
Full-Length Dystrophin Restoration via Targeted Exon Addition in DMD-Patient Specific iPSCs and Cardiomyocytes



Abstract
:1. Introduction
2. Results
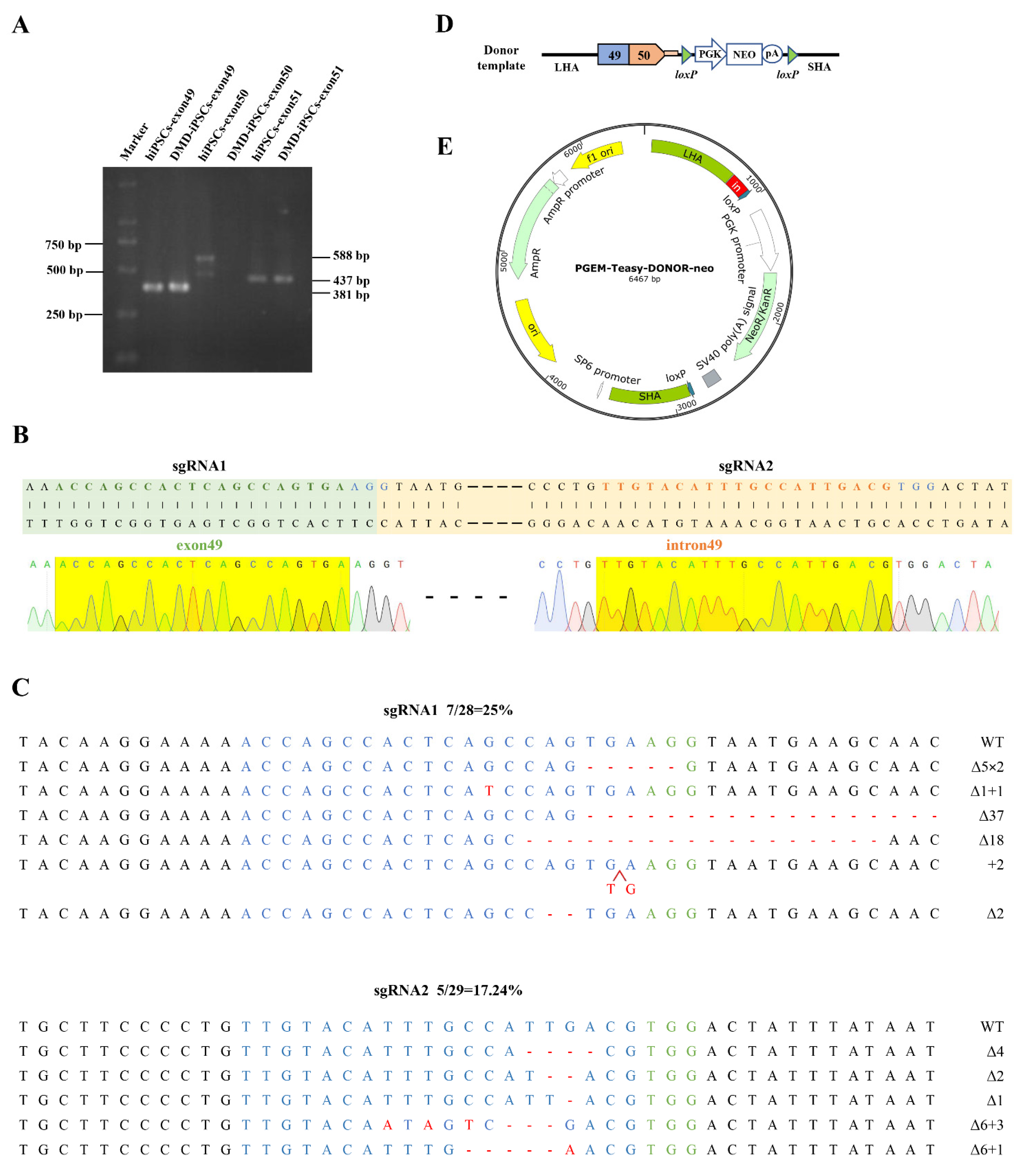
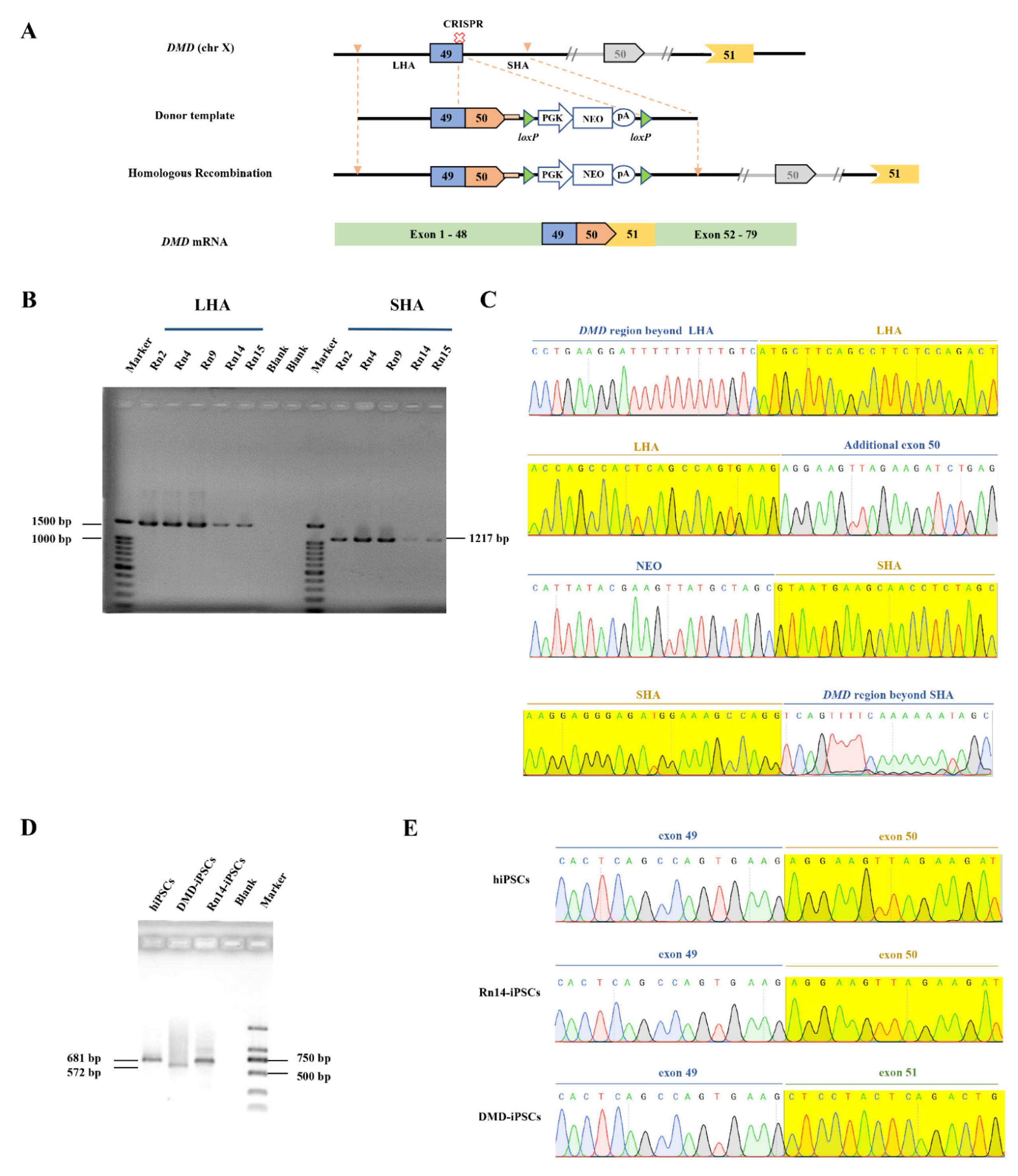
2.1. Design and Construction of CRISPR/Cas9 and Donor Template for In Situ Correction of DMD Mutations
2.2. CRISPR/Cas9-Mediated DMD In Situ Correction in DMD-iPSCs
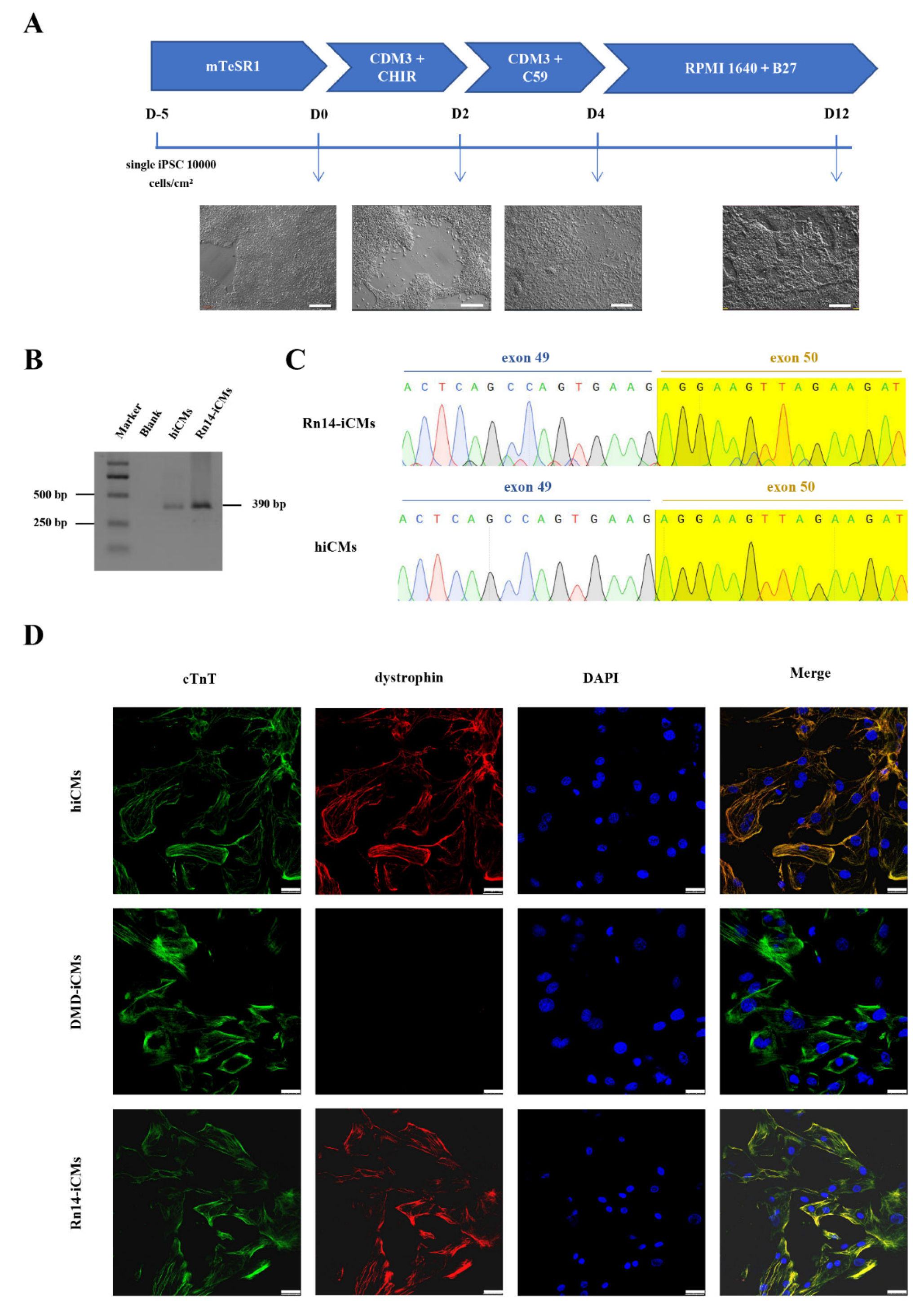
2.3. Dystrophin Expression in the Rn14-iPSCs-Derived Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of CRISPR/Cas9 Plasmids and Detection of Cleavage Activity
4.3. Construction of a Donor Vector for Gene Correction
4.4. In Situ Gene Correction in DMD-iPSCs and Off-Target Analysis
4.5. Karyotyping
4.6. Characterization of iPSCs
4.7. RT-PCR
4.8. iPSC-CM Differentiation
4.9. Immunofluorescence Staining for iCMs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef]
- Politano, L. Read-through approach for stop mutations in Duchenne muscular dystrophy. An Update. Acta Myol. 2021, 40, 43–50. [Google Scholar]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef]
- McNally, E.M.; Wyatt, E.J. Mutation-Based Therapy for Duchenne Muscular Dystrophy: Antisense Treatment Arrives in the Clinic. Circulation 2017, 136, 979–981. [Google Scholar] [CrossRef]
- Rodrigues, M.; Yokota, T. An Overview of Recent Advances and Clinical Applications of Exon Skipping and Splice Modulation for Muscular Dystrophy and Various Genetic Diseases. Methods Mol. Biol. 2018, 1828, 31–55. [Google Scholar]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.-L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef]
- Rowel, L.K.; Rika, M.; Toshifumi, Y. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar]
- Sheikh, O.; Yokota, T. Pharmacology and toxicology of eteplirsen and SRP-5051 for DMD exon 51 skipping: An update. Arch. Toxicol. 2022, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Bolukbasi, M.F.; Gupta, A.; Wolfe, S.A. Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat. Methods 2016, 13, 41–50. [Google Scholar] [CrossRef]
- Manini, A.; Abati, E.; Nuredini, A.; Corti, S.; Comi, G.P. Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence. Front. Neurol. 2021, 12, 814174. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, M.; Iwabuchi, K.A.; Tanaka, M.; Lung, M.S.Y.; Hotta, A. Restoration of Dystrophin Protein Expression by Exon Skipping Utilizing CRISPR-Cas9 in Myoblasts Derived from DMD Patient iPS Cells. Methods. Mol. Biol. 2018, 1828, 191–217. [Google Scholar] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Long, C.; Marschalk, N.; Fine, D.M.; Chen, J.; Duan, D. Cardiac Expression of a Mini-dystrophin That Normalizes Skeletal Muscle Force Only Partially Restores Heart Function in Aged Mdx Mice. Mol. Ther. 2009, 17, 253–261. [Google Scholar] [CrossRef]
- Jin, Y.; Shen, Y.; Su, X.; Weintraub, N.L.; Tang, Y. Effective restoration of dystrophin expression in iPSC Mdx-derived muscle progenitor cells using the CRISPR/Cas9 system and homology-directed repair technology. Comput. Struct. Biotechnol. J. 2020, 18, 765–773. [Google Scholar] [CrossRef]
- Zeng, B.; Zhou, M.; Liu, B.; Shen, F.; Xiao, R.; Su, J.; Hu, Z.; Zhang, Y.; Gu, A.; Wu, L.; et al. Targeted addition of mini-dystrophin into rDNA locus of Duchenne muscular dystrophy patient-derived iPSCs. Biochem. Biophys. Res. Commun. 2021, 545, 40–45. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.; Gambhir, Y.; Bennett, J.; Stedman, H.H. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Mol. Ther. 2020, 28, 1753–1755. [Google Scholar] [CrossRef]
- Wasala, N.B.; Shin, J.-H.; Lai, Y.; Yue, Y.; Montanaro, F.; Duan, D. Cardiac-Specific Expression of ΔH2-R15 Mini-Dystrophin Normalized All Electrocardiogram Abnormalities and the End-Diastolic Volume in a 23-Month-Old Mouse Model of Duchenne Dilated Cardiomyopathy. Hum. Gene Ther. 2018, 29, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Pioner, J.M.; Guan, X.; Klaiman, J.M.; Racca, A.W.; Pabon, L.; Muskheli, V.; Macadangdang, J.; Ferrantini, C.; Hoopmann, M.R.; Moritz, R.L.; et al. Absence of full-length dystrophin impairs normal maturation and contraction of cardiomyocytes derived from human-induced pluripotent stem cells. Cardiovasc. Res. 2020, 116, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef]
- Meng, H.; Leddy, J.J.; Frank, J.; Holland, P.; Tuana, B.S. The Association of Cardiac Dystrophin with Myofibrils/Z-disc Regions in Cardiac Muscle Suggests a Novel Role in the Contractile Apparatus. J. Biol. Chem. 1996, 271, 12364–12371. [Google Scholar] [CrossRef]
- Kaprielian, R.R.; Stevenson, S.; Rothery, S.M.; Cullen, M.J.; Severs, N.J. Distinct Patterns of Dystrophin Organization in Myocyte Sarcolemma and Transverse Tubules of Normal and Diseased Human Myocardium. Circulation 2000, 101, 2586–2594. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Jin, Y.; Shen, Y.; Su, X.; Weintraub, N.; Tang, Y. CRISPR/Cas9 Technology in Restoring Dystrophin Expression in iPSC-Derived Muscle Progenitors. J. Vis. Exp. 2019, 151, e59432. [Google Scholar] [CrossRef]
- Montarras, D.; Morgan, J.; Collins, C.; Relaix, F.; Zaffran, S.; Cumano, A.; Partridge, T.; Buckingham, M. Direct Isolation of Satellite Cells for Skeletal Muscle Regeneration. Science 2005, 309, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wilschut, K.J.; Kouklis, G.; Tian, H.; Hesse, R.; Garland, C.; Sbitany, H.; Hansen, S.; Seth, R.; Knott, P.D.; et al. Human Satellite Cell Transplantation and Regeneration from Diverse Skeletal Muscles. Stem Cell Rep. 2015, 5, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Matthias, N.; Lo, J.; Ortiz-Vitali, J.L.; Shieh, A.W.; Wang, S.H.; Darabi, R. A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 2018, 25, 1966–1981.e4. [Google Scholar] [CrossRef] [PubMed]
- Han, W.M.; Mohiuddin, M.; Anderson, S.E.; García, A.J.; Jang, Y.C. Co-delivery of Wnt7a and muscle stem cells using synthetic bioadhesive hydrogel enhances murine muscle regeneration and cell migration during engraftment. Acta Biomater. 2019, 94, 243–252. [Google Scholar] [CrossRef]
- Gerli, M.F.M.; Moyle, L.A.; Benedetti, S.; Ferrari, G.; Ucuncu, E.; Ragazzi, M.; Constantinou, C.; Louca, I.; Sakai, H.; Ala, P.; et al. Combined Notch and PDGF Signaling Enhances Migration and Expression of Stem Cell Markers while Inducing Perivascular Cell Features in Muscle Satellite Cells. Stem Cell Rep. 2019, 12, 461–473. [Google Scholar] [CrossRef]
- Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Lee, G.K.; Shrager, J.B.; Rando, T.A. Ex Vivo Expansion and In Vivo Self-Renewal of Human Muscle Stem Cells. Stem Cell Rep. 2015, 5, 621–632. [Google Scholar] [CrossRef]
- Shiba, Y.; Gomibuchi, T.; Seto, T.; Wada, Y.; Ichimura, H.; Tanaka, Y.; Ogasawara, T.; Okada, K.; Shiba, N.; Sakamoto, K.; et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 2016, 538, 388–391. [Google Scholar] [CrossRef]
- Bier, A.; Berenstein, P.; Kronfeld, N.; Morgoulis, D.; Ziv-Av, A.; Goldstein, H.; Kazimirsky, G.; Cazacu, S.; Meir, R.; Popovtzer, R.; et al. Placenta-derived mesenchymal stromal cells and their exosomes exert therapeutic effects in Duchenne muscular dystrophy. Biomaterials 2018, 174, 67–78. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Okada, H.; Wada-Maeda, M.; Nakamura, A.; Okada, T.; Takeda, S. Long-term Engraftment of Multipotent Mesenchymal Stromal Cells That Differentiate to Form Myogenic Cells in Dogs with Duchenne Muscular Dystrophy. Mol. Ther. 2012, 20, 168–177. [Google Scholar] [CrossRef]
- Siemionow, M.; Szilagyi, E.; Cwykiel, J.; Domaszewska-Szostek, A.; Heydemann, A.; Martinez, J.; Siemionow, K. Transplantation of Dystrophin Expressing Chimeric Human Cells of Myoblast/Mesenchymal Stem Cell Origin Improves Function in Duchenne Muscular Dystrophy Model. Stem Cells Dev. 2021, 30, 190–202. [Google Scholar] [CrossRef]
- Dakhlallah, D.; Zhang, J.; Yu, L.; Marsh, C.B.; Angelos, M.G.; Khan, M. MicroRNA-133a Engineered Mesenchymal Stem Cells Augment Cardiac Function and Cell Survival in the Infarct Heart. J. Cardiovasc. Pharmacol. 2015, 65, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Miyagawa, S.; Toyofuku, T.; Fukushima, S.; Kawamura, T.; Kawamura, A.; Kashiyama, N.; Nakamura, Y.; Toda, K.; Sawa, Y. Syngeneic Mesenchymal Stem Cells Reduce Immune Rejection After Induced Pluripotent Stem Cell-Derived Allogeneic Cardiomyocyte Transplantation. Sci. Rep. 2020, 10, 4593. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Bae, S.; Kim, J.-S. Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 2015, 31, 4014–4016. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, Z.; Li, Z.; Pang, J.; Feng, M.; Hu, X.; Wang, X.; Lin-Peng, S.; Liu, B.; Chen, F.; et al. In situ genetic correction of F8 intron 22 inversion in hemophilia A patient-specific iPSCs. Sci. Rep. 2016, 6, srep18865. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; del Alamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell–derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Site | Guide Sequence (5′-3′) | Sense Sequence (5′-3′) | Anti-sense Sequence (5′-3′) |
|---|---|---|---|
| exon 49 | CCTTCACTGGCTGAGTGGCTGGT | CACCGACCAGCCACTCAGCCAGTGA | AAACTCACTGGCTGAGTGGCTGGTC |
| intron 49 | TTGTACATTTGCCATTGACGTGG | CACCGTTGTACATTTGCCATTGACG | AAACCGTCAATGGCAAATGTACAAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, R.; Zhou, M.; Wang, P.; Zeng, B.; Wu, L.; Hu, Z.; Liang, D. Full-Length Dystrophin Restoration via Targeted Exon Addition in DMD-Patient Specific iPSCs and Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 9176. https://doi.org/10.3390/ijms23169176
Xiao R, Zhou M, Wang P, Zeng B, Wu L, Hu Z, Liang D. Full-Length Dystrophin Restoration via Targeted Exon Addition in DMD-Patient Specific iPSCs and Cardiomyocytes. International Journal of Molecular Sciences. 2022; 23(16):9176. https://doi.org/10.3390/ijms23169176
Chicago/Turabian StyleXiao, Rou, Miaojin Zhou, Peiyun Wang, Baitao Zeng, Lingqian Wu, Zhiqing Hu, and Desheng Liang. 2022. "Full-Length Dystrophin Restoration via Targeted Exon Addition in DMD-Patient Specific iPSCs and Cardiomyocytes" International Journal of Molecular Sciences 23, no. 16: 9176. https://doi.org/10.3390/ijms23169176
APA StyleXiao, R., Zhou, M., Wang, P., Zeng, B., Wu, L., Hu, Z., & Liang, D. (2022). Full-Length Dystrophin Restoration via Targeted Exon Addition in DMD-Patient Specific iPSCs and Cardiomyocytes. International Journal of Molecular Sciences, 23(16), 9176. https://doi.org/10.3390/ijms23169176