
Functional Crosstalk between PCSK9 Internalization and Pro-Inflammatory Activation in Human Macrophages: Role of Reactive Oxygen Species Release

 ,
, 
Abstract
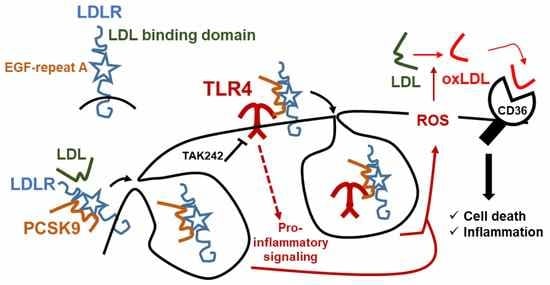
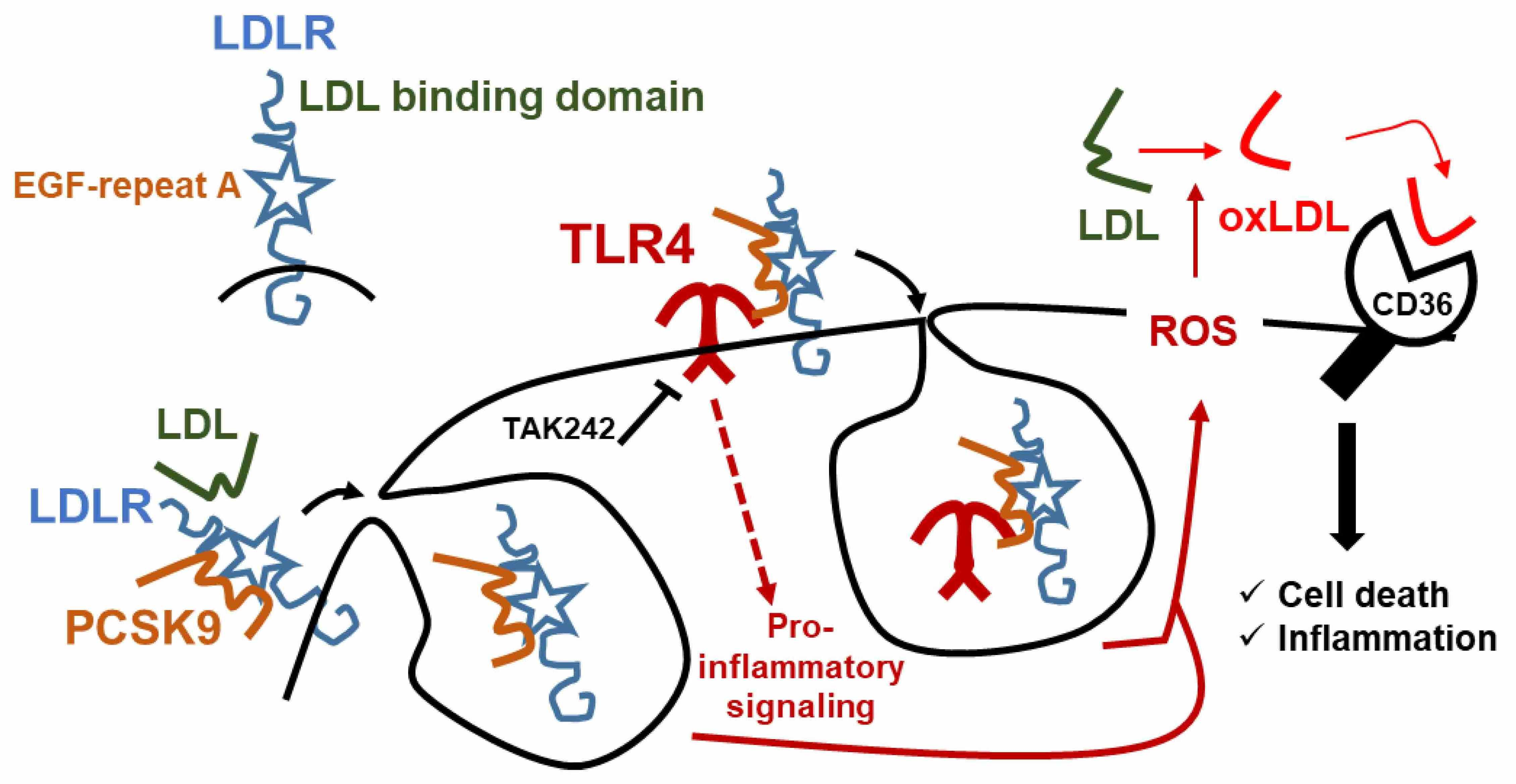
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
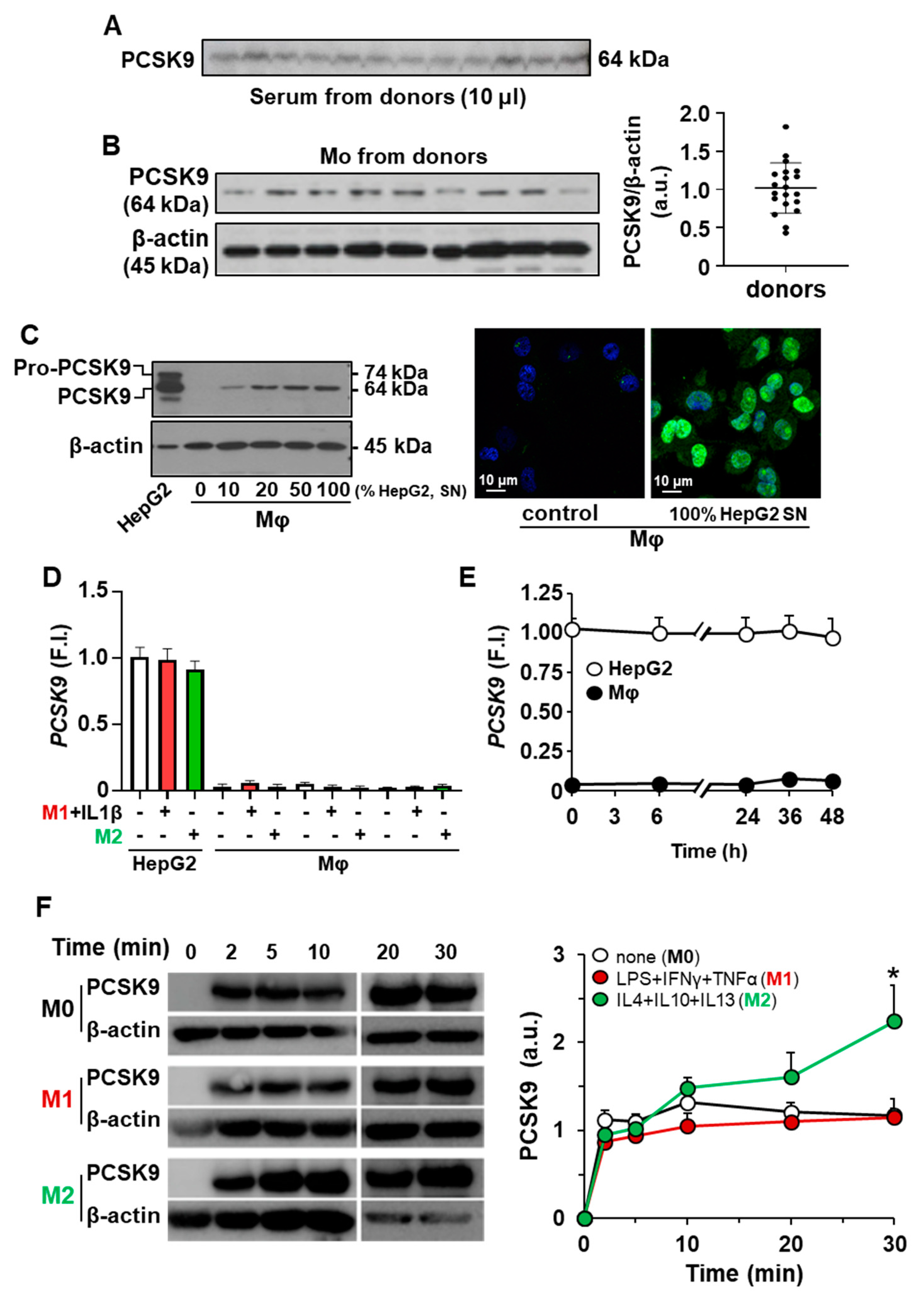
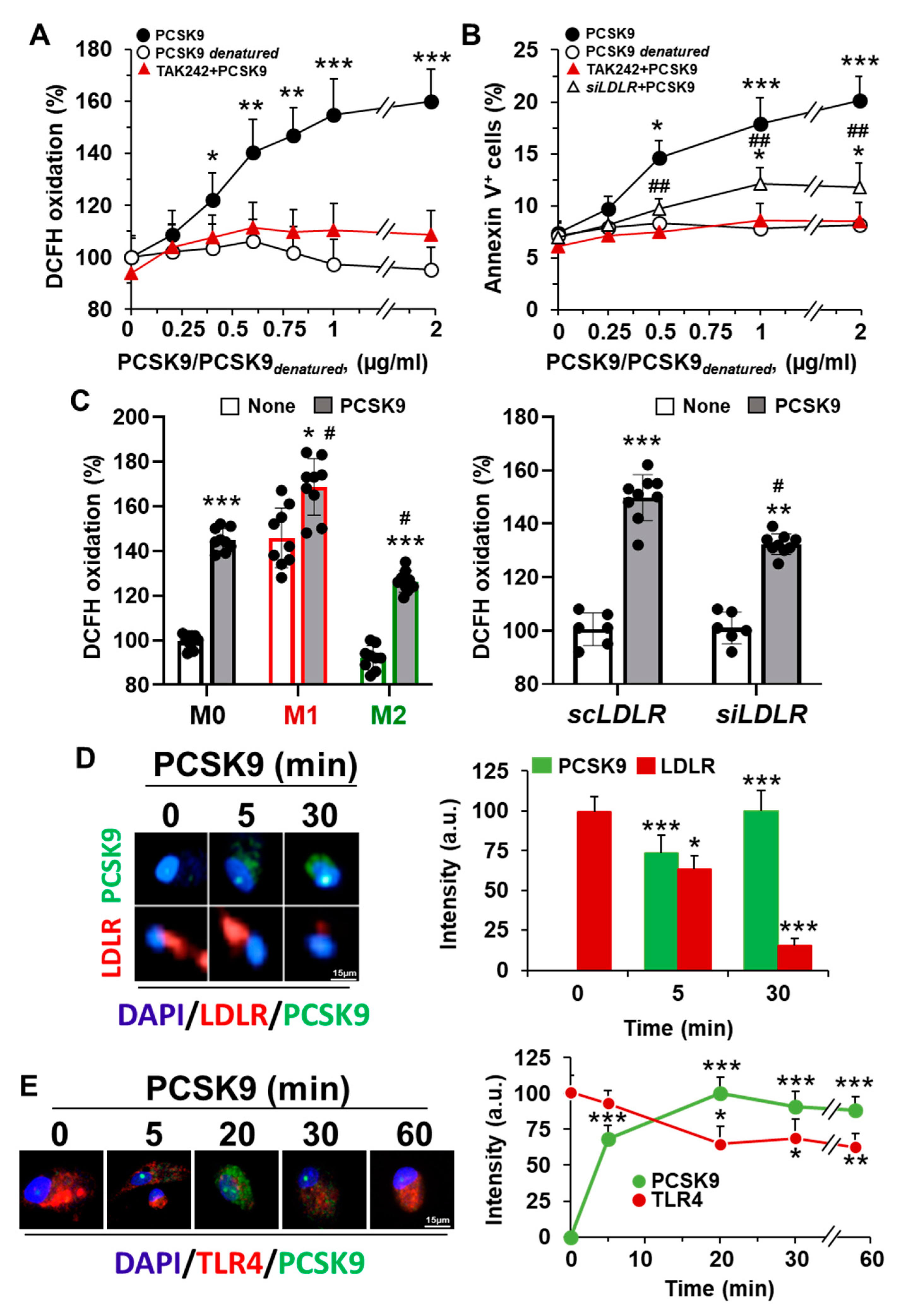
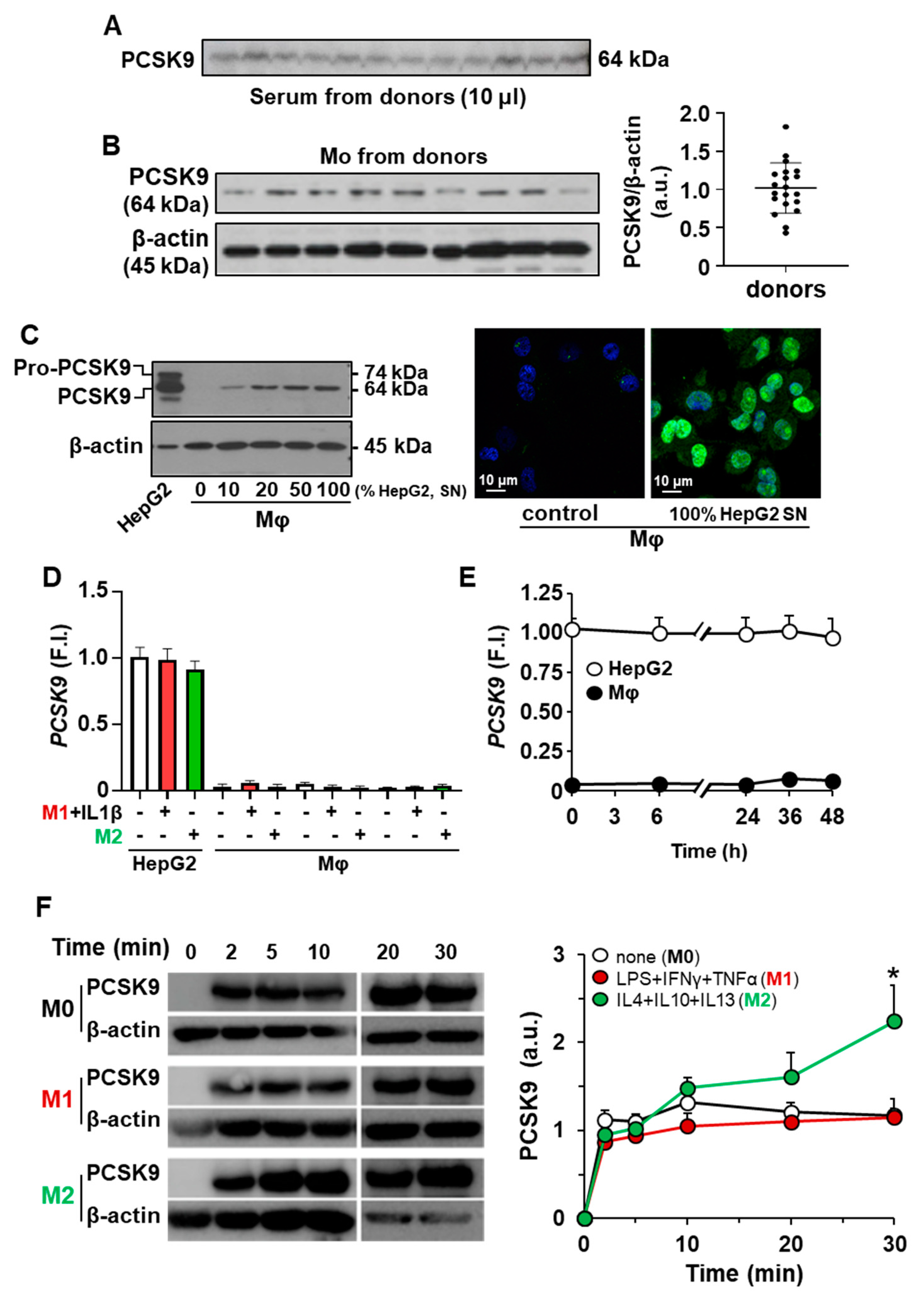
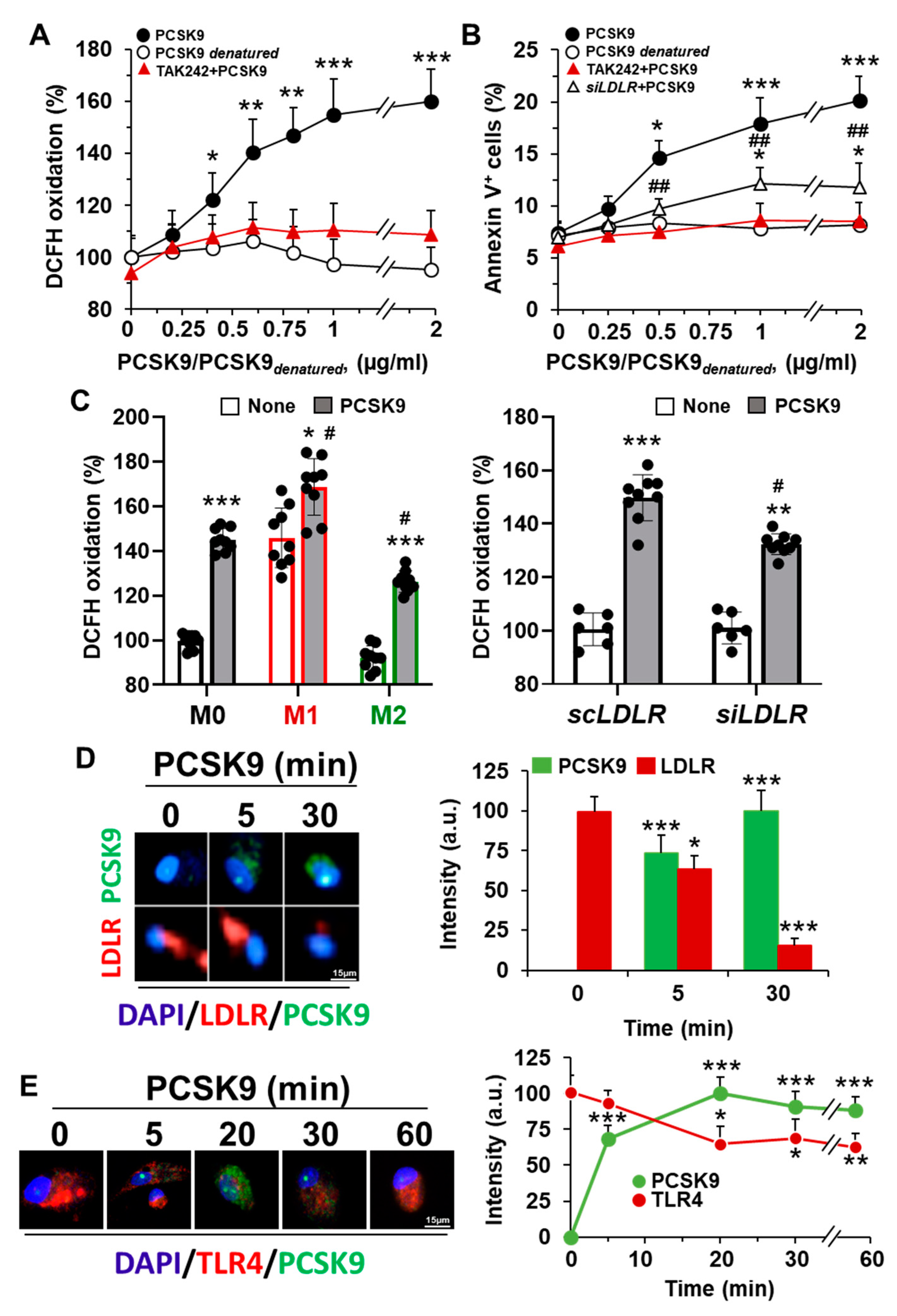
2.1. Incorporation of PCSK9 in Human Macrophages Enhances ROS Synthesis and Reduces Cell Viability
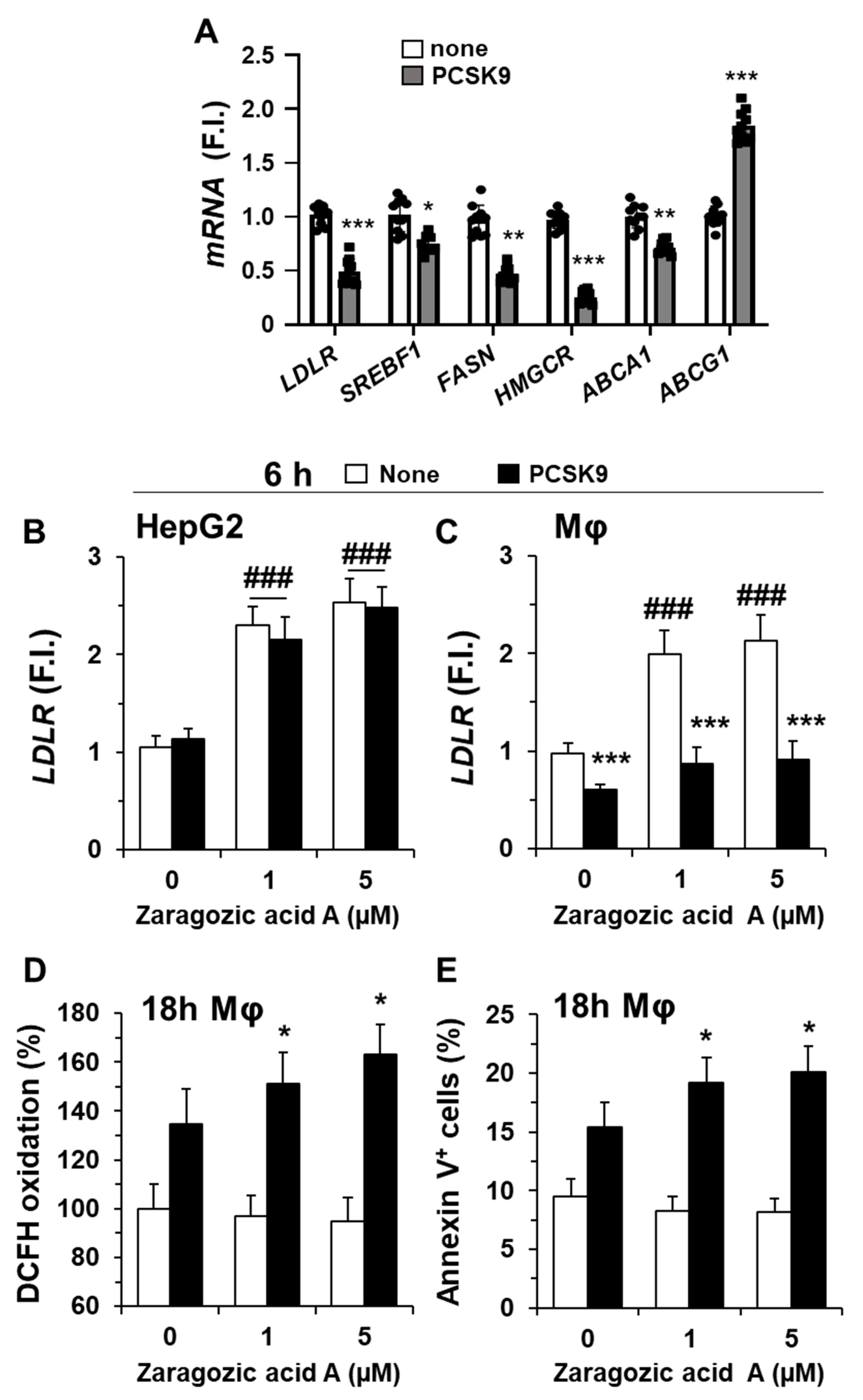
2.2. Incorporation of PCSK9 in Human Macrophages Reduces LDLR Content and Decreases the mRNA Levels of Genes Involved in Lipid and Cholesterol Metabolism
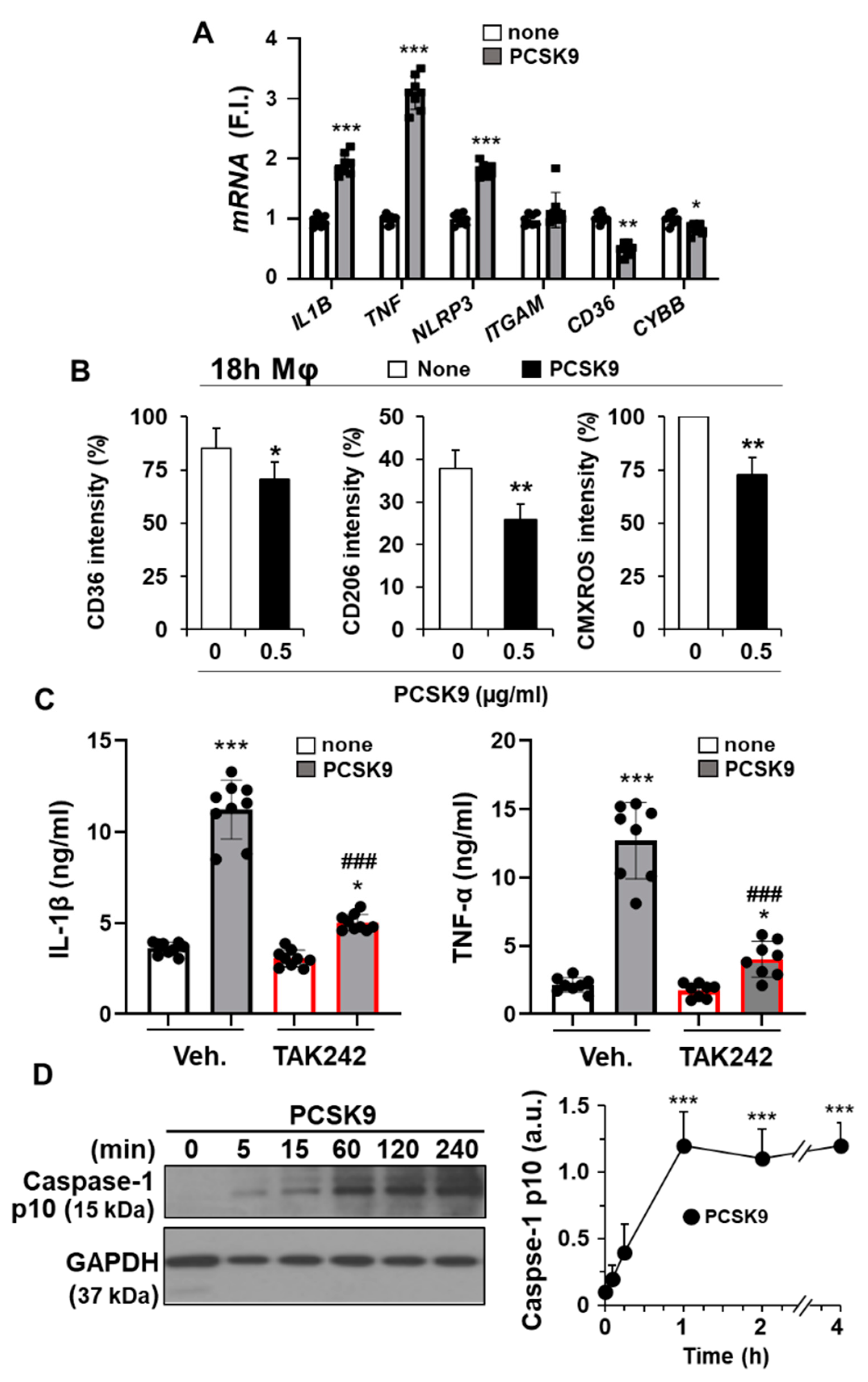
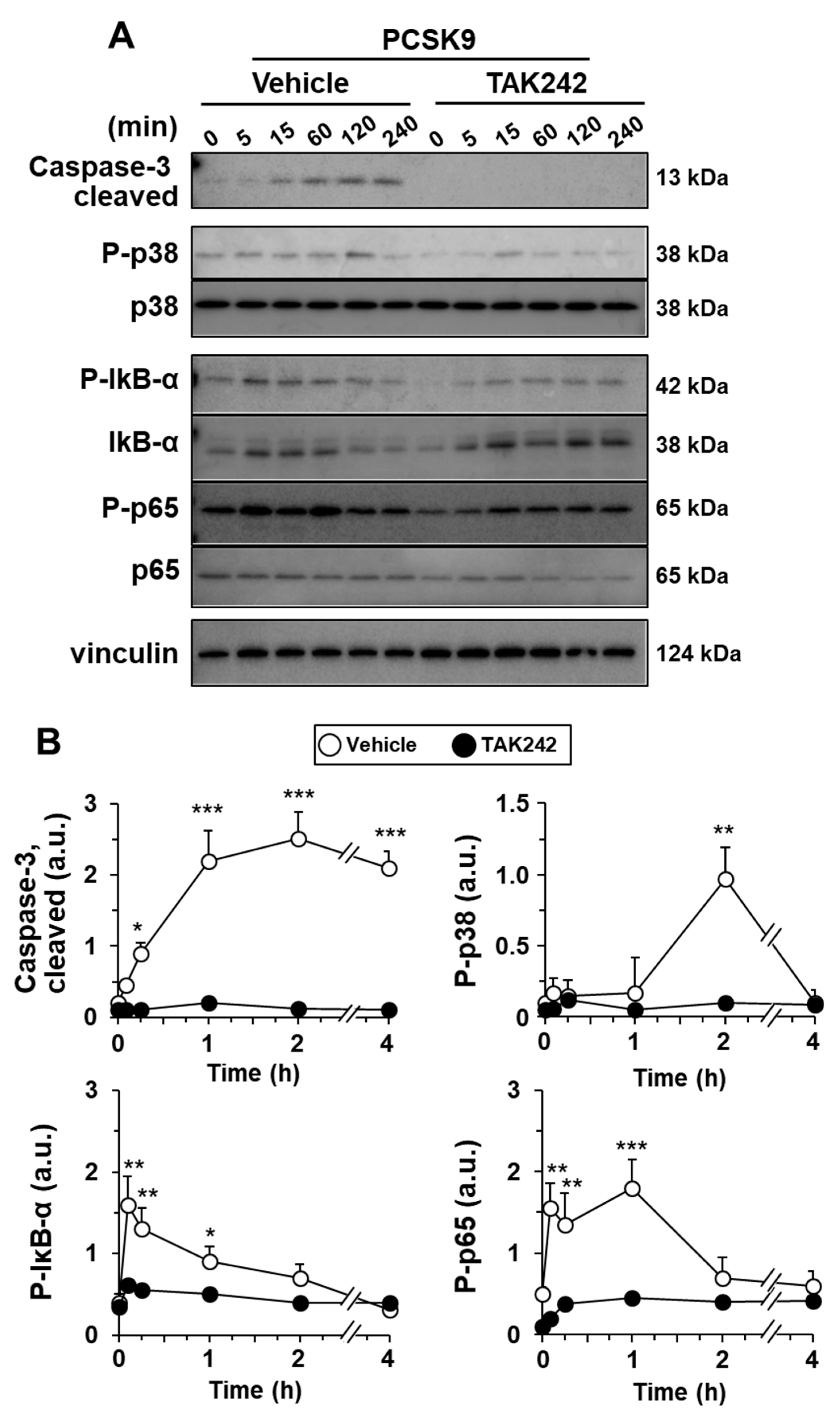
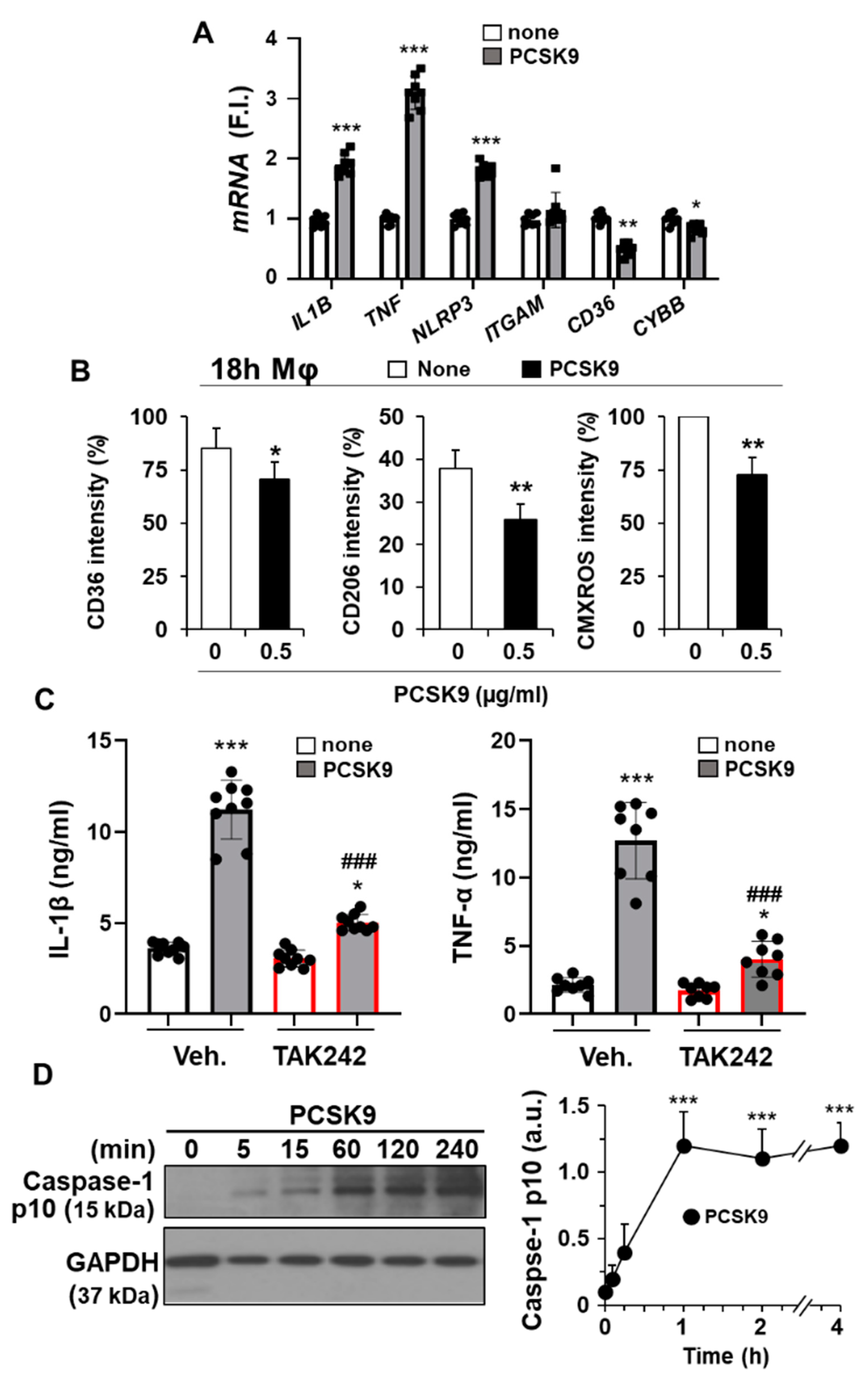
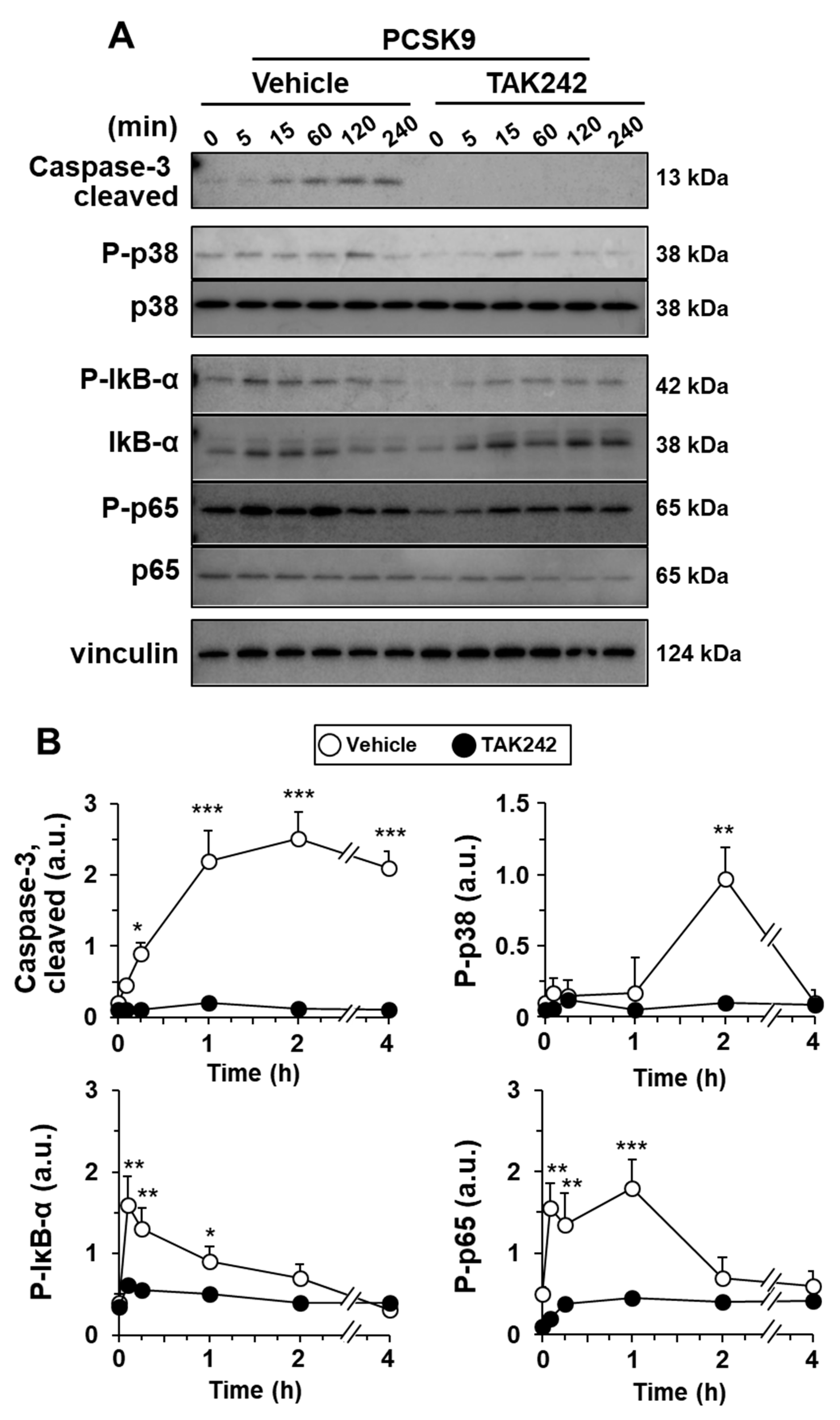
2.3. PCSK9 Activates Pro-Inflammatory and Pro-Apoptotic Signaling Pathways in Human Macrophages
2.4. The Pro-Inflammatory and Pro-Apoptotic Effects after Incorporation of PCSK9 in Human Macrophages Are TLR4 Dependent
3. Discussion
4. Methods and Materials
4.1. Materials
4.2. Isolation of Human Monocytes and Preparation of Human Macrophages
4.3. Preparation of Elicited Peritoneal Mice Macrophages
4.4. Differentiation of Macrophages into Pro-Inflammatory and Alternatively Activated Cells
4.5. Cell Treatments
4.6. Human LDLR Silencing
4.7. Measurement of ROS Production
4.8. Measurement of Cell Membrane Receptors
4.9. Evaluation of Mitochondrial Inner Membrane Potential
4.10. Quantification of Annexin V+ Cells
4.11. Protein Analysis by Western Blot
4.12. RNA Isolation and Analysis
4.13. Immunofluorescence Analysis
4.14. Quantification of IL-1β and TNF-α
4.15. Statistical Analysis
5. Conclusions
- PCSK9 incorporation in macrophages enhances ROS production and decreases cell viability.
- LDLR-dependent incorporation of PCSK9 decreases the expression of ABCA1.
- The pro-inflammatory activation after PCSK9 uptake is dependent on TLR4 signaling.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| LDL | Low-density lipoprotein |
| LDLR | LDL receptor |
| oxLDL | Oxidized LDL |
| ROS | Reactive oxygen species |
| PAMPS/DAMPS | Pathogen-associated and damage-associated molecular patterns |
| ABC | ATP-binding cassette transporter |
| hMφ | Human macrophage |
References
- Luquero, A.; Badimon, L.; Borrell-Pages, M. PCSK9 functions in atherosclerosis are not limited to plasmatic LDL-Cholesterol regulation. Front. Cardiovasc. Med. 2021, 8, 639727. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, M.; Hosseini-Fard, R.; Najafi, M. Circulating low density lipoprotein (LDL). Horm. Mol. Biol. Clin. Investig. 2018, 35, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/Kexin type 9 to epidermal growth factor-like repeat a of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Peng, Z.; Gu, H.; Wang, M.; Wang, G.; Zhang, D. Regulation of PCSK9 expression and function: Mechanisms and therapeutic implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Hamouda, H.A.; Villeneuve, L.; Demers, A.; Mayer, G. Trafficking dynamics of PCSK9-Induced LDLR degradation: Focus on human PCSK9 mutations and C-Terminal domain. PLoS ONE 2016, 11, e0157230. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Sabouri-Rad, S.; Gotto, A.M.; Pirro, M.; Banach, M.; Awan, Z.; Barreto, G.E.; Sahebkar, A. PCSK9 and inflammation: A review of experimental and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Pothineni, N.V.K.; Goel, A.; Lüscher, T.F.; Mehta, J.L. PCSK9 and inflammation: Role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc. Res. 2020, 116, 908–915. [Google Scholar] [CrossRef]
- Mentrup, T.; Cabrera-Cabrera, F.; Schröder, B. Proteolytic regulation of the Lectin-like oxidized lipoprotein receptor LOX-1. Front. Cardiovasc. Med. 2020, 7, 594441. [Google Scholar] [CrossRef] [PubMed]
- Jay, A.G.; Hamilton, J.A. The enigmatic membrane fatty acid transporter CD36: New insights into fatty acid binding and their effects on uptake of oxidized LDL. Prostaglandins Leukot. Essent. Fat. Acids 2018, 138, 64–70. [Google Scholar] [CrossRef]
- Jin, M.S.; Lee, J.O. Structures of the toll-like receptor family and its ligand complexes. Immunity 2008, 29, 182–191. [Google Scholar] [CrossRef]
- Tang, Z.-H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.-T.; Li, T.-H.; Wang, Z.; Wei, D.-H.; Liu, L.-S.; Zheng, X.-L.; et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Scalise, V.; Sanguinetti, C.; Neri, T.; Cianchetti, S.; Lai, M.; Carnicelli, V.; Celi, A.; Pedrinelli, R. PCSK9 Induces tissue factor expression by activation of TLR4/NFkB signaling. Int. J. Mol. Sci. 2021, 22, 12640. [Google Scholar] [CrossRef]
- Laird, M.H.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Luquero, A.; Crespo, J.; Peña, E.; Borrell-Pages, M. PCSK9 and LRP5 in macrophage lipid internalization and inflammation. Cardiovasc. Res. 2021, 117, 2054–2068. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, X.; Zhang, P.; Wang, X.; Fan, Y.; Zhou, S.; Mu, S.; Mehta, J.L.; Ding, Z. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovasc. Res. 2020, 116, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB signaling in macrophages: Dynamics, crosstalk, and signal integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Castrillo, A.; Díaz-Guerra, M.J.; Hortelano, S.; Martín-Sanz, P.; Boscá, L. Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-Delta (12,14)-prostaglandin J (2) in activated murine macrophages. Mol. Cell. Biol. 2000, 20, 1692–1698. [Google Scholar] [CrossRef]
- Zhang, Y.; Bliska, J.B. Role of toll-like receptor signaling in the apoptotic response of macrophages to yersinia infection. Infect. Immun. 2003, 71, 1513–1519. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K. TLR4 Cross-Talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Arditi, M. Innate immunity and toll-like receptors: Clinical implications of basic science research. J. Pediatr. 2004, 144, 421–429. [Google Scholar] [CrossRef]
- Choi, S.-H.; Sviridov, D.; Miller, Y.I. Oxidized cholesteryl esters and inflammation. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 393–397. [Google Scholar] [CrossRef]
- Seimon, T.A.; Obstfeld, A.; Moore, K.J.; Golenbock, D.T.; Tabas, I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc. Natl. Acad. Sci. USA 2006, 103, 19794–19799. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.; Tabas, I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Mayer, G.; Benjannet, S.; Bergeron, E.; Marcinkiewicz, J.; Nassoury, N.; Mayer, H.; Nimpf, J.; Prat, A.; Seidah, N.G. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 2008, 283, 2363–2372. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; Döring, Y.; van der Vorst, E.P.C. PCSK9: A Multi-Faceted protein that is involved in cardiovascular biology. Biomedicines 2021, 9, 793. [Google Scholar] [CrossRef]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Ferré, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. Paediatr. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Westerterp, M. ABC transporters, cholesterol efflux, and implications for cardiovascular diseases. Adv. Exp. Med. Biol. 2020, 1276, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Pagler, T.A.; Wang, M.; Mondal, M.; Murphy, A.J.; Westerterp, M.; Moore, K.J.; Maxfield, F.R.; Tall, A.R. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ. Res. 2011, 108, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Out, R.; Hoekstra, M.; Habets, K.; Meurs, I.; de Waard, V.; Hildebrand, R.B.; Wang, Y.; Chimini, G.; Kuiper, J.; Van Berkel, T.J.C.; et al. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arter. Thromb. Vasc. Biol. 2008, 28, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.D.; Kurtz, M.M.; Rew, D.J.; Amend, A.M.; Karkas, J.D.; Bostedor, R.G.; Bansal, V.S.; Dufresne, C.; VanMiddlesworth, F.L.; Hensens, O.D. Zaragozic acids: A family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 80–84. [Google Scholar] [CrossRef]
- Ruscica, M.; Ferri, N.; Corsini, A.; Sirtori, C.R. PCSK9 antagonists and inflammation. Atherosclerosis 2018, 268, 235–236. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2018, 113, 5. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Bhargava, P.; Stanya, K.J.; Alexander, R.K.; Liou, Y.H.; Jacobi, D.; Knudsen, N.H.; Hyde, A.; Gangl, M.R.; Liu, S.; et al. Macrophage alternative activation confers protection against lipotoxicity-induced cell death. Mol. Metab. 2017, 6, 1186–1197. [Google Scholar] [CrossRef]
- Yurtseven, E.; Ural, D.; Baysal, K.; Tokgözoğlu, L. An update on the role of PCSK9 in atherosclerosis. J. Atheroscler. Thromb. 2020, 27, 909–918. [Google Scholar] [CrossRef]
- Leren, T.P. Sorting an LDL receptor with bound PCSK9 to intracellular degradation. Atherosclerosis 2014, 237, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.-Q.; Shi, H.-W.; Li, J.-J. Proprotein convertase Subtilisin/Kexin Type 9 and inflammation: An updated review. Front. Cardiovasc. Med. 2022, 9, 763516. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tang, Z.-H.; Peng, J.; Liao, L.; Pan, L.-H.; Wu, C.-Y.; Jiang, Z.-S.; Wang, G.-X.; Liu, L.-S. The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer’s disease progression (Review). Biomed. Rep. 2014, 2, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, W.; Sun, S.; Zhang, Y.; Chen, Z. PCSK9: A potential therapeutic target for sepsis. J. Immunol. Res. 2020, 2020, 2687692. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef]
- Ragusa, R.; Basta, G.; Neglia, D.; De Caterina, R.; Del Turco, S.; Caselli, C. PCSK9 and atherosclerosis: Looking beyond LDL regulation. Eur. J. Clin. Investig. 2021, 51, e13459. [Google Scholar] [CrossRef]
- Karagiannis, A.D.; Liu, M.; Toth, P.P.; Zhao, S.; Agrawal, D.K.; Libby, P.; Chatzizisis, Y.S. Pleiotropic anti-atherosclerotic effects of PCSK9 inhibitors from molecular biology to clinical translation. Curr. Atheroscler. Rep. 2018, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A. PCSK9 and LDLR degradation. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Pandzic, E.; Gelissen, I.C.; Whan, R.; Barter, P.J.; Sviridov, D.; Gaus, K.; Rye, K.-A.; Cochran, B.J. The ATP binding cassette transporter, ABCG1, localizes to cortical actin filaments. Sci. Rep. 2017, 7, 42025. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Bradley, M.N.; Castrillo, A.; Bruhn, K.W.; Mak, P.A.; Pei, L.; Hogenesch, J.; O’Connell, R.M.; Cheng, G.; Saez, E.; et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 2004, 119, 299–309. [Google Scholar] [CrossRef]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu. Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef]
- Yang, C.; McDonald, J.G.; Patel, A.; Zhang, Y.; Umetani, M.; Xu, F.; Westover, E.J.; Covey, D.F.; Mangelsdorf, D.J.; Cohen, J.C.; et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J. Biol. Chem. 2006, 281, 27816–27826. [Google Scholar] [CrossRef]
- Wiciński, M.; Żak, J.; Malinowski, B.; Popek, G.; Grześk, G. PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 2017, 8, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.P.; Bennett, B.L.; Manning, A.M.; Brenner, D.A.; Jobin, C. Differential requirement for NF-kB-inducing kinase in the induction of NF-kB by IL-1b, TNF-a, and Fas. Am. J. Physiol. Cell Physiol. 2002, 283, C347–C357. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; de Las Heras, B.; Hortelano, S.; Rodriguez, B.; Villar, A.; Bosca, L. Inhibition of the nuclear factor kappa B (NF-kappa B) pathway by tetracyclic kaurene diterpenes in macrophages. Specific effects on NF-kappa B-inducing kinase activity and on the coordinate activation of ERK and p38 MAPK. J. Biol. Chem. 2001, 276, 15854–15860. [Google Scholar] [CrossRef] [PubMed]
- Pradère, J.-P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-κB p65 activity by serine 536 phosphorylation. Sci. Signal. 2016, 9, ra85. [Google Scholar] [CrossRef] [PubMed]
- Persson, J.; Nilsson, J.; Lindholm, M.W. Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol. 2008, 9, 70. [Google Scholar] [CrossRef]
- Hampton, E.N.; Knuth, M.W.; Li, J.; Harris, J.L.; Lesley, S.A.; Spraggon, G. The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. USA 2007, 104, 14604–14609. [Google Scholar] [CrossRef]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Wu, H.; Wu, Q.; Duan, M.; Deng, W.; Tang, Q. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019, 24, 101215. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Sen, G.C. IRF-3 and Bax: A deadly affair. Cell Cycle 2010, 9, 2479–2480. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, S.W.; Harshfield, E.L.; Hemerich, D.; Stacey, D.; Wood, A.M.; Asselbergs, F.W. From lipid locus to drug target through human genomics. Cardiovasc. Res. 2018, 114, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Sharma, G.; Fisher, E.A. Atherosclerosis: Making a U turn. Annu. Rev. Med. 2020, 71, 191–201. [Google Scholar] [CrossRef]
- Giglio, R.V.; Pantea Stoian, A.; Al-Rasadi, K.; Banach, M.; Patti, A.M.; Ciaccio, M.; Rizvi, A.A.; Rizzo, M. Novel therapeutical approaches to managing atherosclerotic risk. Int. J. Mol. Sci. 2021, 22, 4633. [Google Scholar] [CrossRef]
- Rashid, S.; Curtis, D.E.; Garuti, R.; Anderson, N.N.; Bashmakov, Y.; Ho, Y.K.; Hammer, R.E.; Moon, Y.-A.; Horton, J.D. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. USA 2005, 102, 5374–5379. [Google Scholar] [CrossRef]
- Povo-Retana, A.; Mojena, M.; Stremtan, A.B.; Fernández-García, V.B.; Gómez-Sáez, A.; Nuevo-Tapioles, C.; Molina-Guijarro, J.M.; Avendaño-Ortiz, J.; Cuezva, J.M.; López-Collazo, E.; et al. Specific effects of trabectedin and lurbinectedin on human macrophage function and fate—Novel Insights. Cancers 2020, 12, 3060. [Google Scholar] [CrossRef]
- Povo-Retana, A.; Mojena, M.; Boscá, A.; Pedrós, J.; Peraza, D.A.; Valenzuela, C.; Laparra, J.M.; Calle, F.; Boscá, L. Graphene particles interfere with pro-inflammatory polarization of human macrophages: Functional and electrophysiological evidence. Adv. Biol. 2021, 5, 2100882. [Google Scholar] [CrossRef]
- Hortelano, S.; Alvarez, A.M.; Boscá, L. Nitric oxide induces tyrosine nitration and release of cytochrome c preceding an increase of mitochondrial transmembrane potential in macrophages. FASEB J. 1999, 13, 2311–2317. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaén, R.I.; Povo-Retana, A.; Rosales-Mendoza, C.; Capillas-Herrero, P.; Sánchez-García, S.; Martín-Sanz, P.; Mojena, M.; Prieto, P.; Boscá, L. Functional Crosstalk between PCSK9 Internalization and Pro-Inflammatory Activation in Human Macrophages: Role of Reactive Oxygen Species Release. Int. J. Mol. Sci. 2022, 23, 9114. https://doi.org/10.3390/ijms23169114
Jaén RI, Povo-Retana A, Rosales-Mendoza C, Capillas-Herrero P, Sánchez-García S, Martín-Sanz P, Mojena M, Prieto P, Boscá L. Functional Crosstalk between PCSK9 Internalization and Pro-Inflammatory Activation in Human Macrophages: Role of Reactive Oxygen Species Release. International Journal of Molecular Sciences. 2022; 23(16):9114. https://doi.org/10.3390/ijms23169114
Chicago/Turabian StyleJaén, Rafael I., Adrián Povo-Retana, César Rosales-Mendoza, Patricia Capillas-Herrero, Sergio Sánchez-García, Paloma Martín-Sanz, Marina Mojena, Patricia Prieto, and Lisardo Boscá. 2022. "Functional Crosstalk between PCSK9 Internalization and Pro-Inflammatory Activation in Human Macrophages: Role of Reactive Oxygen Species Release" International Journal of Molecular Sciences 23, no. 16: 9114. https://doi.org/10.3390/ijms23169114
APA StyleJaén, R. I., Povo-Retana, A., Rosales-Mendoza, C., Capillas-Herrero, P., Sánchez-García, S., Martín-Sanz, P., Mojena, M., Prieto, P., & Boscá, L. (2022). Functional Crosstalk between PCSK9 Internalization and Pro-Inflammatory Activation in Human Macrophages: Role of Reactive Oxygen Species Release. International Journal of Molecular Sciences, 23(16), 9114. https://doi.org/10.3390/ijms23169114