
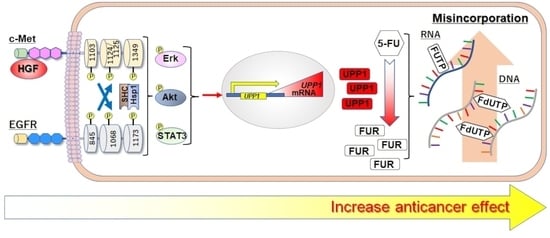
Hepatocyte Growth Factor Enhances Antineoplastic Effect of 5-Fluorouracil by Increasing UPP1 Expression in HepG2 Cells

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
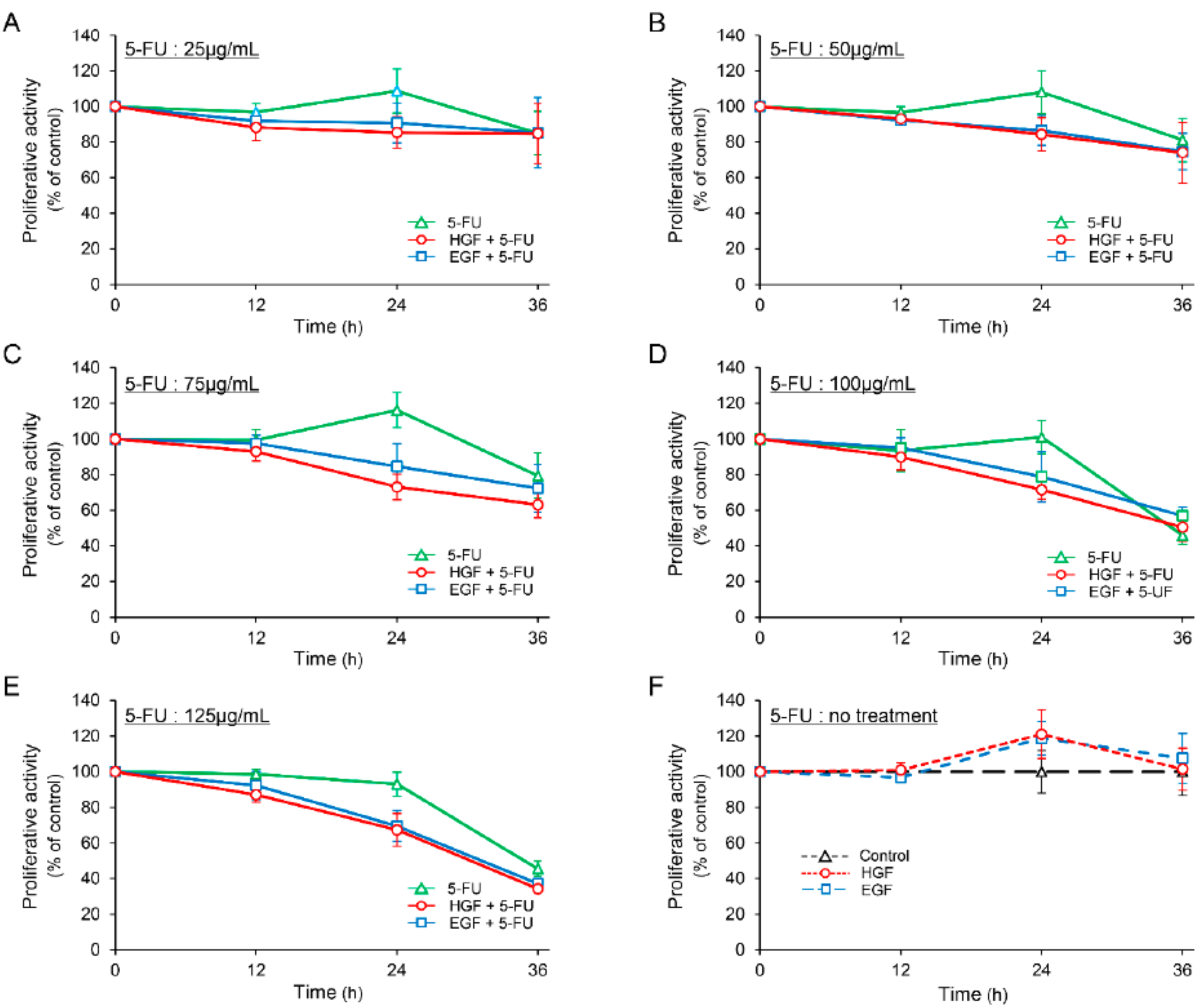
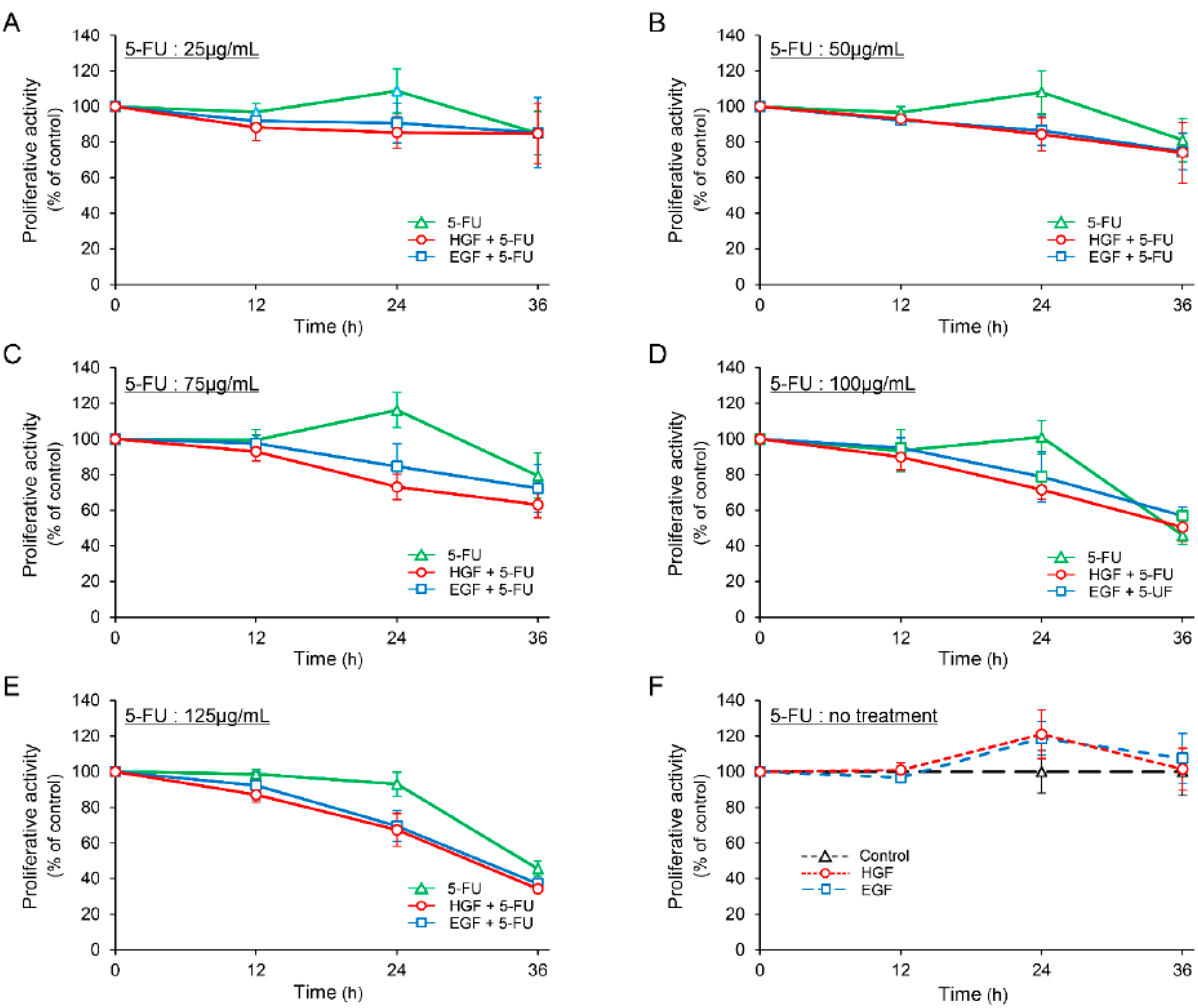
2.1. HGF and Epidermal Growth Factor (EGF) Enhances the Antineoplastic Effect of 5-FU
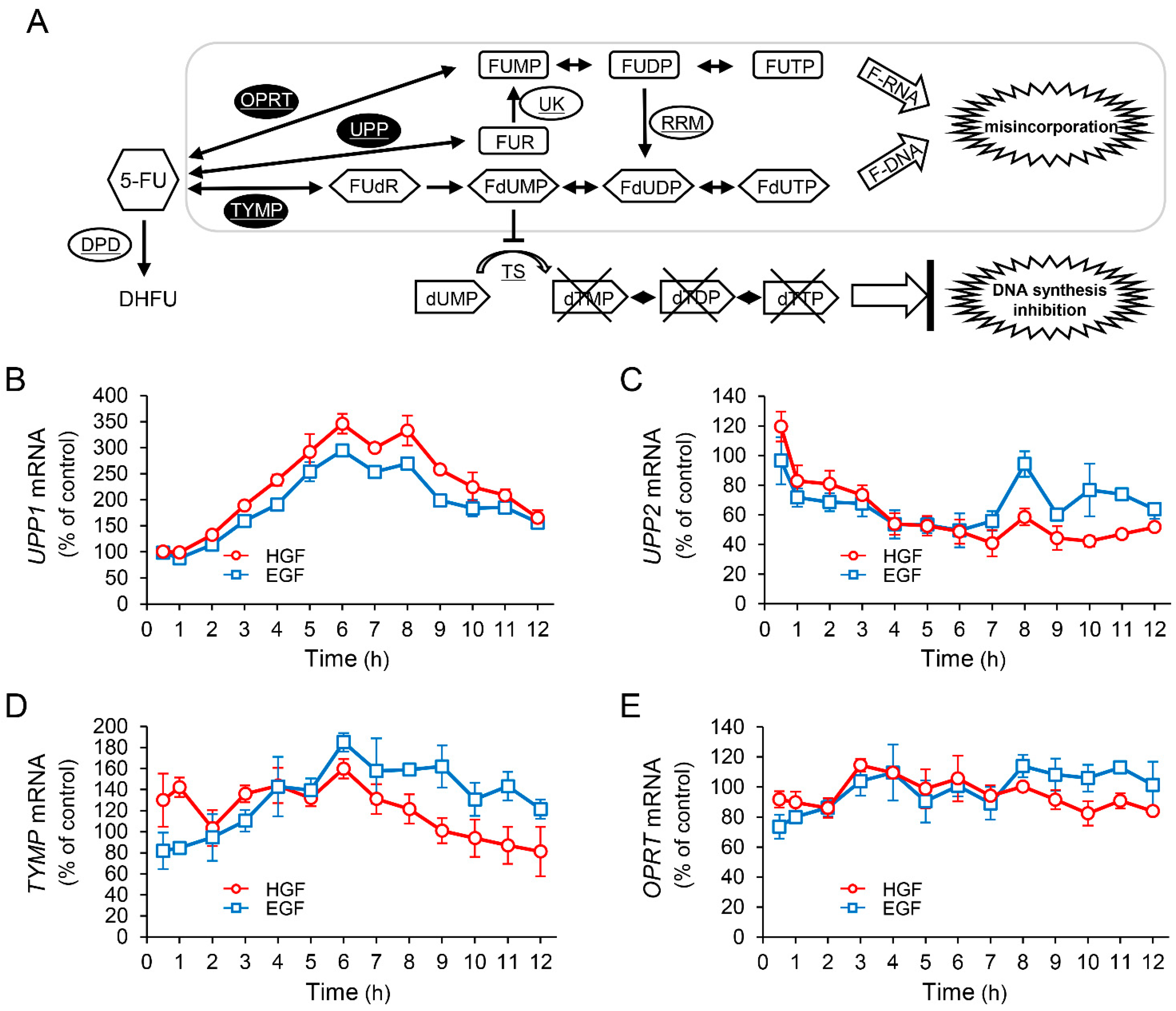
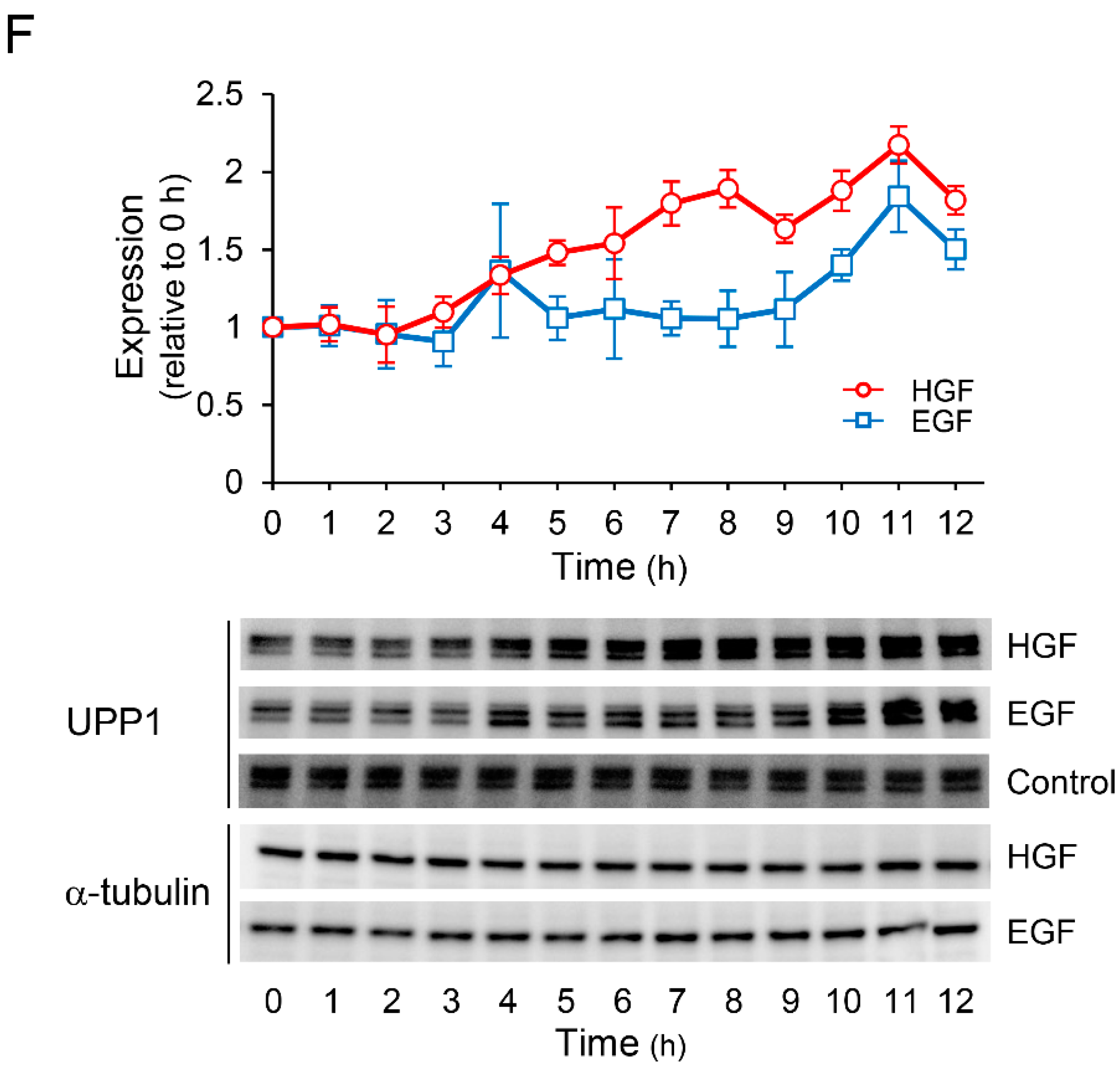
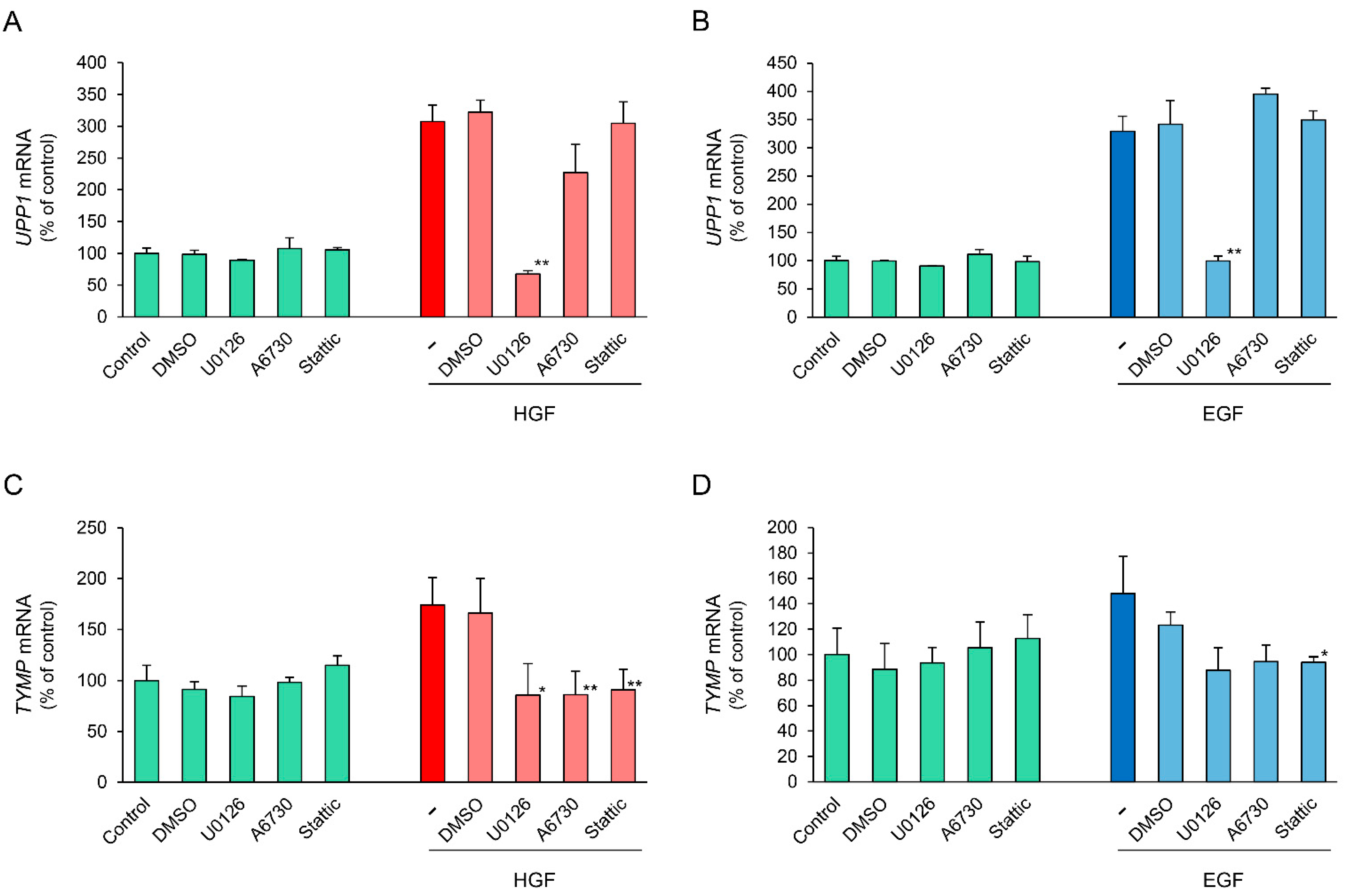
2.2. HGF and EGF Increase UPP1 Expression
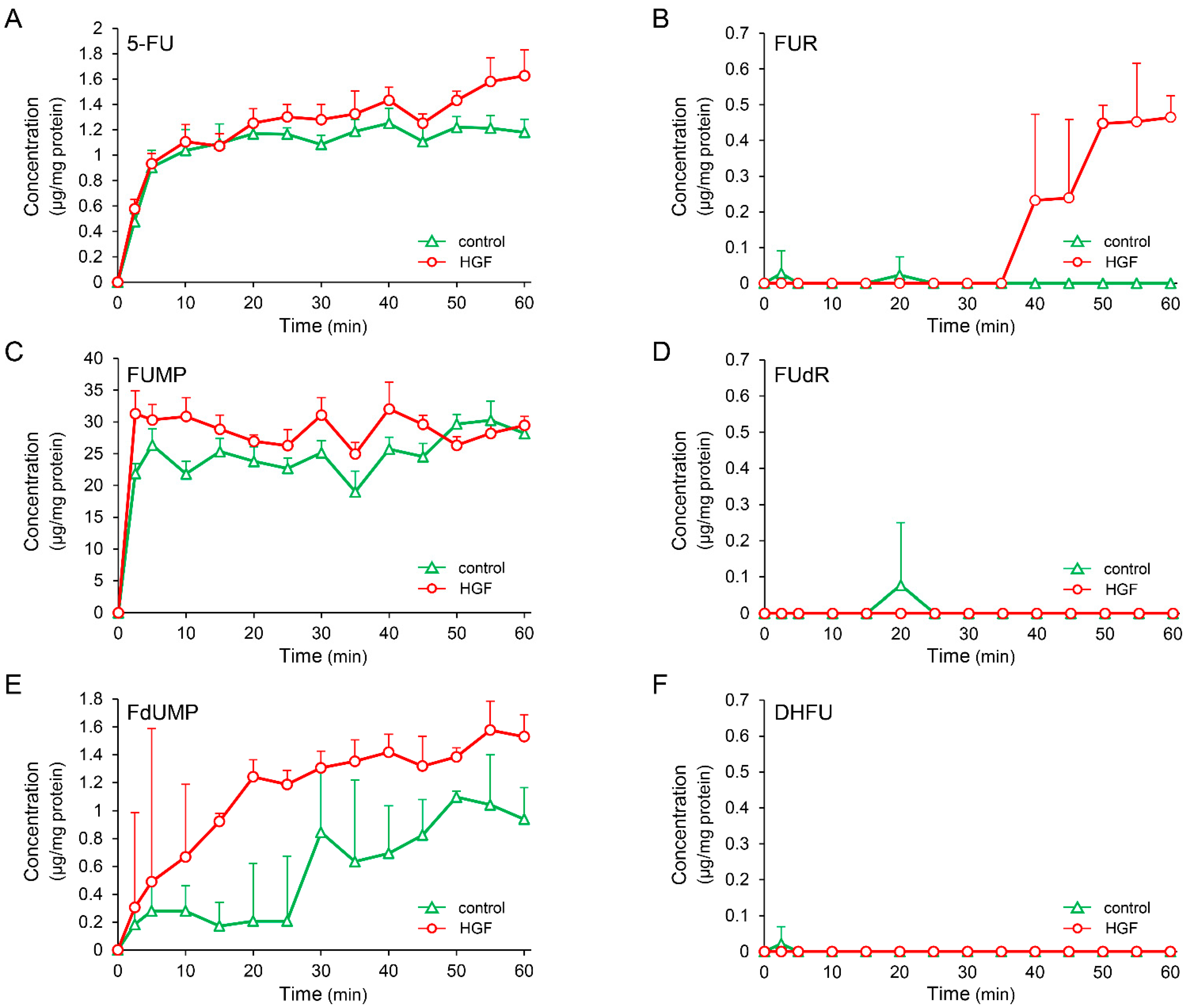
2.3. HGF Increases Intracellular Concentration of 5-Fluorouridine (FUR), an Initial Anabolite of 5-FU
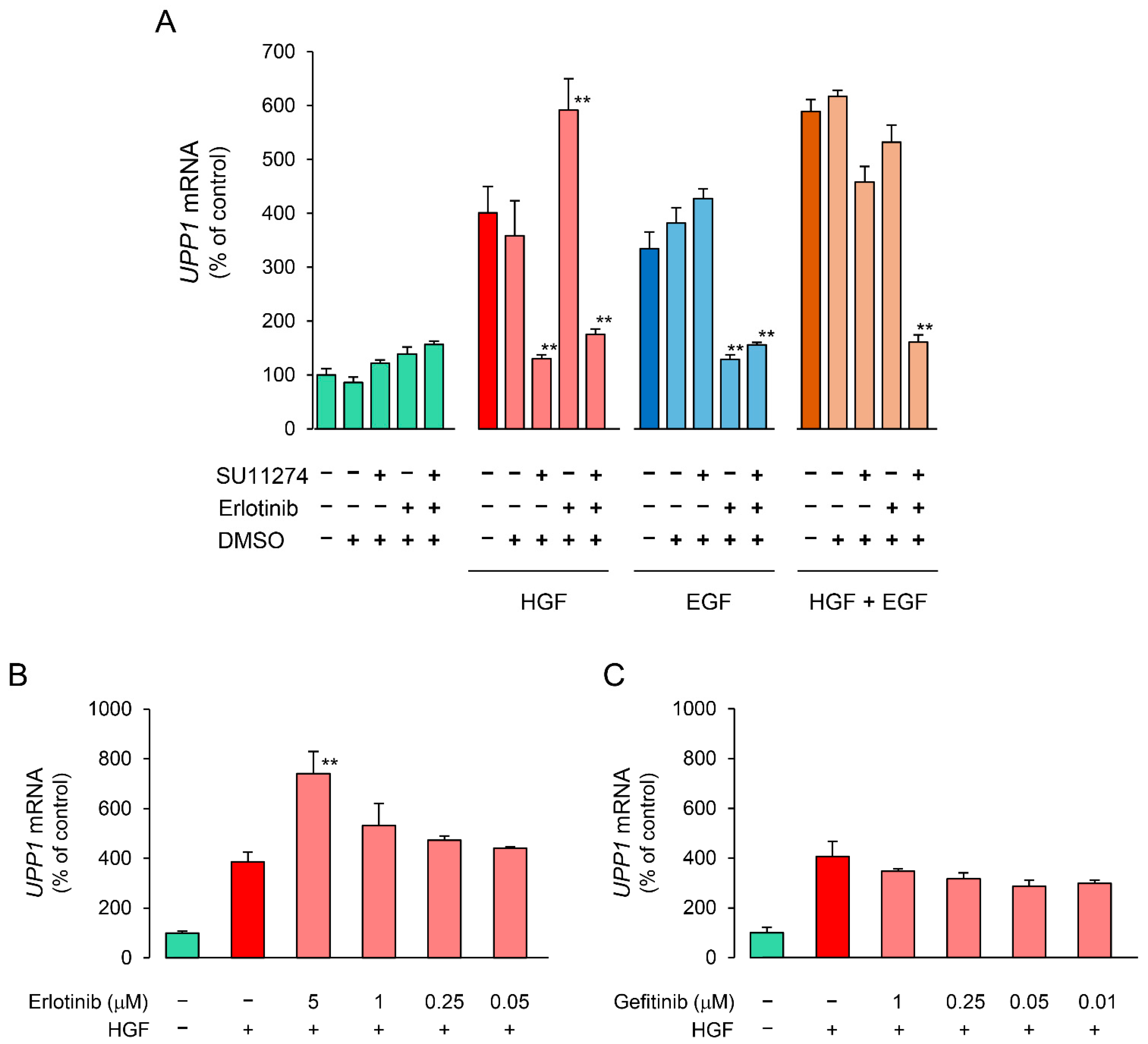
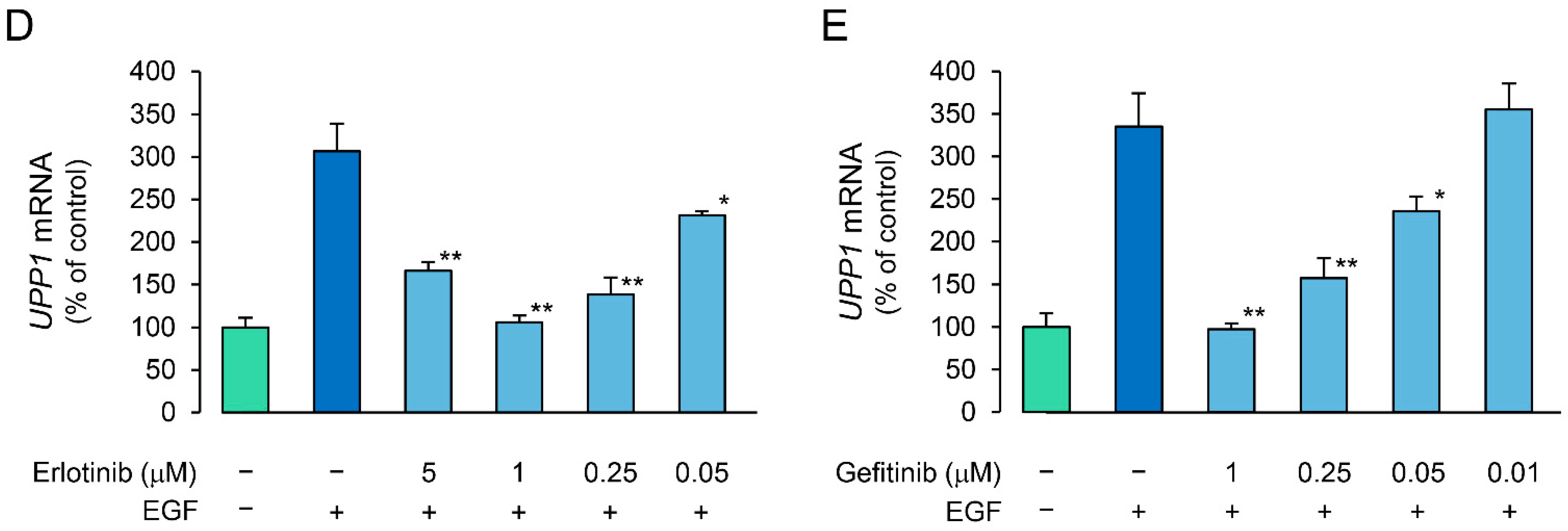
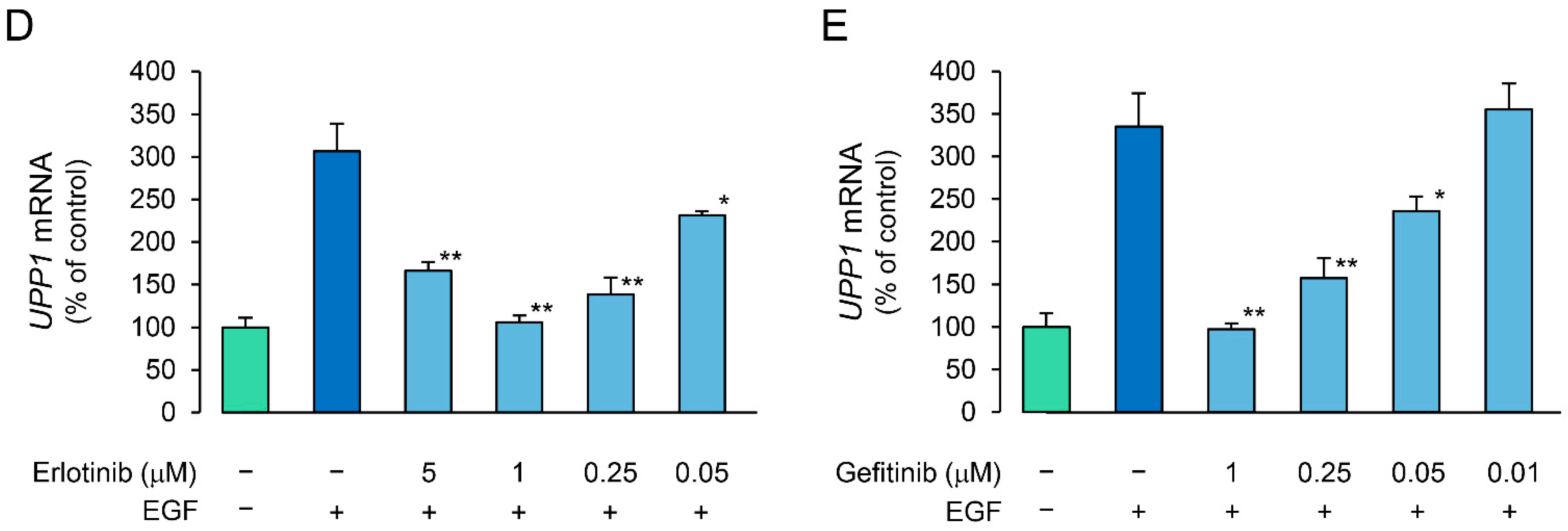
2.4. Effects of Erlotinib and Gefitinib on HGF-Induced Increase in Expression of UPP1 mRNA Are Antithetical
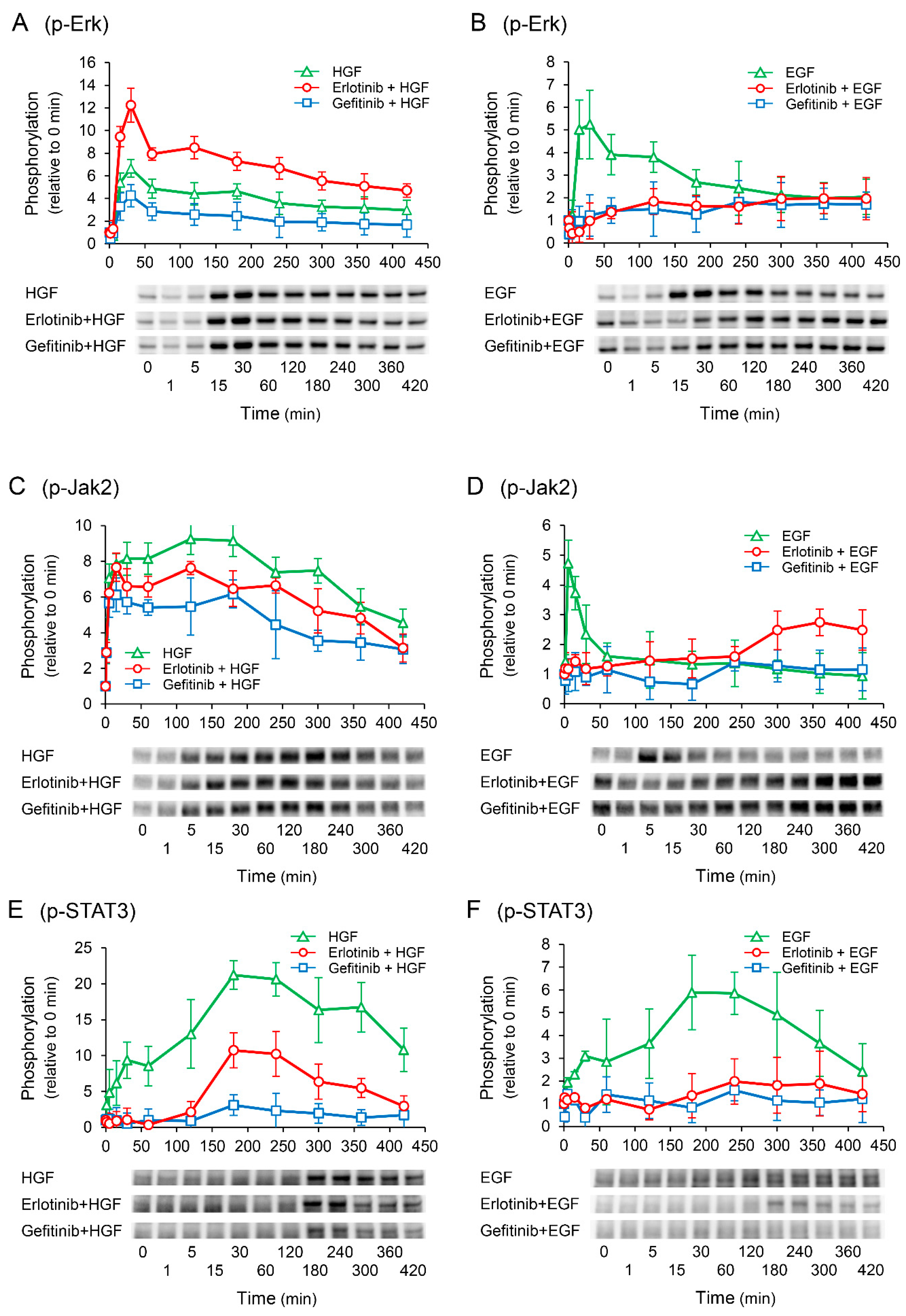
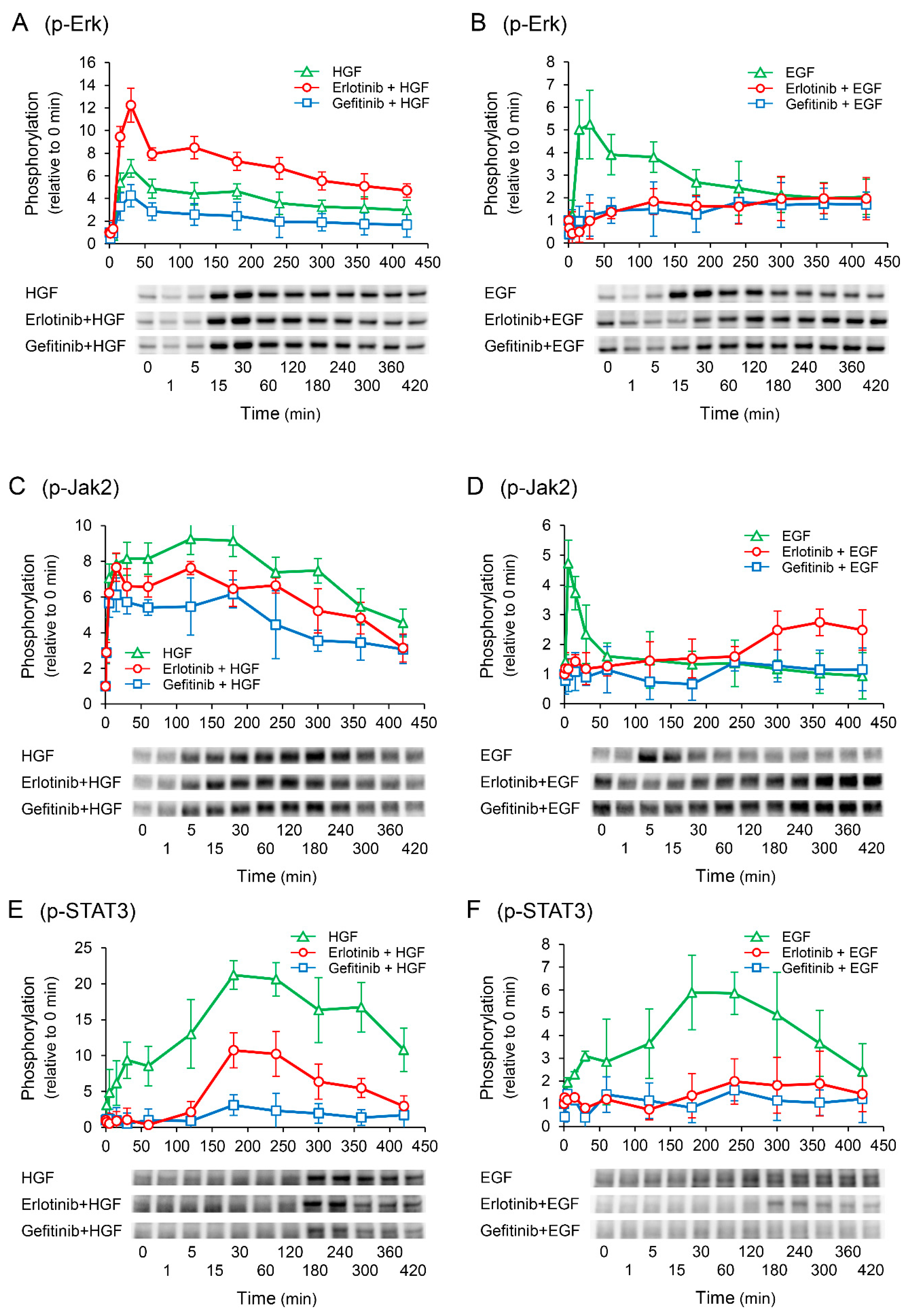
2.5. Erlotinib Enhances HGF-Induced Increase in Extracellular Signal-Regulated Kinase (Erk) Phosphorylation
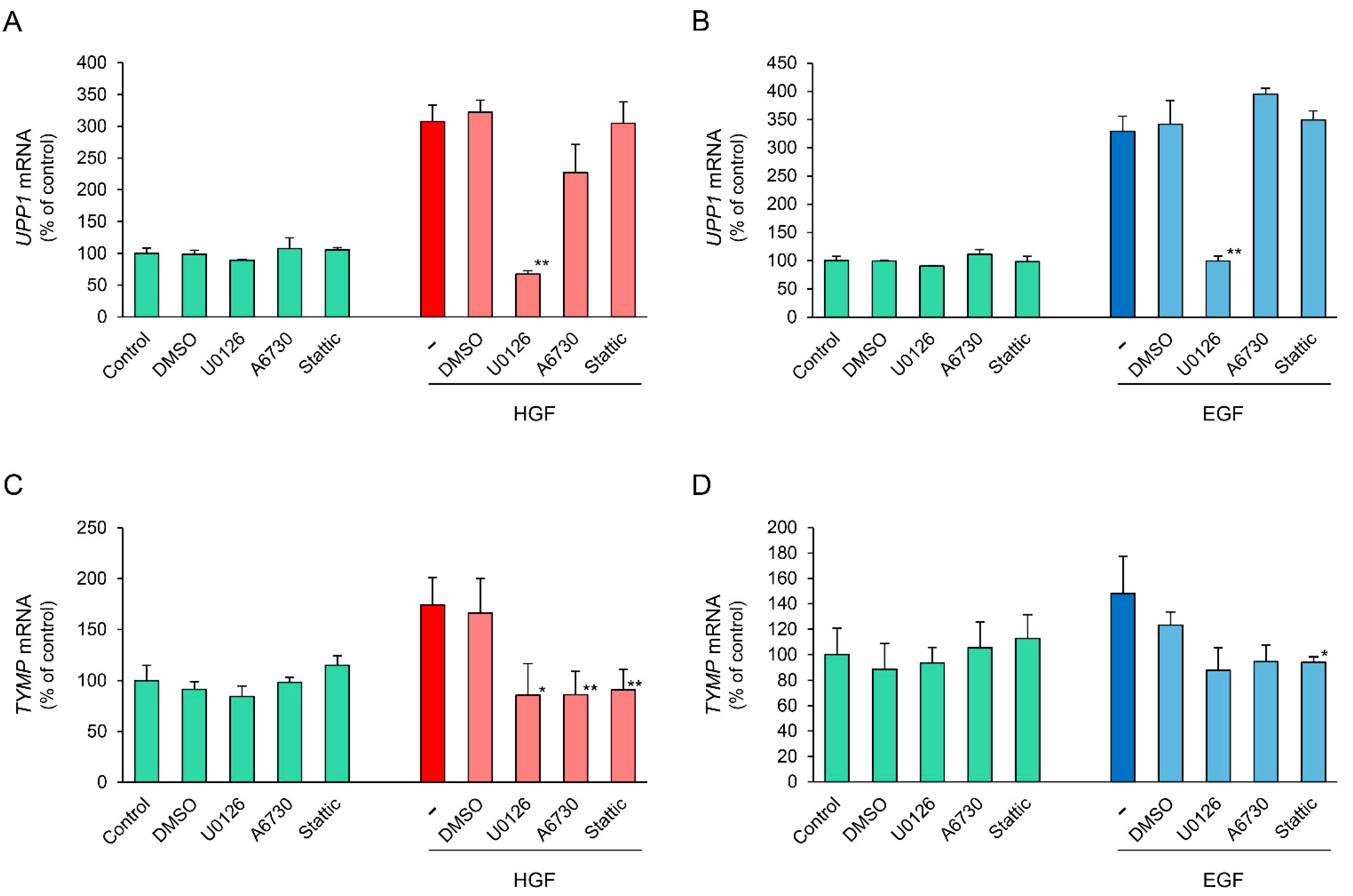
2.6. Effects of Specific Inhibitors of Downstream Signaling Molecules on HGF- and EGF-Induced Increase in UPP1 mRNA Expression
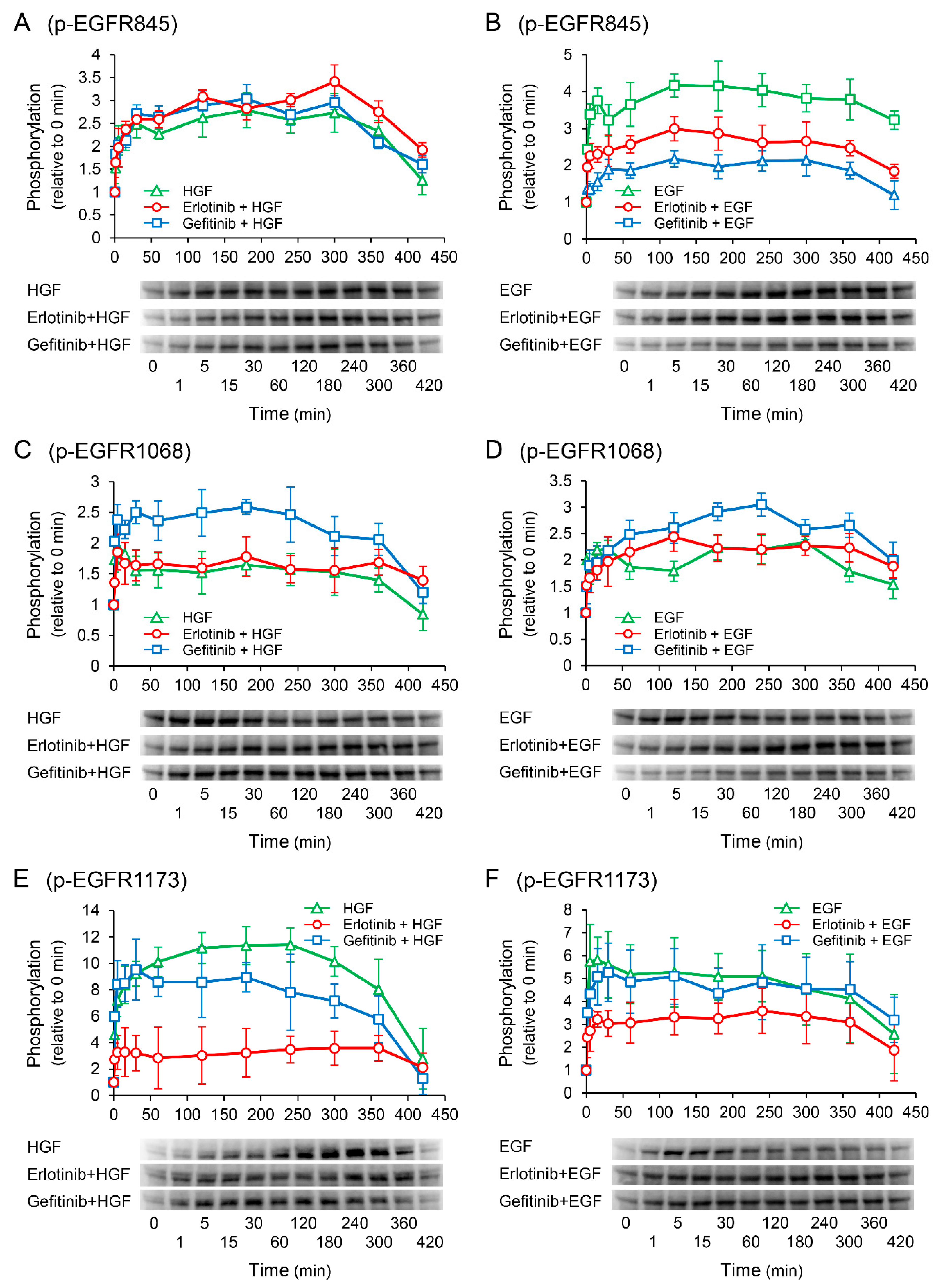
2.7. Erlotinib Markedly Suppresses the HGF-Induced Increase in EGFR Phosphorylation at the Tyr1173 Sight
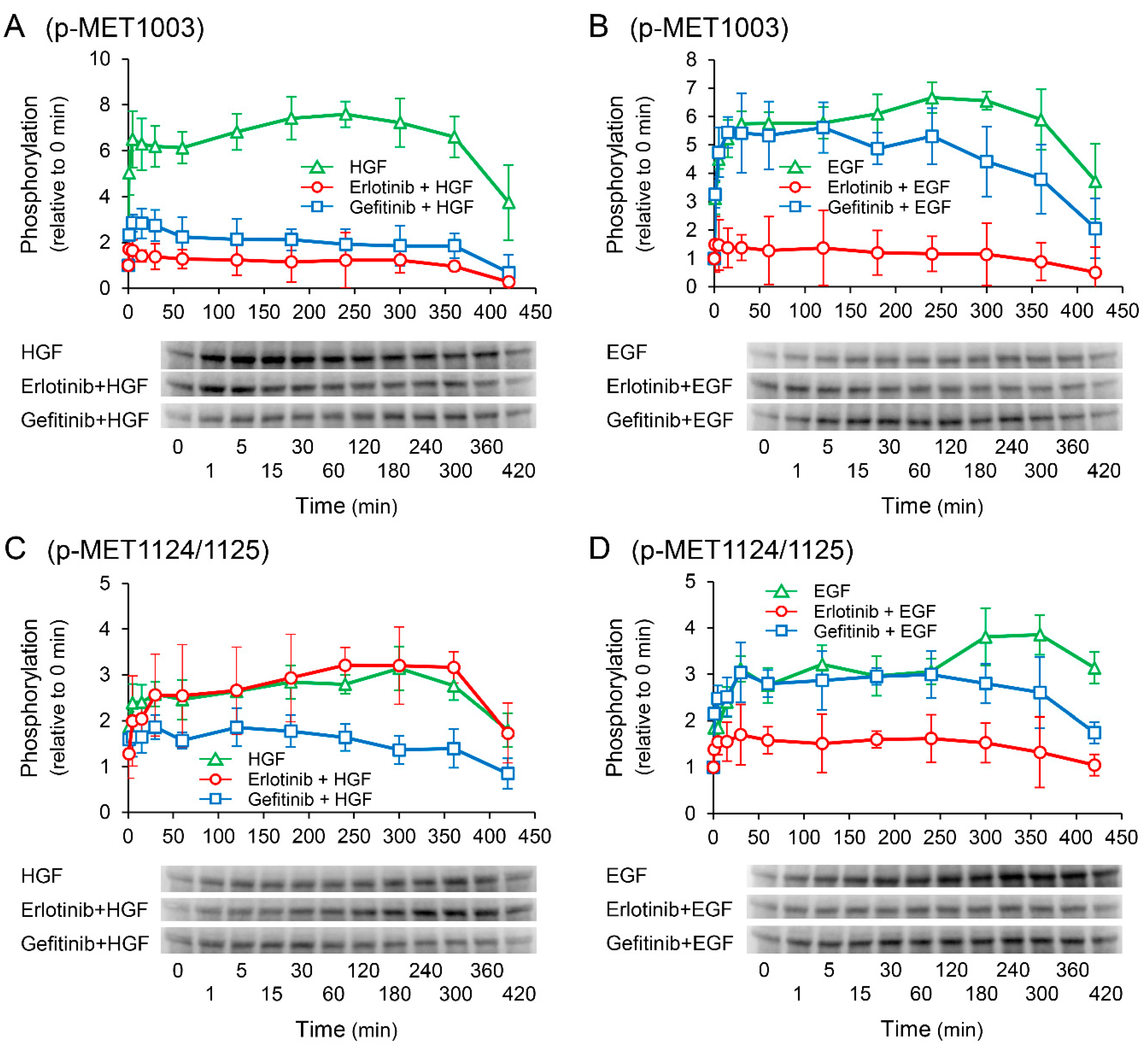
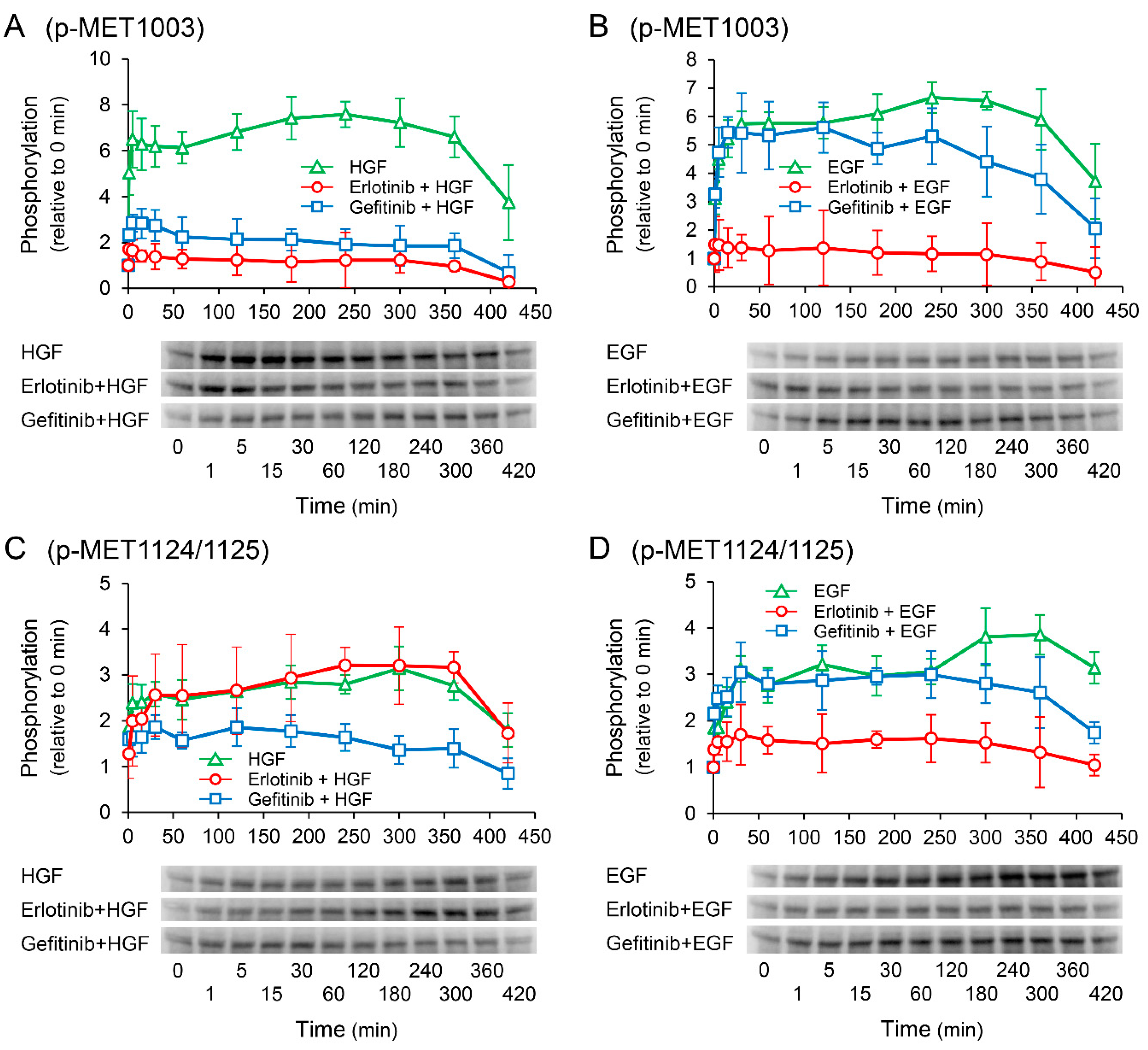
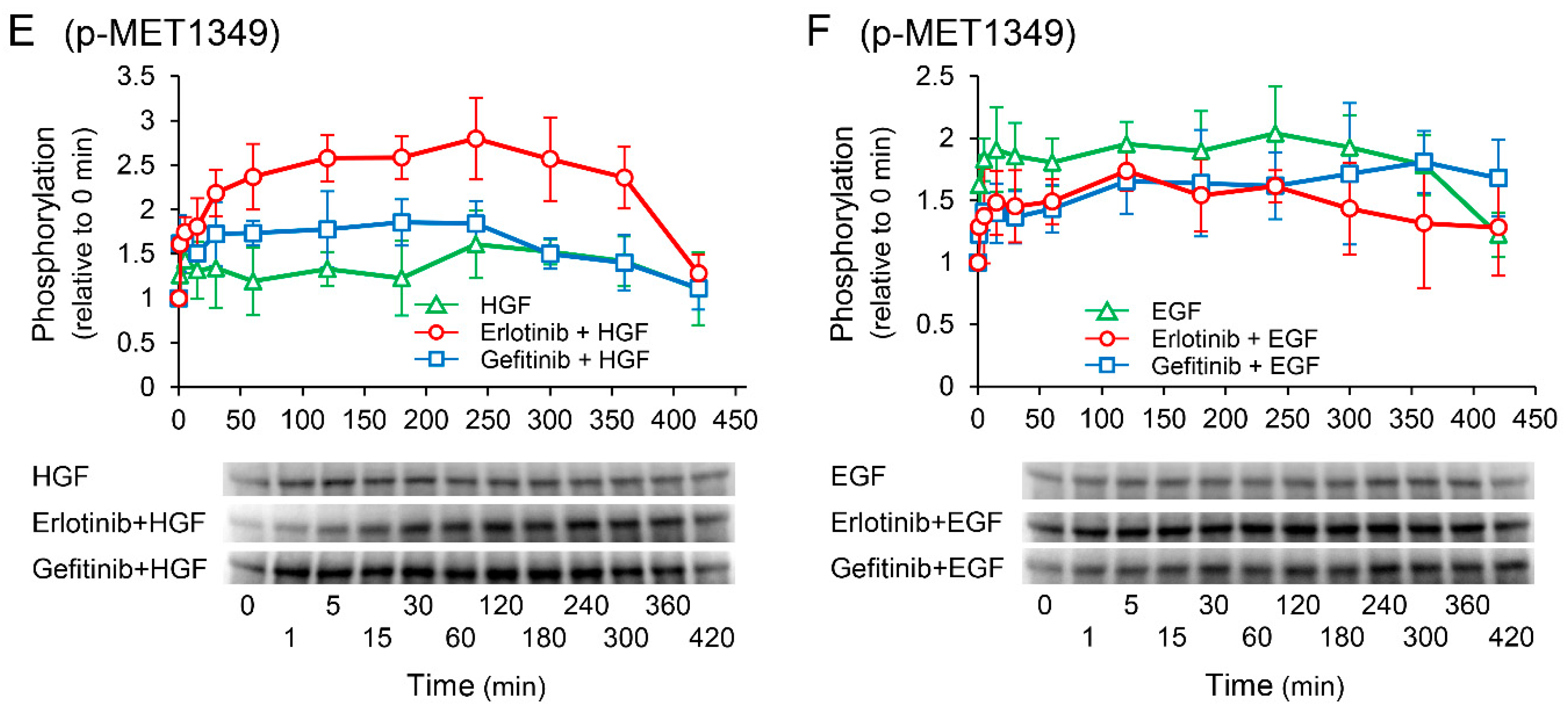
2.8. Effects of Receptor Tyrosine Kinase Inhibitors on Growth Factor-Induced Phosphorylation of c-Met
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.5. Western Blot Analysis
4.6. Measurement of 5-FU and Its Metabolites in HepG2 Cells
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olivero, M.; Rizzo, M.; Madeddu, R.; Casadio, C.; Pennacchietti, S.; Nicotra, M.R.; Prat, M.; Maggi, G.; Arena, N.; Natali, P.G.; et al. Overexpression and activation of hepatocyte growth factor/scatter factor in human non-small-cell lung carcinomas. Br. J. Cancer 1996, 74, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Shen, D.; Yin, X.; Gavine, P.; Zhang, T.; Su, X.; Zhan, P.; Xu, Y.; Lv, J.; Qian, J.; et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br. J. Cancer 2014, 110, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.W.; Shi, G.M.; Zhou, J.; Wu, F.Z.; Ding, Z.B.; Hu, M.Y.; Xu, Y.; Song, Z.J.; Wang, Z.J.; Wu, J.C.; et al. Role of overexpression of CD151 and/or c-Met in predicting prognosis of hepatocellular carcinoma. Hepatology 2009, 49, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Kawakami, H.; Okamoto, I.; Arao, T.; Okamoto, W.; Matsumoto, K.; Taniguchi, H.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K.; et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget 2013, 4, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lim, H.Y.; Shi, S.; Lee, J.; Deng, S.; Xie, T.; Zhu, Z.; Wang, Y.; Pocalyko, D.; Yang, W.J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yonemura, Y.; Nojima, N.; Hirono, Y.; Fushida, S.; Fujimura, T.; Miwa, K.; Endo, Y.; Yamamoto, H.; Watanabe, H. The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c-met), autocrine motility factor receptor, and urokinase-type plasminogen activator receptor. Cancer 1998, 82, 2112–2122. [Google Scholar] [CrossRef]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Puccini, A.; Marin-Ramos, N.I.; Bergamo, F.; Schirripa, M.; Lonardi, S.; Lenz, H.J.; Loupakis, F.; Battaglin, F. Safety and Tolerability of c-MET Inhibitors in Cancer. Drug Saf. 2019, 42, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Neijssen, J.; Cardoso, R.M.F.; Chevalier, K.M.; Wiegman, L.; Valerius, T.; Anderson, G.M.; Moores, S.L.; Schuurman, J.; Parren, P.; Strohl, W.R.; et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J. Biol. Chem. 2021, 296, 100641. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I.; et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar] [CrossRef]
- Malka, D.; Francois, E.; Penault-Llorca, F.; Castan, F.; Bouche, O.; Bennouna, J.; Ghiringhelli, F.; Fouchardiere, C.; Borg, C.; Samalin, E.; et al. FOLFOX alone or combined with rilotumumab or panitumumab as first-line treatment for patients with advanced gastroesophageal adenocarcinoma (PRODIGE 17-ACCORD 20-MEGA): A randomised, open-label, three-arm phase II trial. Eur. J. Cancer 2019, 115, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T.; Gomez-Lechon, M.J.; Jover, R.; Nakamura, T.; Castell, J.V. Human hepatocyte growth factor down-regulates the expression of cytochrome P450 isozymes in human hepatocytes in primary culture. J. Pharmacol. Exp. Ther. 1998, 284, 760–767. [Google Scholar] [PubMed]
- Okumura, M.; Iwakiri, T.; Takagi, A.; Hirabara, Y.; Kawano, Y.; Arimori, K. Hepatocyte growth factor suppresses the anticancer effect of irinotecan by decreasing the level of active metabolite in HepG2 cells. Biochem. Pharmacol. 2011, 82, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Maroun, C.R.; Rowlands, T. The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014, 142, 316–338. [Google Scholar] [CrossRef]
- Im, Y.S.; Shin, H.K.; Kim, H.R.; Jeong, S.H.; Kim, S.R.; Kim, Y.M.; Lee, D.H.; Jeon, S.H.; Lee, H.W.; Choi, J.K. Enhanced cytotoxicity of 5-FU by bFGF through up-regulation of uridine phosphorylase 1. Mol. Cells 2009, 28, 119–124. [Google Scholar] [CrossRef]
- Cao, D.; Ziemba, A.; McCabe, J.; Yan, R.; Wan, L.; Kim, B.; Gach, M.; Flynn, S.; Pizzorno, G. Differential expression of uridine phosphorylase in tumors contributes to an improved fluoropyrimidine therapeutic activity. Mol. Cancer Ther. 2011, 10, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Nakashiro, K.; Tanaka, H.; Tokuzen, N.; Hamakawa, H. Ribonucleotide reductase M2 is a promising molecular target for the treatment of oral squamous cell carcinoma. Int. J. Oncol. 2015, 46, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Breindel, J.L.; Haskins, J.W.; Cowell, E.P.; Zhao, M.; Nguyen, D.X.; Stern, D.F. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013, 73, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef]
- Rozakis-Adcock, M.; McGlade, J.; Mbamalu, G.; Pelicci, G.; Daly, R.; Li, W.; Batzer, A.; Thomas, S.; Brugge, J.; Pelicci, P.G.; et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 1992, 360, 689–692. [Google Scholar] [CrossRef]
- Okabayashi, Y.; Kido, Y.; Okutani, T.; Sugimoto, Y.; Sakaguchi, K.; Kasuga, M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J. Biol. Chem. 1994, 269, 18674–18678. [Google Scholar] [CrossRef]
- Keilhack, H.; Tenev, T.; Nyakatura, E.; Godovac-Zimmermann, J.; Nielsen, L.; Seedorf, K.; Bohmer, F.D. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J. Biol. Chem. 1998, 273, 24839–24846. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Abella, J.V.; Peschard, P.; Naujokas, M.A.; Lin, T.; Saucier, C.; Urbe, S.; Park, M. Met/Hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol. Cell. Biol. 2005, 25, 9632–9645. [Google Scholar] [CrossRef] [PubMed]
- Furge, K.A.; Zhang, Y.W.; Vande Woude, G.F. Met receptor tyrosine kinase: Enhanced signaling through adapter proteins. Oncogene 2000, 19, 5582–5589. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Loos, W.J.; de Bruijn, P.; van Zuylen, L.; Verweij, J.; Nooter, K.; Stoter, G.; Sparreboom, A. Determination of 5-fluorouracil in microvolumes of human plasma by solvent extraction and high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1999, 735, 293–297. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okumura, M.; Iwakiri, T.; Yoshikawa, N.; Nagatomo, T.; Ayabe, T.; Tsuneyoshi, I.; Ikeda, R. Hepatocyte Growth Factor Enhances Antineoplastic Effect of 5-Fluorouracil by Increasing UPP1 Expression in HepG2 Cells. Int. J. Mol. Sci. 2022, 23, 9108. https://doi.org/10.3390/ijms23169108
Okumura M, Iwakiri T, Yoshikawa N, Nagatomo T, Ayabe T, Tsuneyoshi I, Ikeda R. Hepatocyte Growth Factor Enhances Antineoplastic Effect of 5-Fluorouracil by Increasing UPP1 Expression in HepG2 Cells. International Journal of Molecular Sciences. 2022; 23(16):9108. https://doi.org/10.3390/ijms23169108
Chicago/Turabian StyleOkumura, Manabu, Tomomi Iwakiri, Naoki Yoshikawa, Takao Nagatomo, Takanori Ayabe, Isao Tsuneyoshi, and Ryuji Ikeda. 2022. "Hepatocyte Growth Factor Enhances Antineoplastic Effect of 5-Fluorouracil by Increasing UPP1 Expression in HepG2 Cells" International Journal of Molecular Sciences 23, no. 16: 9108. https://doi.org/10.3390/ijms23169108
APA StyleOkumura, M., Iwakiri, T., Yoshikawa, N., Nagatomo, T., Ayabe, T., Tsuneyoshi, I., & Ikeda, R. (2022). Hepatocyte Growth Factor Enhances Antineoplastic Effect of 5-Fluorouracil by Increasing UPP1 Expression in HepG2 Cells. International Journal of Molecular Sciences, 23(16), 9108. https://doi.org/10.3390/ijms23169108