sICAM-1, IL-1β, MIF, and su-PAR with 3-month mortality in acute exacerbation of idiopathic pulmonary fibrosis. Mediators Inflamm. 2020, 2020, 4534272. [Google Scholar] [CrossRef]
- Tsitoura, E.; Trachalaki, A.; Vasarmidi, E.; Mastrodemou, S.; Margaritopoulos, G.A.; Kokosi, M.; Fanidis, D.; Galaris, A.; Aidinis, V.; Renzoni, E.; et al. Collagen 1a1 expression by airway macrophages increases in fibrotic ILDs and is associated with FVC decline and increased mortality. Front. Immunol. 2021, 12, 645548. [Google Scholar] [CrossRef]
- Marzolla, V.; Feraco, A.; Limana, F.; Kolkhof, P.; Armani, A.; Caprio, M. Class-specific responses of brown adipose tissue to steroidal and nonsteroidal mineralocorticoid receptor antagonists. J. Endocrinol. Investig. 2022, 45, 215–220. [Google Scholar] [CrossRef]
- Gomez-Marcos, M.A.; Blazquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; Garcia-Ortiz, L.; Martinez-Salgado, C. Serum superoxide dismutase is associated with vascular structure and function in hypertensive and diabetic patients. Oxid. Med. Cell. Longev. 2016, 2016, 9124676. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Klarin, D.; Natarajan, P.; Zekavat, S.M.; Nomura, A.; Haas, M.; Aragam, K.; Ardissino, D.; Wilson, J.G.; et al. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 2018, 137, 222–232. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Food ingredients involved in white-to-brown adipose tissue conversion and in calorie burning. Front. Physiol. 2019, 9, 1954. [Google Scholar] [CrossRef]
- Kaisanlahti, A.; Glumoff, T. Browning of white fat: Agents and implications for beige adipose tissue to type 2 diabetes. J. Physiol. Biochem. 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. The role of aldosterone in obesity-related hypertension. Am. J. Hypertens. 2016, 29, 415–423. [Google Scholar] [CrossRef]
- Deliyanti, D.; Miller, A.G.; Tan, G.; Binger, K.J.; Samson, A.L.; Wilkinson-Berka, J.L. Neovascularization is attenuated with aldosterone synthase inhibition in rats with retinopathy. Hypertension 2012, 59, 607–613. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Tan, G.; Jaworski, K.; Harbig, J.; Miller, A.G. Identification of a retinal aldosterone system and the protective effects of mineralocorticoid receptor antagonism on retinal vascular pathology. Circ. Res. 2009, 104, 124–133. [Google Scholar] [CrossRef]
- Zhao, M.; Valamanesh, F.; Celerier, I.; Savoldelli, M.; Jonet, L.; Jeanny, J.C.; Jaisser, F.; Farman, N.; Behar-Cohen, F. The neuroretina is a novel mineralocorticoid target: Aldosterone up-regulates ion and water channels in Müller glial cells. FASEB J. 2010, 24, 3405–3415. [Google Scholar] [CrossRef]
- Golestaneh, N.; De Kozak, Y.; Klein, C.; Mirshahi, M. Epithelial sodium channel and the mineralocorticoid receptor in cultured rat Müller glial cells. Glia 2001, 33, 160–168. [Google Scholar] [CrossRef]
- Deliyanti, D.; Armani, R.; Casely, D.; Figgett, W.A.; Agrotis, A.; Wilkinson-Berka, J.L. Retinal vasculopathy is reduced by dietary salt restriction: Involvement of Glia, ENaCα, and the renin-angiotensin-aldosterone system. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2033–2041. [Google Scholar] [CrossRef]
- Canonica, J.; Mehanna, C.; Bonnard, B.; Jonet, L.; Gelize, E.; Jais, J.P.; Jaisser, F.; Zhao, M.; Behar-Cohen, F. Effect of acute and chronic aldosterone exposure on the retinal pigment epithelium-choroid complex in rodents. Exp. Eye Res. 2019, 187, 107747. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Célérier, I.; Bousquet, E.; Jeanny, J.C.; Jonet, L.; Savoldelli, M.; Offret, O.; Curan, A.; Farman, N.; Jaisser, F.; et al. Mineralocorticoid receptor is involved in rat and human ocular chorioretinopathy. J. Clin. Investig. 2012, 122, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Gelize, E.; Levy, R.; Moulin, A.; Azan, F.; Berdugo, M.; Naud, M.C.; Guegan, J.; Delaunay, K.; Pussard, E.; et al. Mineralocorticoid receptor pathway and its antagonism in a model of diabetic retinopathy. Diabetes 2021, 70, 2668–2682. [Google Scholar] [CrossRef]
- Rossing, P.; Anker, S.; Garweg, J.; Osonoi, T.; Pitt, B.; Rosas, S.E.; Ruilope, L.; Zhu, D.; Brinker, M.; Finis, D.; et al. Effect of Finerenone on Occurrence of Vision-Threatening Events in Patients with DR. Diabetes 2022, 71 (Suppl. 1), 826-P. [Google Scholar] [CrossRef]
- Kraus, B.J.; Weir, M.R.; Bakris, G.L.; Mattheus, M.; Cherney, D.Z.I.; Sattar, N.; Heerspink, H.J.L.; Ritter, I.; von Eynatten, M.; Zinman, B.; et al. Characterization and implications of the initial estimated glomerular filtration rate ‘dip’ upon sodium-glucose cotransporter-2 inhibition with empagliflozin in the EMPA-REG OUTCOME trial. Kidney Int. 2021, 99, 750–762. [Google Scholar] [CrossRef]
- Bakris, G.; Weir, M.R. Initial drops in glomerular filtration rate with certain drug classes retard kidney disease progression. Am. J. Nephrol. 2022, 53, 513–515. [Google Scholar] [CrossRef]
- Ruilope, L.; Agarwal, R.; Anker, S.; Filippatos, G.; Pitt, B.; Rossing, P.; Sarafidis, P.; Schmieder, R.E.; Joseph, A.; Mentenich, N.; et al. Effects of finerenone on cardiorenal outcomes in blood pressure subgroups in patients with CKD and T2D. Nephrol. Dial. Transplant. 2021, 36 (Suppl. 1), i65–i66. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Pitt, B.; Anker, S.D.; Rossing, P.; Kovesdy, C.P.; Pecoits-Filho, R.; Pergola, P.; Joseph, A.; Lage, A.; Mentenich, N.; et al. Kidney outcomes with finerenone: An analysis from the FIGARO-DKD study. Nephrol. Dial. Transplant. 2022, gfac157. [Google Scholar] [CrossRef]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Agarwal, R.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Schloemer, P.; Tornus, I.; Joseph, A.; et al. Finerenone and cardiovascular outcomes in patients with chronic kidney disease and type 2 diabetes. Circulation 2021, 143, 540–552. [Google Scholar] [CrossRef]
- Filippatos, G.; Bakris, G.L.; Pitt, B.; Agarwal, R.; Rossing, P.; Ruilope, L.M.; Butler, J.; Lam, C.S.P.; Kolkhof, P.; Roberts, L.; et al. Finerenone reduces new-onset atrial fibrillation in patients with chronic kidney disease and type 2 diabetes. J. Am. Coll. Cardiol. 2021, 78, 142–152. [Google Scholar] [CrossRef]
- Filippatos, G.; Bakris, G.; Agarwal, A.; Anker, S.; Ruilope, L.; Pitt, B. Finerenone in Chronic Kidney Disease and Type 2 Diabetes: A FIDELITY Analysis of Left Ventricle Hypertrophy; ESC-HF: Madrid, Spain, 2022; Available online: https://esc365.escardio.org/presentation/250291?query=finerenone (accessed on 20 July 2022).
- Ferrannini, E. Sodium-glucose co-transporters and their inhibition: Clinical physiology. Cell. Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef]
- Bonora, B.M.; Avogaro, A.; Fadini, G.P. Extraglycemic effects of SGLT2 inhibitors: A review of the evidence. Diabetes Metab. Syndr. Obes. 2020, 13, 161–174. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular mechanisms of SGLT2 inhibitor on cardiorenal protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: A state-of-the-art review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Chertow, G.M.; Vart, P.; Jongs, N.; Toto, R.D.; Gorriz, J.L.; Hou, F.F.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; Sjöström, C.D.; et al. Effects of dapagliflozin in stage 4 chronic kidney disease. J. Am. Soc. Nephrol. 2021, 32, 2352–2361. [Google Scholar] [CrossRef]
- Bakris, G.; Oshima, M.; Mahaffey, K.W.; Agarwal, R.; Cannon, C.P.; Capuano, G.; Charytan, D.M.; de Zeeuw, D.; Edwards, R.; Greene, T.; et al. Effects of canagliflozin in patients with baseline eGFR <30 mL/min per 1.73 m2: Subgroup analysis of the randomized CREDENCE trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1705–1714. [Google Scholar] [CrossRef]
- Rossing, P.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Chan, J.C.N.; Kooy, A.; McCafferty, K.; Schernthaner, G.; et al. Finerenone in predominantly advanced CKD and type 2 diabetes with or without sodium-glucose cotransporter-2 inhibitor therapy. Kidney Int. Rep. 2021, 7, 36–45. [Google Scholar] [CrossRef]
- Rossing, P.; Anker, S.D.; Filippatos, G.; Pitt, B.; Ruilope, L.; Birkenfeld, A.L.; McGill, J.B.; Rosas, S.E.; Joseph, A.; Gebel, M.; et al. Finerenone in patients with chronic kidney disease and type 2 diabetes by sodium-glucose co-transporter-2 inhibitor treatment: The FIDELITY analysis. Diabetes Care 2022, dc220294. [Google Scholar] [CrossRef]
- Provenzano, M.; Puchades, M.; Garofalo, C.; Jongs, N.; D’Marco, L.; Andreucci, M.; De Nicola, L.; Gorriz, J.; Heerspink, H. Albuminuria-lowering effect of dapagliflozin, eplerenone, and their combination in patients with chronic kidney disease: A randomized cross-over clinical trial. J. Am. Soc. Nephrol. 2022, 33, 1569–1580. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Zannad, F.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Jamal, W.; Steubl, D.; Schueler, E.; et al. Interplay of mineralocorticoid receptor antagonists and empagliflozin in heart failure: EMPEROR-Reduced. J. Am. Coll. Cardiol. 2021, 77, 1397–1407. [Google Scholar] [CrossRef]
- Shen, L.; Kristensen, S.L.; Bengtsson, O.; Bohm, M.; de Boer, R.A.; Docherty, K.F.; Inzucchi, S.E.; Katova, T.; Kober, L.; Kosiborod, M.N.; et al. Dapagliflozin in HFrEF patients treated with mineralocorticoid receptor antagonists: An analysis of DAPA-HF. JACC Heart Fail. 2021, 9, 254–264. [Google Scholar] [CrossRef]
- Filippatos, G.; Anker, S.D.; Bohm, M.; Gheorghiade, M.; Kober, L.; Krum, H.; Maggioni, A.P.; Ponikowski, P.; Voors, A.A.; Zannad, F.; et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur. Heart J. 2016, 37, 2105–2114. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Marker | Effect of Finerenone in Preclinical Models | Function/Role | Evidence for Clinical Association |
|---|---|---|---|
| Kidney | |||
| Fibronectin | ↓ Kidney mRNA expression and protein levels in model of CKD progression in T2D [34] | Glycoprotein in the glomerular mesangial ECM | CKD progression [44] |
| KIM-1 | ↓ Kidney expression in rat model of AKI [43] | Kidney injury molecule-1 (marker of tubule cell injury) | Acute kidney injury [45] |
| MCP-1 (CCL-2) | ↓ Kidney mRNA expression in DOCA-salt model of CKD [46] and in model of CKD progression in T2D [34] | Pro-inflammatory cytokine (regulating monocyte/macrophage recruitment) | CKD/CKD progression [47,48,49,50,51] |
| MMP-2 | ↓ Kidney mRNA expression in DOCA-salt model of CKD [46] ↓ Plasma activities in nondiabetic CKD model [52] | ECM homeostasis | CKD/CKD progression [51,53] |
| MMP-9 | ↓ Plasma activities in nondiabetic CKD model [52] | ECM homeostasis | CKD/CKD progression [53,54] |
| NGAL (LCN2) | ↓ Kidney mRNA expression [25,26] | Involved in innate immunity and in response to tubular injury | CKD/CKD progression [55,56] |
| NKD2 | ↓ Kidney expression in mouse models of kidney fibrosis [57] | Pro-fibrotic cytokine | Kidney fibrosis [58] |
| OPN (=Spp1) | ↓ Kidney mRNA expression in DOCA-salt models of CKD [46,59] | Pro-inflammatory cytokine involved in chronic inflammation Key cytokine-regulating tissue repair, promoting collagen organization and regulating ECM and myofibroblast interactions | CKD/CKD progression [60] |
| PAI-1 | ↓ Kidney mRNA expression in DOCA-salt model of CKD [46], two models of kidney fibrosis [57] and model of CKD progression in T2D [34] but ↑ mRNA expression in macrophages from kidney tissue in IR injury model [61] | Serine protease inhibitor, which limits fibrinolysis; marker of inflammation and remodeling | CKD progression [62], kidney fibrosis [51] |
| Sgk1 | ↓ Kidney mRNA expression and protein levels in model of CKD progression in T2D [34] | Promotes inflammation and fibrosis | - |
| TGF-β | ↓ Kidney mRNA expression in model of AKI-mediated CKD [26] | Pro-fibrotic cytokine | CKD/CKD progression [51,63] |
| COL1A1 | ↓ Kidney mRNA expression in model of AKI-mediated CKD [26], a nondiabetic hypertensive cardiorenal disease model [64], and two models of kidney fibrosis [57] | ECM molecule | - |
| E-cadherin | ↑ Protein expression in model of AKI-mediated CKD [26] | Cell adhesion molecule | - |
| Nrf2 | ↑ mRNA expression in model of AKI-mediated CKD [26] | Regulator of antioxidant defense | CKD progression [65] |
| SOD-3 | ↑ Protein expression in model of AKI-mediated CKD [26] | Antioxidant enzyme | SOD-3 is depleted from human CKD kidneys [66] |
| Endothelin-B receptor | Prevents cysteine sulfenic acid modification of ET-B receptor in model of IR-induced AKI [26,43] | Regulator of vascular function | - |
| MDA | Kidney levels in model of IR-induced AKI [26] | Oxidative stress marker | - |
| 8-OHdG | Plasma levels in model of IR-induced AKI [26] | Oxidative stress marker | - |
| IL-6 | ↓ Kidney mRNA expression in IR injury model [61] | Pro-fibrotic and pro-inflammatory cytokine | CKD progression [67] |
| IL-1β | ↓ Kidney mRNA expression in IR injury model [61] | Pro-inflammatory cytokine and M1 macrophage marker | CKD progression [67] |
| TNF-α | ↓ Kidney mRNA expression in IR injury model [61] | Pro-inflammatory cytokine | CKD progression [47,67,68] |
| Mannose receptor | ↑ mRNA expression in macrophages from kidney tissue in IR injury model [61] | Anti-inflammatory marker | - |
| PPAR-γ | ↑ mRNA expression in macrophages from kidney tissue in IR injury model [61] | Anti-inflammatory marker | - |
| IL-10 | ↑ mRNA expression in macrophages from kidney tissue in IR injury model [61] | Anti-inflammatory cytokine | - |
| Arginase 1 | ↑ mRNA expression in macrophages from kidney tissue in IR injury model [61] | Anti-inflammatory marker | CKD progression |
| IL-34 | ↓ Kidney mRNA expression in DOCA-salt model of CKD [59] | Monocyte growth and survival | Worsening of CKD and severity of renal dysfunction [69] |
| Marker | Effect of Finerenone in Preclinical Models | Function/Role | Evidence for Clinical Association |
|---|---|---|---|
| Cardiac | |||
| CTGF | ↓ Protein expression in cardiac fibroblasts [72] | Pro-fibrotic cytokine that induces collagen production and subsequent pro-fibrotic enzymes | Cardiac fibrosis and dysfunction [73] |
| Fibronectin | ↓ Protein expression in cardiac fibroblasts [72] | Glycoprotein in fibrotic cardiac tissue | - |
| Galectin 3 | ↓ Cardiac mRNA expression after isoproterenol treatment [70] | Implicated in cardiac and renal inflammation and fibrosis | CKD progression [74,75] |
| COL1A1 | ↓ Cardiac mRNA expression after isoproterenol treatment [70] | ECM molecule | Heart failure progression [76] |
| LOX | ↓ Protein expression in cardiac fibroblasts [72] | Downstream mediator of CTGF, important for collagen cross-linking | Cardiac fibrosis [77] |
| NGAL (LCN2) | ↓ Protein expression in human cardiac fibroblasts and ↓ cardiac NGAL expression in mice post-MI [78] | Involved in innate immunity and cardiovascular extracellular matrix remodeling after MR activation | Serum NGAL levels were associated with lower 6-month LV ejection fraction recovery in post-MI patients [78] |
| Nox2 | ↓ Cardiac mRNA expression after isoproterenol treatment [70] | ROS-generating enzyme | Adverse myocardial remodeling in end-stage DCM [79] |
| TGF-β | ↓ Cardiac expression [72] after isoproterenol treatment [70] | Pro-fibrotic cytokine | - |
| Tnnt2 | ↓ Cardiac mRNA expression [80] | Contractile protein | - |
| Tenascin-X | ↓ Cardiac mRNA expression after isoproterenol treatment [70] | Pro-fibrotic cytokine | - |
| Marker | Effect of Finerenone in Preclinical Models | Function/Role | Evidence for Clinical Association |
|---|---|---|---|
| Other | |||
| IL-10 | ↓ Pulmonary expression [105] | Anti-inflammatory cytokine M2 macrophage marker/wound-healing phenotype | Pulmonary fibrosis [107] |
| TNF-α | ↓ Pulmonary expression [105] | Pro-inflammatory cytokine; involved in innate immune response | - |
| IL-1β | ↓ Pulmonary expression [105] | Pro-inflammatory cytokine and M1 macrophage marker | Mortality in acute exacerbations of idiopathic pulmonary fibrosis [108] |
| COL1A1 | ↓ Pulmonary expression [105] | ECM molecule | Pulmonary fibrosis [109] |
| IL-6 | ↓ Pulmonary expression [105] | Pro-fibrotic and pro-inflammatory cytokine | - |
| IL-12 | ↓ Pulmonary expression [105] | Pro-fibrotic and pro-inflammatory cytokine | Pulmonary fibrosis [107] |
| PGC1-α | ↑ Expression in interscapular brown adipose tissue [110] | Thermogenesis | - |
| Adrb3 | ↑ Expression in interscapular brown adipose tissue [110] | Thermogenesis | - |
| UCP-1 | ↑ Expression in interscapular brown adipose tissue [110] | Thermogenesis | - |
| SOD | ↑ Vasculature expression [81] | Antioxidant | Vascular dysfunction [111] |
| eNOS | ↑ Vasculature expression [81] | Catalyst for production of NO | Enhanced NO signaling is associated with reduced risks of coronary heart disease, peripheral arterial disease, and stroke [112] |
| Trial Name | NCT Number | Indication | Planned Enrollment | Primary Endpoint | Estimated Study Completion |
|---|---|---|---|---|---|
| FINE-REAL | NCT05348733 | CKD and T2D | 4000 | Treatment patterns * | February 2026 |
| CONFIDENCE | NCT05254002 | CKD and T2D | 807 | Relative change in UACR from baseline to 180 days | January 2024 |
| FIND-CKD | NCT05047263 | Nondiabetic CKD | 1580 | Mean rate change of total eGFR slope from baseline to month 32 | December 2025 |
| FIONA | NCT05196035 | Pediatric CKD | 219 | ≥30% UPCR reduction from baseline to day 180 | September 2026 |
| FINEARTS-HF | NCT04435626 | HF with LVEF ≥40% | 5500 | CV death and HF events | May 2024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolkhof, P.; Lawatscheck, R.; Filippatos, G.; Bakris, G.L. Nonsteroidal Mineralocorticoid Receptor Antagonism by Finerenone—Translational Aspects and Clinical Perspectives across Multiple Organ Systems. Int. J. Mol. Sci. 2022, 23, 9243. https://doi.org/10.3390/ijms23169243
Kolkhof P, Lawatscheck R, Filippatos G, Bakris GL. Nonsteroidal Mineralocorticoid Receptor Antagonism by Finerenone—Translational Aspects and Clinical Perspectives across Multiple Organ Systems. International Journal of Molecular Sciences. 2022; 23(16):9243. https://doi.org/10.3390/ijms23169243
Chicago/Turabian StyleKolkhof, Peter, Robert Lawatscheck, Gerasimos Filippatos, and George L. Bakris. 2022. "Nonsteroidal Mineralocorticoid Receptor Antagonism by Finerenone—Translational Aspects and Clinical Perspectives across Multiple Organ Systems" International Journal of Molecular Sciences 23, no. 16: 9243. https://doi.org/10.3390/ijms23169243
APA StyleKolkhof, P., Lawatscheck, R., Filippatos, G., & Bakris, G. L. (2022). Nonsteroidal Mineralocorticoid Receptor Antagonism by Finerenone—Translational Aspects and Clinical Perspectives across Multiple Organ Systems. International Journal of Molecular Sciences, 23(16), 9243. https://doi.org/10.3390/ijms23169243