
Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
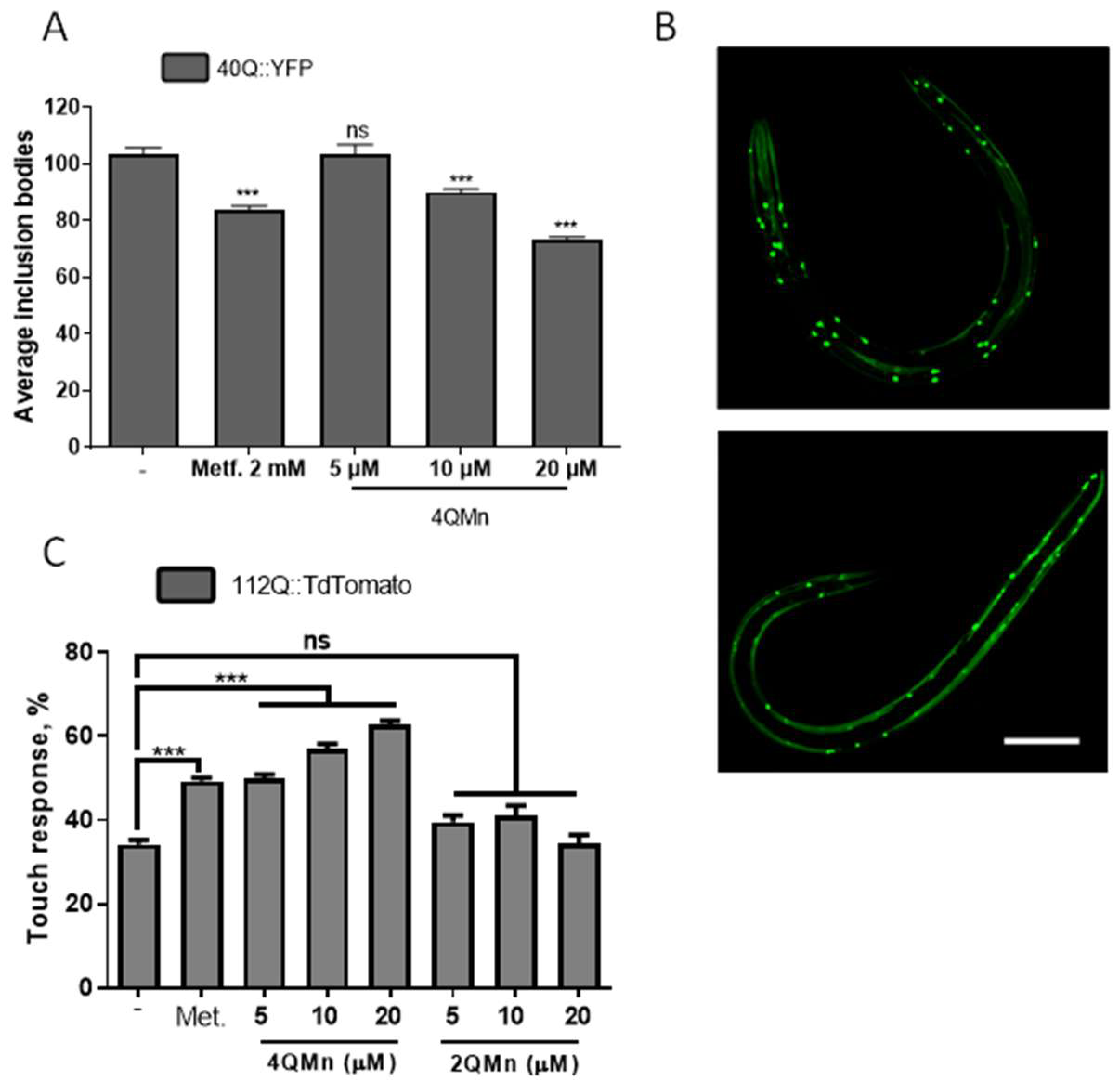
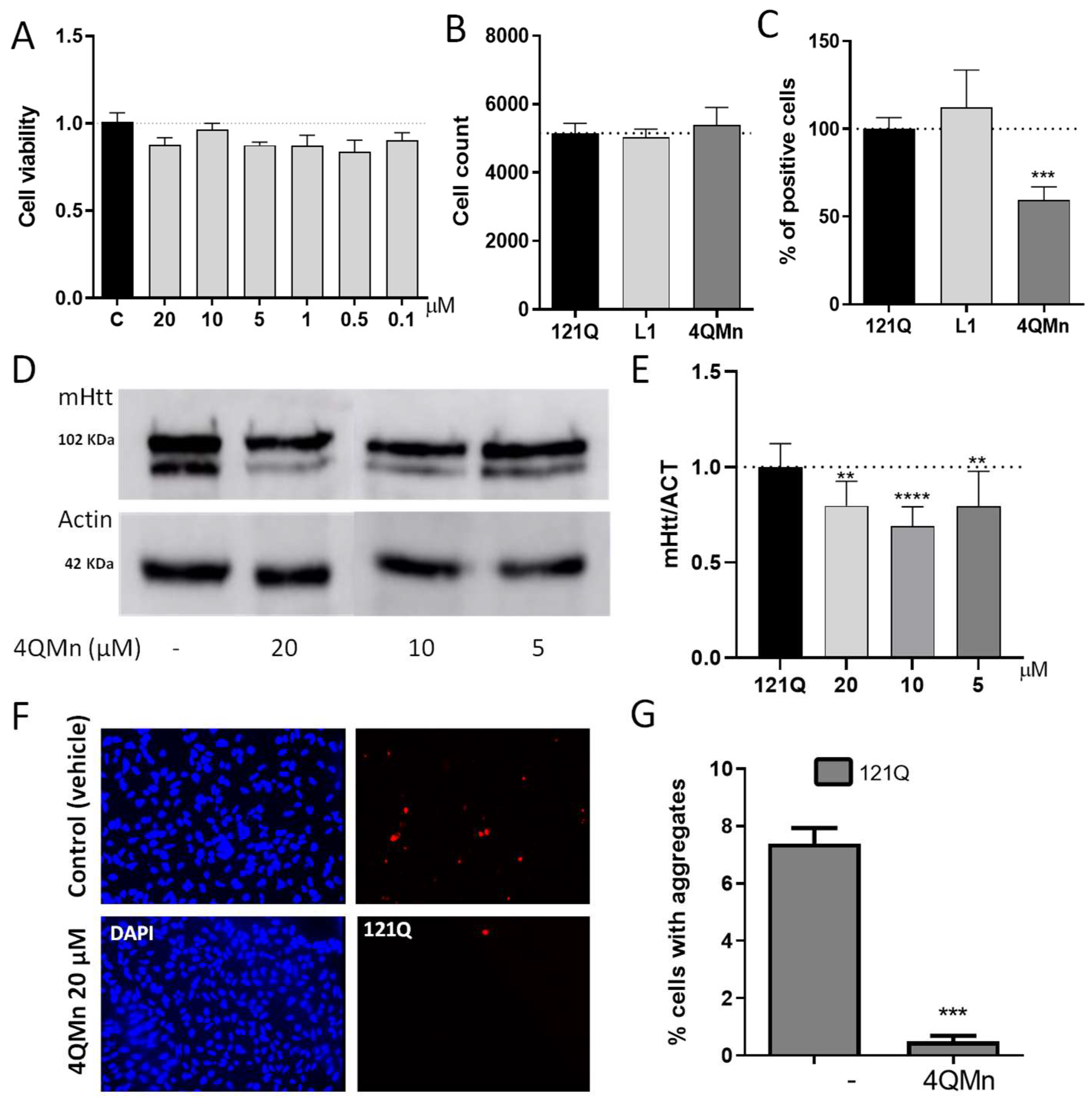
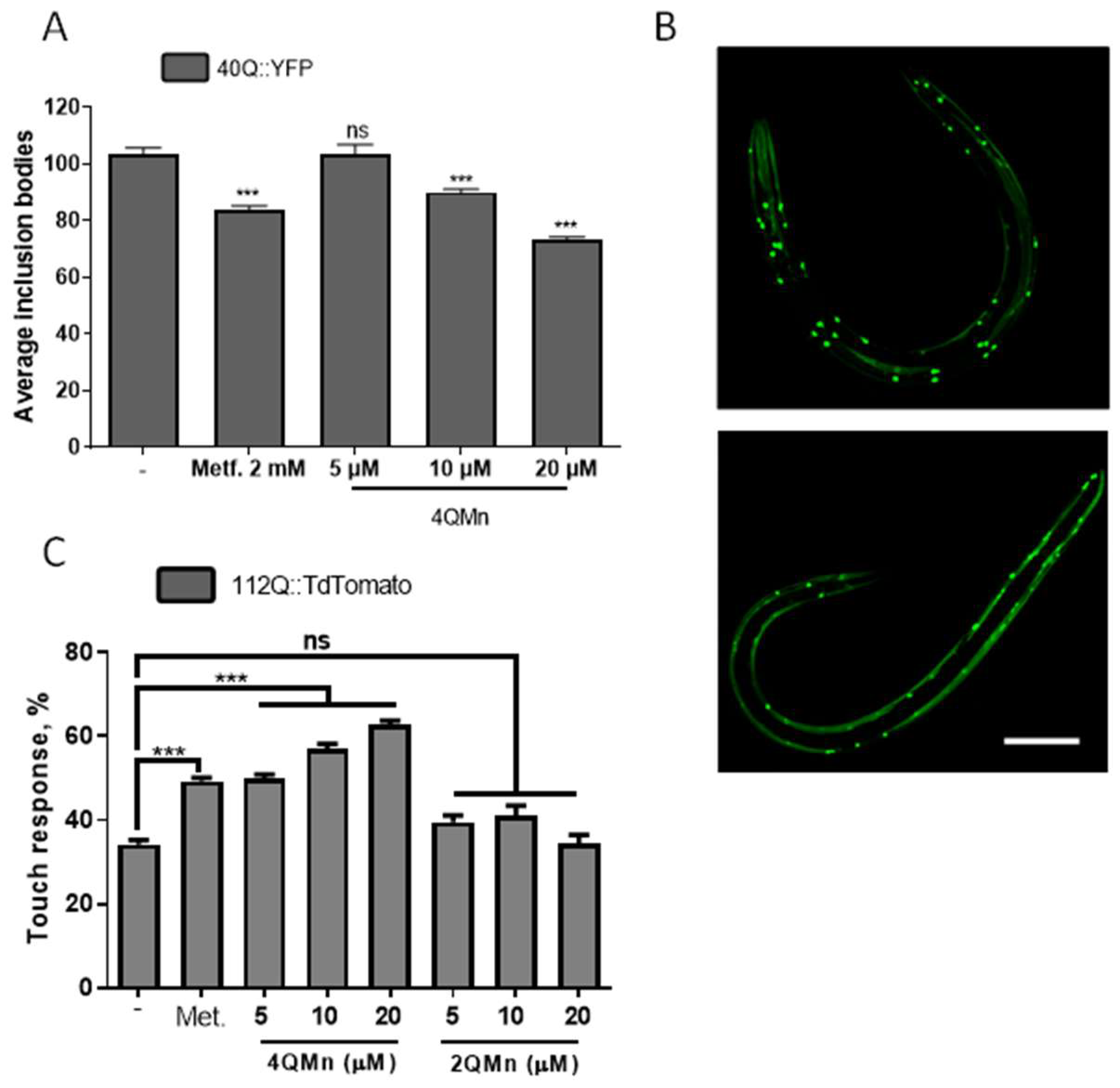
2.1. Treating In Vitro and In Vivo Models of HD with 4QMn Reduces Protein Aggregation
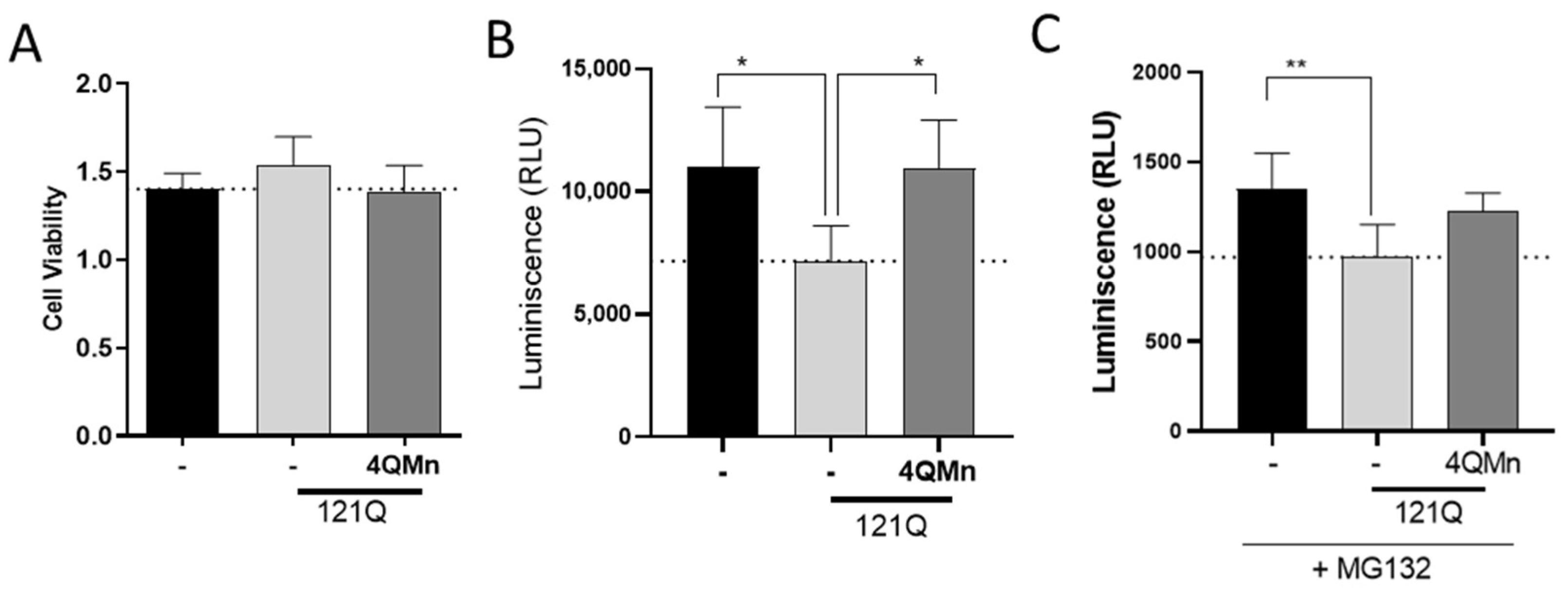
2.2. The Proteasomal System Is Impaired by mHtt and Treatment with 4QMn Restores Its Activity
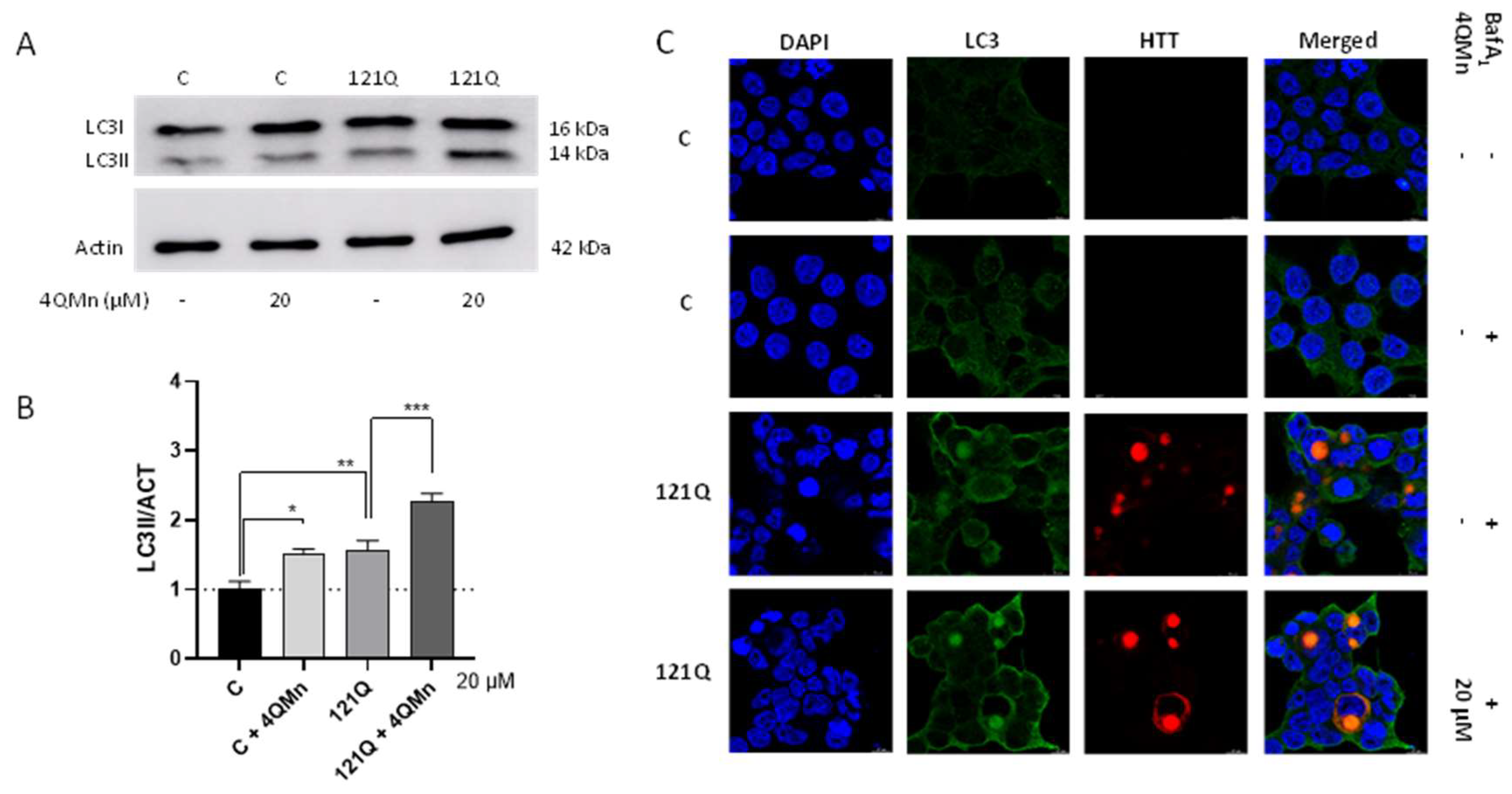
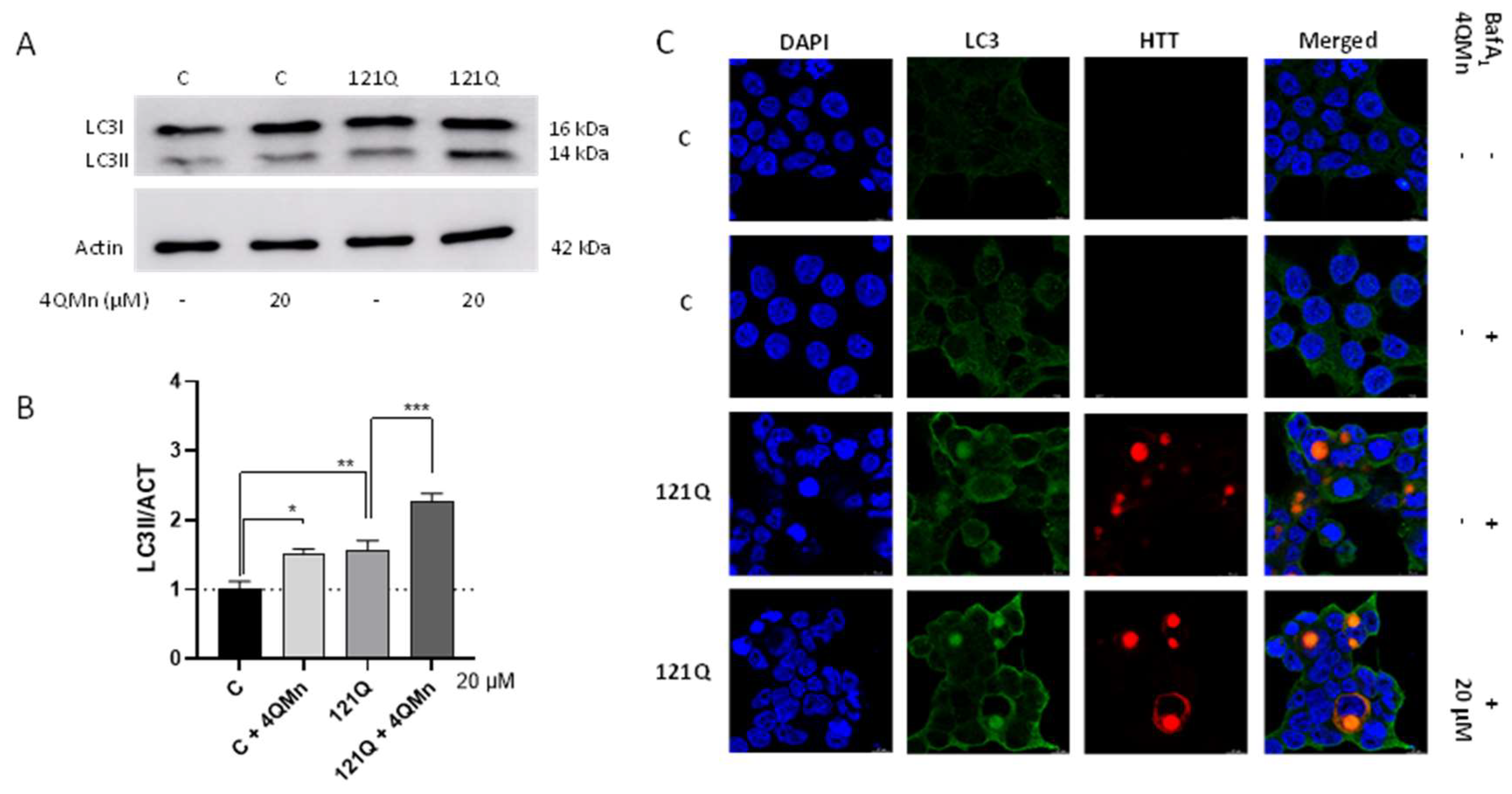
2.3. 4QMn Promotes Autophagy in Cells Stressed by mHtt
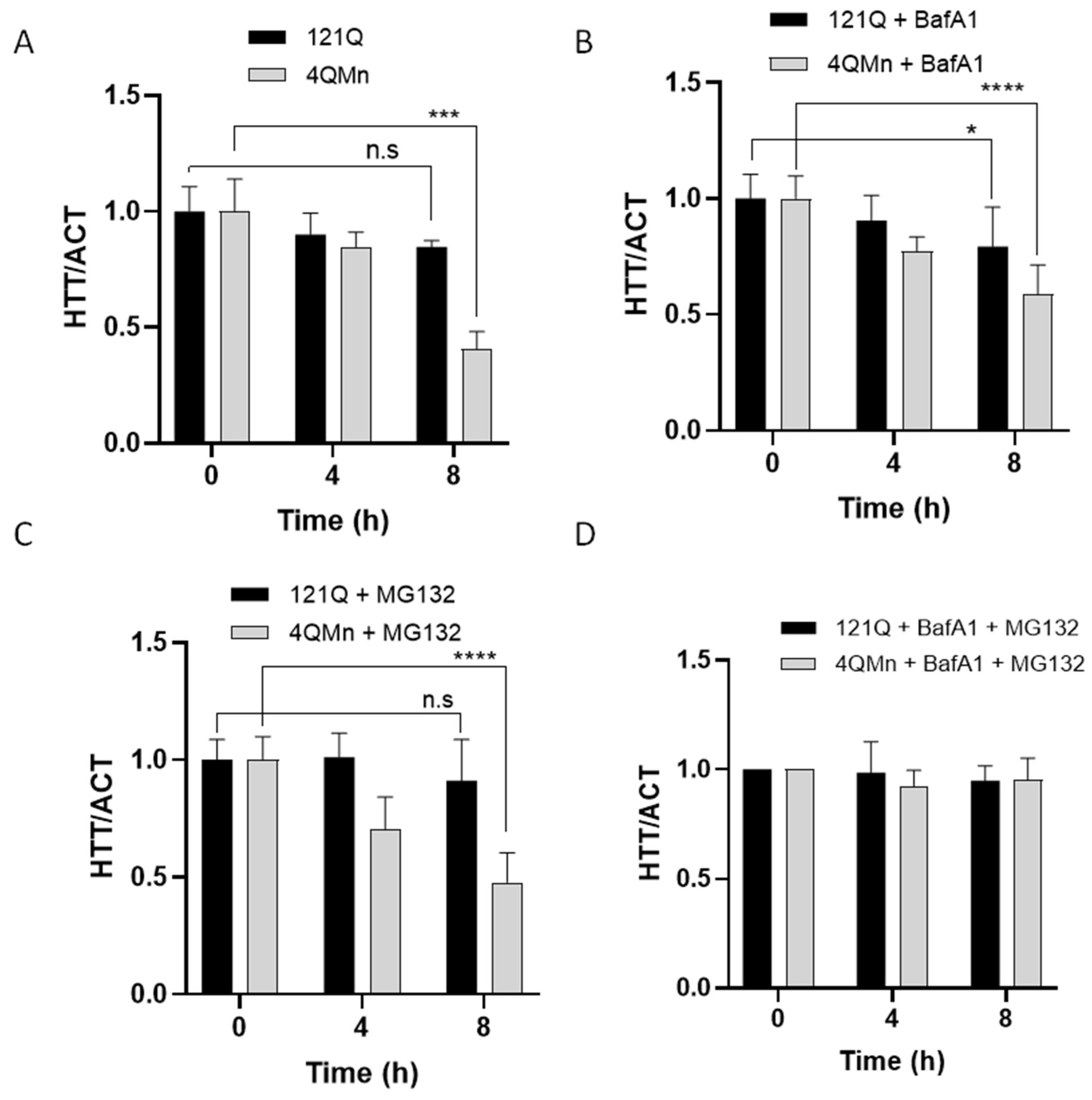
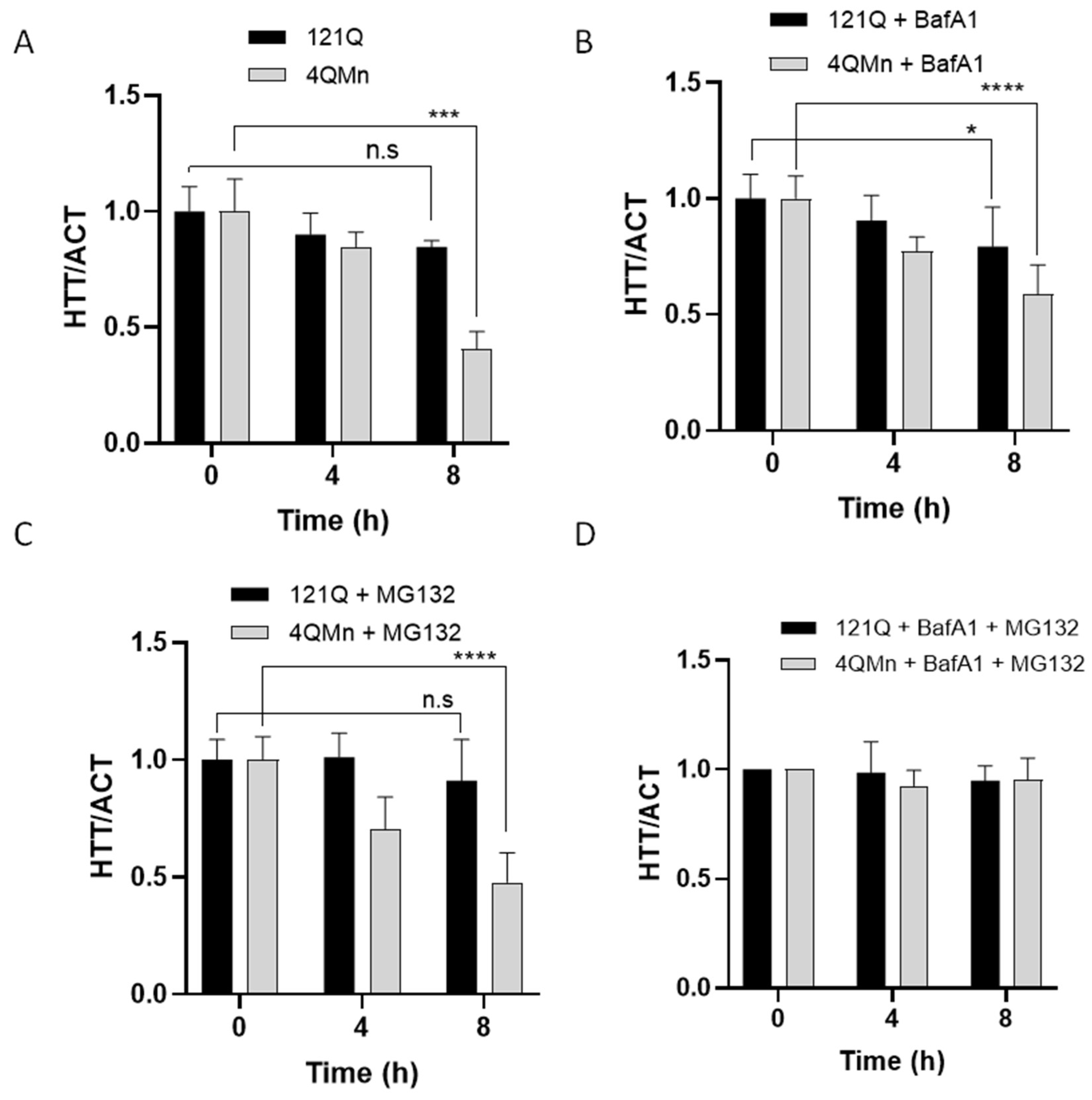
2.4. 4QMn Activates UPS and Autophagy for mHtt Degradation
3. Materials and Methods
3.1. Cell Culture and Transfection
3.2. Worm Culture and Manipulation
3.3. MTT Assay
3.4. Immunofluorescence Quantification through In-Cell Analyzer
3.5. Western Blot Analysis
3.6. Proteasome Activity
3.7. Autophagy Markers Study
3.8. Immunostaining
3.9. Cycloheximide Chase Assay (CHX)
3.10. Statistical Analysis
4. Conclusions and Final Remarks
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tam, S.; Spiess, C.; Auyeung, W.; Joachimiak, L.; Chen, B.; Poirier, M.A.; Frydman, J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 2009, 16, 1279–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millán, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to treat Huntington disease: A pleiotropic drug against a multi-system disorder. Mech. Ageing Dev. 2022, 204, 111670. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; You, Y.; Kordower, J.H.; Brady, S.T.; Morfini, G.A. Differential vulnerability of neurons in Huntington’s disease: The role of cell type-specific features. J. Neurochem. 2010, 113, 1073–1091. [Google Scholar] [CrossRef] [Green Version]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Hong, Y.; Li, X.-J.; Li, S.-H. Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front. Mol. Neurosci. 2016, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.N.V.; Luk, H.W.; Chan, H.Y.E.; Lau, K.-F. Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem. J. 2012, 443, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Ortega, Z.; Lucas, J.J. Ubiquitin-proteasome system involvement in Huntington’s disease. Front. Mol. Neurosci. 2014, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Harding, R.J.; Tong, Y.-F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, J.D.; Fão, L.; Vilaça, R.; Cardoso, S.M.; Rego, A.C. Macroautophagy and Mitophagy in Neurodegenerative Disorders: Focus on Therapeutic Interventions. Biomedicines 2021, 9, 1625. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Escribano, A.P.; Bono-Yagüe, J.; García-Gimeno, M.A.; Sequedo, M.D.; Hervás, D.; Fornés-Ferrer, V.; Torres-Sánchez, S.C.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Synergistic activation of AMPK prevents from polyglutamine-induced toxicity in Caenorhabditis elegans. Pharmacol. Res. 2020, 161, 105105. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Clares, M.P.; Serena, C.; Blasco, S.; Nebot, A.; del Castillo, L.; Soriano, C.; Domènech, A.; Sánchez-Sánchez, A.V.; Soler-Calero, L.; Mullor, J.L.; et al. Mn(II) complexes of scorpiand-like ligands. A model for the MnSOD active centre with high in vitro and in vivo activity. J. Inorg. Biochem. 2015, 143, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Serena, C.; Calvo, E.; Clares, M.P.; Diaz, M.L.; Chicote, J.U.; Beltrán-Debon, R.; Fontova, R.; Rodriguez, A.; García-España, E.; García-España, A. Significant in vivo anti-inflammatory activity of Pytren4Q-Mn a superoxide dismutase 2 (SOD2) mimetic scorpiand-like Mn (II) complex. PLoS ONE 2015, 10, e0119102. [Google Scholar] [CrossRef] [PubMed]
- Clares, M.P.; Blasco, S.; Inclán, M.; del Castillo Agudo, L.; Verdejo, B.; Soriano, C.; Doménech, A.; Latorre, J.; García-España, E. Manganese(II) complexes of scorpiand-like azamacrocycles as MnSOD mimics. Chem. Commun. Camb. Engl. 2011, 47, 5988–5990. [Google Scholar] [CrossRef]
- Weiss, K.R.; Kimura, Y.; Lee, W.-C.M.; Littleton, J.T. Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington’s disease model. Genetics 2012, 190, 581–600. [Google Scholar] [CrossRef] [Green Version]
- Sontag, E.M.; Lotz, G.P.; Agrawal, N.; Tran, A.; Aron, R.; Yang, G.; Necula, M.; Lau, A.; Finkbeiner, S.; Glabe, C.; et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 11109–11119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Camarena, Á.; Merino, M.; Sánchez-Sánchez, A.V.; Blasco, S.; Llinares, J.M.; Mullor, J.L.; García-España, E. An antioxidant boehmite amino-nanozyme able to disaggregate Huntington’s inclusion bodies. Chem. Commun. Camb. Engl. 2022, 58, 5021–5024. [Google Scholar] [CrossRef]
- Muñoz-Lobato, F.; Rodríguez-Palero, M.J.; Naranjo-Galindo, F.J.; Shephard, F.; Gaffney, C.J.; Szewczyk, N.J.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Link, C.D.; et al. Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid. Redox Signal. 2014, 20, 217–235. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiela, E.; Dues, D.J.; Senchuk, M.M.; Van Raamsdonk, J.M. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 2016, 96, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schafer, W.R. Mechanosensory molecules and circuits in C. elegans. Pflugers Arch. 2015, 467, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.A.; Connolly, J.B.; Wellington, C.; Hayden, M.; Dausset, J.; Neri, C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Vazquez-Manrique, R.P.; Tourette, C.; Farina, F.; Offner, N.; Mukhopadhyay, A.; Orfila, A.-M.; Darbois, A.; Menet, S.; Tissenbaum, H.A.; et al. Integration of β-Catenin, Sirtuin, and FOXO Signaling Protects from Mutant Huntingtin Toxicity. J. Neurosci. 2012, 32, 12630–12640. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Tsvetkov, A.S.; Finkbeiner, S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J. Biol. Chem. 2009, 284, 4398–4403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.; Cho, I.-H. Sulforaphane Ameliorates 3-Nitropropionic Acid-Induced Striatal Toxicity by Activating the Keap1-Nrf2-ARE Pathway and Inhibiting the MAPKs and NF-κB Pathways. Mol. Neurobiol. 2016, 53, 2619–2635. [Google Scholar] [CrossRef]
- Luis-García, E.; Limon Pacheco, J.; Serrano-García, N.; Hernández-Pérez, A.; Pedraza-Chaverri, J.; Orozco-Ibarra, M. Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 2016, 31, e21837. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Moser, R.; Régulier, E.; Breuillaud, L.; Dixon, M.; Beesen, A.A.; Elliston, L.; Santos, M.D.F.S.; Kim, J.; Jones, L.; et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2313–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarch, Z.; Giampà, C.; Patassini, S.; Bernardi, G.; Fusco, F.R. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2008, 30, 375–387. [Google Scholar] [CrossRef]
- Wong, H.K.; Bauer, P.O.; Kurosawa, M.; Goswami, A.; Washizu, C.; Machida, Y.; Tosaki, A.; Yamada, M.; Knöpfel, T.; Nakamura, T.; et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum. Mol. Genet. 2008, 17, 3223–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-J.; Li, S. Proteasomal dysfunction in aging and Huntington disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-J.; Li, H.; Li, S. Clearance of mutant huntingtin. Autophagy 2010, 6, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Rubinsztein, D.C. Huntington’s disease: Degradation of mutant huntingtin by autophagy. FEBS J. 2008, 275, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. BioEssays News Rev. Mol. Cell. Dev. Biol. 2020, 42, e2000122. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. In Methods in Enzymology; Autophagy in Mammalian Systems, Part B; Academic Press: Cambridge, MA, USA, 2009; Volume 452, pp. 181–197. [Google Scholar]
- Lee, H.; Noh, J.-Y.; Oh, Y.; Kim, Y.; Chang, J.-W.; Chung, C.-W.; Lee, S.-T.; Kim, M.; Ryu, H.; Jung, Y.-K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2012, 21, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, S.; Renna, M.; Tomas-Zapico, C.; Brown, S.D.M.; Lucas, J.J.; Rubinsztein, D.C.; Acevedo-Arozena, A. α-synuclein levels affect autophagosome numbers in vivo and modulate Huntington disease pathology. Autophagy 2012, 8, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, A.S.; Miller, J.; Arrasate, M.; Wong, J.S.; Pleiss, M.A.; Finkbeiner, S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 2010, 107, 16982–16987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Qin, Z.-H. Degradation of misfolded proteins by autophagy: Is it a strategy for Huntington’s disease treatment? J. Huntingt. Dis. 2013, 2, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, M.; García-Gimeno, M.A.; Sanchis, A.; Bono-Yagüe, J.; Cumella, J.; Lagartera, L.; Pérez, C.; Priego, E.-M.; Campos, A.; Sanz, P.; et al. Neuroprotective Effect of IND1316, an Indole-Based AMPK Activator, in Animal Models of Huntington Disease. ACS Chem. Neurosci. 2022, 13, 275–287. [Google Scholar] [CrossRef]
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2022, 36, 105–119. [Google Scholar] [CrossRef]
- Lane, R.M.; Smith, A.; Baumann, T.; Gleichmann, M.; Norris, D.; Bennett, C.F.; Kordasiewicz, H. Translating Antisense Technology into a Treatment for Huntington’s Disease. Methods Mol. Biol. Clifton NJ 2018, 1780, 497–523. [Google Scholar] [CrossRef]
- Imbert, M.; Blandel, F.; Leumann, C.; Garcia, L.; Goyenvalle, A. Lowering Mutant Huntingtin Using Tricyclo-DNA Antisense Oligonucleotides As a Therapeutic Approach for Huntington’s Disease. Nucleic Acid Ther. 2019, 29, 256–265. [Google Scholar] [CrossRef]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef]
- Lemprière, S. Antisense oligonucleotide reduces mutant protein in patients with HD. Nat. Rev. Neurol. 2019, 15, 435. [Google Scholar] [CrossRef]
- Ionis Pharmaceuticals, Inc. A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Multiple Ascending Doses of Intrathecally Administered ISIS 443139 in Patients with Early Manifest Huntington’s Disease. Clinicaltrials.gov, Clinical Trial Registration NCT02519036; May 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT02519036 (accessed on 30 May 2022).
- Wave Life Sciences Ltd. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 1b/2a Study of WVE-120101 Administered Intrathecally in Patients with Huntington’s Disease. Clinicaltrials.gov, Clinical Trial Registration NCT03225833; February 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03225833 (accessed on 30 May 2022).
- Wave Life Sciences Ltd. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 1b/2a Study of WVE-120102 Administered Intrathecally in Patients with Huntington’s Disease. Clinicaltrials.gov, Clinical Trial Registration NCT03225846; March 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03225846 (accessed on 30 May 2022).
- Hoffmann-La Roche An Open-Label Extension Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of RO7234292 (ISIS 443139) in Huntington’s Disease Patients Who Participated in Prior Investigational Studies of RO7234292 (ISIS 443139). Clinicaltrials.gov, Clinical Trial Registration NCT03342053; March 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03342053 (accessed on 30 May 2022).
- Fields, E.; Vaughan, E.; Tripu, D.; Lim, I.; Shrout, K.; Conway, J.; Salib, N.; Lee, Y.; Dhamsania, A.; Jacobsen, M.; et al. Gene targeting techniques for Huntington’s disease. Ageing Res. Rev. 2021, 70, 101385. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino, M.; Sequedo, M.D.; Sánchez-Sánchez, A.V.; Clares, M.P.; García-España, E.; Vázquez-Manrique, R.P.; Mullor, J.L. Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 8936. https://doi.org/10.3390/ijms23168936
Merino M, Sequedo MD, Sánchez-Sánchez AV, Clares MP, García-España E, Vázquez-Manrique RP, Mullor JL. Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease. International Journal of Molecular Sciences. 2022; 23(16):8936. https://doi.org/10.3390/ijms23168936
Chicago/Turabian StyleMerino, Marián, María Dolores Sequedo, Ana Virginia Sánchez-Sánchez, Mª Paz Clares, Enrique García-España, Rafael P. Vázquez-Manrique, and José L. Mullor. 2022. "Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease" International Journal of Molecular Sciences 23, no. 16: 8936. https://doi.org/10.3390/ijms23168936
APA StyleMerino, M., Sequedo, M. D., Sánchez-Sánchez, A. V., Clares, M. P., García-España, E., Vázquez-Manrique, R. P., & Mullor, J. L. (2022). Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease. International Journal of Molecular Sciences, 23(16), 8936. https://doi.org/10.3390/ijms23168936