1. Introduction
The massive and often indiscriminate use of antibiotics for decades in human and animal healthcare as well as in agriculture gives rise to antibiotic multi-resistance in bacterial pathogens; this has become a major concern worldwide as it leads to an increasing number of therapeutic failures. According to current projections reported by the World Health Organization (WHO), infectious diseases could once again become one of the leading causes of death in the world by 2050 and cause dramatic damage to the world economy [
1]. The “One Health” concept recognizes the interdependency between the environment, human health and animal health; it is promoted by various international organizations such as the WHO, the Organization for Animal Health (OIE), and the Food and Agriculture Organization (FAO) (see for a definition the FAO website at
https://www.fao.org/one-health/en) (accessed on 14 June 2022). One Health aims, among other objectives, to promote the rational use of antibiotics and the development of alternative strategies to tackle bacterial infections.
Bacteriophages (or phages) are viruses that infect bacteria and thus represent, as anti-bacterial agents, a promising alternative (or complement) to antibiotics. A given bacteriophage specifically infects a more or less narrow range of bacteria, sometimes down to the species or serotype level. Furthermore, bacteriophages are very diverse and generally easy to isolate and produce. Bacteriophages were independently discovered during the First World War by Frederick W. Twort in 1915 [
2] and by Félix d’Hérelle in 1917 [
3], who coined the term “bacteriophage” (see Félix d’Hérelle seeding article presented in English by Dr. Roux [
4]). Very rapidly in 1918, when antibiotics did not even exist, Félix d’Hérelle foresaw how bacteriophages could be used against bacterial infections and successfully utilized them to treat dysentery [
5]. Phage therapy was born and developed to treat both humans and animals. However, in the 1940s, phage therapy was supplanted by the emergence of antibiotics and was abandoned in the Western world, but it was kept in use until today in the former Eastern Bloc countries and former USSR member states [
6]. Nowadays, there is a renewed interest worldwide for bacteriophages and phage therapy in the search for an alternative to antibiotics to tackle multi-resistant bacteria and develop targeted antibacterial therapies.
Salmonella enterica is a Gram-negative zoonotic bacterium and a major cause of diarrhea worldwide. Avian breeding farms are the main reservoirs for the
Salmonella species, transmitted to humans by food, especially eggs. In the concept of “One Health”, treating these reservoirs would have a positive impact on human health. Isolation and characterization of phages targeting
Salmonella are thus important to develop
S. enterica biocontrol strategies and prevent massive antibiotic resistance in animal reservoirs. Phage therapy relies essentially on the formulation of a cocktail, regrouping several different phages in order to avoid the emergence of bacterial resistance, especially cross-resistance, and to eventually target as wide a range of bacterial serotypes as possible. Another beneficial property of bacteriophage cocktails is that they can be designed to target specific pathogens, leaving the rest of the microbiota unaffected, which is not the case with antibiotics and their indiscriminate action against a wide range of bacteria. A recent and relevant study by Nale et al. highlights the synergistic effects of a three-phage cocktail against prevalent
Salmonella serotypes on poultry and pigs [
7]. Using
Galleria mellonella as a model that correlates to large-scale animal models, the authors proved that, in combination, their 3 phages could lyse about 99.97% of the 22 serotypes they investigated. They also showed in vivo in the
Galleria mellonella model that their cocktail was very efficient as a prophylactic agent. Closer to human health issues is the recent report of the successful treatment of a patient infected by a multi-drug resistant
Mycobacterium abscessus strain by a patient-tailored three-phage cocktail [
8]. However, the authors cautiously warn that a generalized approach is far from being a reality.
Just like antibiotherapy, phage therapy collides with bacterial resistance to phage infection. Its success relies on the evaluation of such a probability, implying that we should not reproduce the errors we made with antibiotics. Indeed, the co-evolution of bacteria and their viruses for eons led to the adaptation and acquisition of a high diversity of bacterial anti-phage strategies. The “pan-immune system” of bacteria comprising anti-phage defenses has been nicely reviewed by Bernheim and Sorek [
9]. These strategies evolved by bacteria can be as simple as selecting for mutations in the phage receptor and preventing phage adsorption by modifying and adapting its lipopolysaccharide (LPS). Of greater concern are mutations in non-receptor host factors that can lead to phage cross-resistance, as was shown recently in
S. enterica serovar Typhimurium [
10]. Bacteria can also resort to elegant, more complex and specialized defense systems once the phage genome has been injected, such as Restriction-Modification (RM) [
11], CRISPR-Cas [
12], abortive infection (Abi) [
13] comprising the recently described cyclic-oligonucleotide-based anti-phage signaling system (CBASS) [
14], and bacteriophage Exclusion (BREX) [
15,
16,
17] systems to name but a few. These systems are often passed on between bacteria via horizontal gene transfers. As we keep discovering new anti-phage defense systems, the selection of phages and the formulation of a cocktail should take bacterial defense against phage infection into consideration. Undoubtedly, co-evolution works both ways, and phages, therefore, evolved their own arsenal to counter cellular defenses. One can cite anti-RM or anti-CRISPR proteins [
18] as well as the newly discovered anti-CBASS and anti-Pycsar proteins [
19,
20]. Given the diversity of anti-phage defense systems, a matching diversity of yet unknown anti-cellular defense functions is probably buried in the phage genomic “dark-matter” awaiting discovery. The phage “dark-matter” encompasses all those predicted genes in phage genomes and metaviromes with unknown functions [
21]. It is important to collect as much information as possible on the phage genomes to select the best candidates when foreseeing phage therapy and to keep a balance between killing efficiency, susceptibility against cellular defenses, and potential antagonistic interactions between the therapeutical phages. A thorough genomic characterization of phage genomes, and especially functional annotation, are thus of crucial importance for the successful deployment of phage-based biocontrol strategies.
In this study, we sequenced and analyzed 10 new bacteriophage genomes infecting
Salmonella enterica subsp.
enterica serovar Typhimurium strain ATCC 14028S. These phages were isolated from wastewater and fresh water pounds in the Sevilla area in Spain. Most phages originate from a study by Olivenza et al. [
22] aimed at developing epigenetic phage biosensors used to identify and select phages from different natural environments. Others belong to the personal collections of Dr. M. Ansaldi and Prof. J. Casadesús. To improve phage gene detection and annotation, we benchmarked five different gene callers for syntaxic annotation. We could ascribe a taxonomic affiliation for each phage at the genus level, thanks to genome-wide comparative analyses enabled by vContact2 [
23] (based on the gene-sharing network) and VICTOR (based on genome-wide phylogeny) [
24]. For functional annotation of the predicted ORFs, we combined complementary approaches based on remote homologies detection using Hidden Markov Model (HMM) protein profiles comparisons with PHROG [
25], a newly published database of viral protein clusters, and the pdb70 database at the Protein Data Bank [
26]. Thirdly, we specifically searched for anti-CRISPR proteins (Acr) using the integrated online platform AcrHub [
27], relying on machine learning for Acr prediction. Acr are indeed valuable assets when foreseeing phage therapy. We further mined our annotated genomes for functions that may favor or, on the contrary, disfavor the selection of a phage for phage therapy.
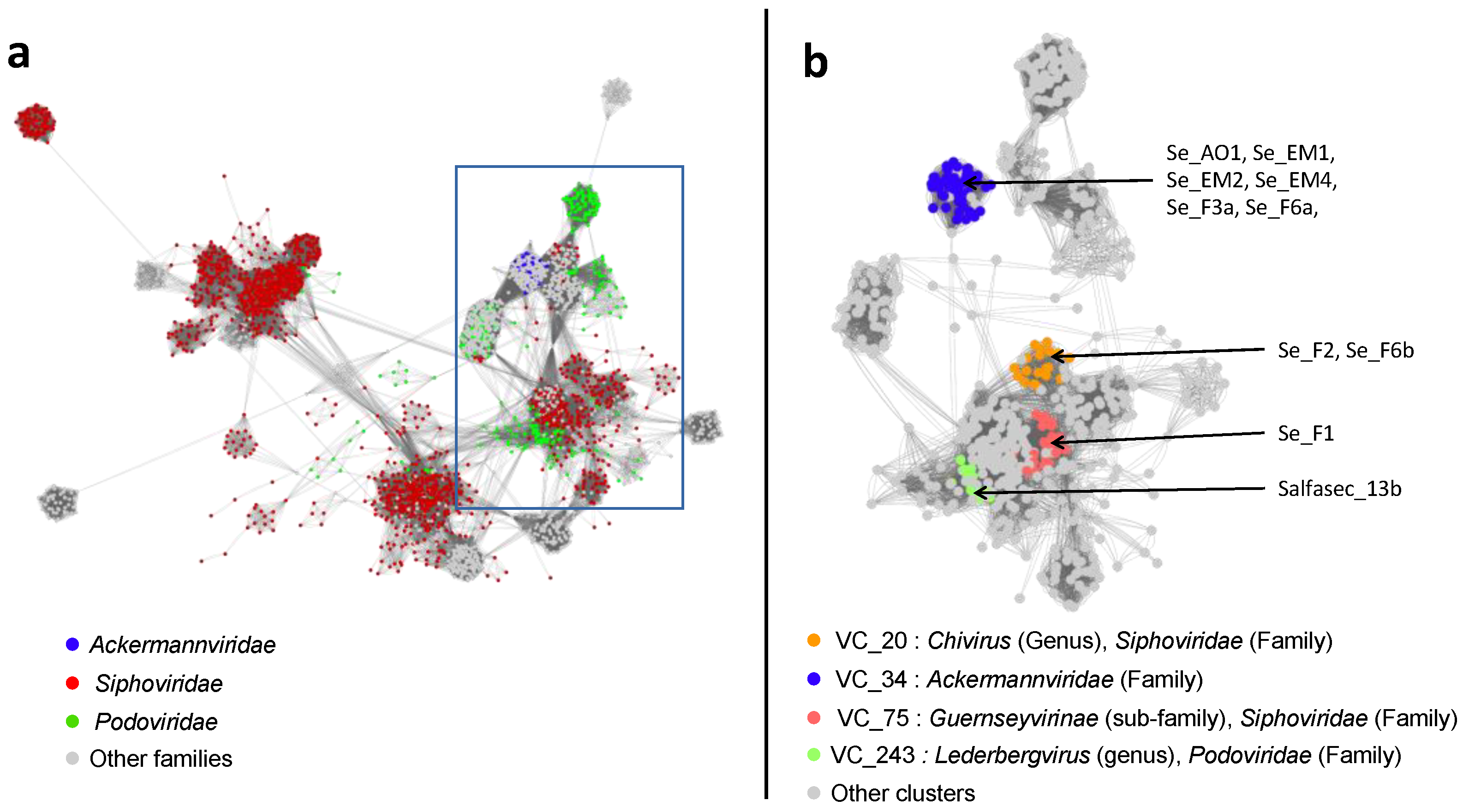
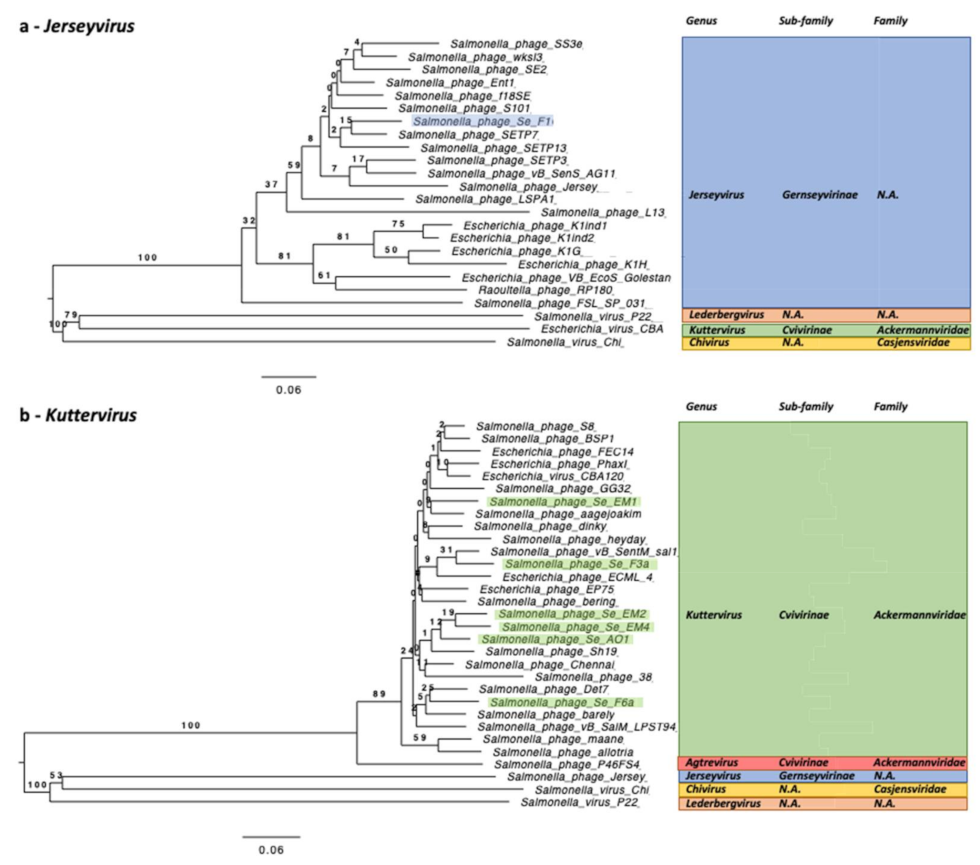
The 10 newly isolated phage strains studied here all belong to the Caudoviricetes and are spread among four genera (Kuttervirus, Chivirus, Jerseyvirus, and Ledergbervirus) with a majority of six Kuttervirus phages isolated (Ackermannviridae). Eight of these ten strains are new species. One phage, Salmonella phage Salfasec13b (Lederbergvirus), was predicted as temperate, which is not a desired property for phage therapy. Among the most interesting features, we found phage-encoded proteins targeting the cellular defenses against phage infections, such as the Restriction-Modification, CRISPR-Cas, and Abortive infection systems. It is noteworthy that, Salmonella phage SeF3a (Kuttervirus) is likely to code for a novel anti-Abi system derived from the bacterial Phage Shock Protein A (PspA), making this phage an interesting candidate for phage therapy with two different anti-Abi systems, one found in all our Kutterviruses previously known to allow bacteriophage T4 to resist the Rex Abi system encoded in some prophages, and the second found in Salmonella phage SeF3a hypothesized in this study. We incidentally could propose functional annotations for 24 protein families in the PHROG database currently annotated “unknown function”. Altogether, this work highlights the need to design specific pipelines for phage genomic analyses to unravel the phage genomic “dark matter” and illustrates the importance of prior knowledge of functions encoded in phage genomes for an educated selection of phage candidates for therapeutic cocktails.
3. Materials and Methods
3.1. Phage Lysates and Bacterial Strain
Se_AO1, Se_EM1, Se_EM2, Se_EM3, Se_EM4, Se_F1, Se_F2, Se_F3, Se_F6, Salfasec_9, Salfasec_10, Salfasec_11, and Salfasec_13 culture lysates were kindly provided by Dr. Olivenza (Departamento de Genética, Facultad de Biologia, Universidad de Sevilla, 41012 Sevilla, Spain). All phages were isolated in the Sevilla area (Spain) from wastewater or freshwater pounds. When required, Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028S (ATCC 14028S) was used to propagate and titrate bacteriophages. Cells were grown aerobically in either Lysogeny Broth (LB) at 37 °C under shaking (180 rpm) for liquid cultures or on LB-1% agar plates incubated at 37 °C.
3.2. Phage Propagation, Purification, and Titration
To propagate phages from culture lysates, 200 mL Erlenmeyer flasks containing 25 mL of LB were inoculated with an overnight (ON) culture of ATCC 14028S at an optical density measured at 600 nm (OD
600nm) of 0.04. Cells were grown until OD
600nm reached 0.4, then inoculated with 100 µL of 0.22 µm-filtered and chloroform-treated culture lysate. The culture was further incubated for about 4 h. Cells were lysed by the addition of 10% chloroform (vortexed for 15 s and incubated for 5 min at room temperature, repeated twice). The aqueous supernatant (phage lysate) was recovered after centrifugation (7500×
g, 15 min) to pellet the cell debris and separate the aqueous phase from the solvent. The phage lysate was then 0.22 µm-filtered and stored at 4 °C. Phage titration by double agar overlay plaque assay was performed according to Kropinski et al. [
61]. Phage titers are expressed in Plaque Forming Unit per mL (PFU/mL). For further phage purification and concentration, all centrifugation steps were done at 4 °C, 20,800×
g for 1 h, and ice-cold buffers were used. A total of 4 mL of phage lysate (titer between 1.10
10 and 3.10
11 PFU/mL) were centrifuged, and the pellet was resuspended in 2 mL of 0.22 µm-filtered Phage Buffer (PB: 10 mM Tris-HCl pH 7.5, 100 mM NaCl, 25 mM MgCl
2, 1 mM CaCl
2). Phages were pelleted once again and resuspended in 200 µL of PB. Purified phage suspensions were stored at 4 °C.
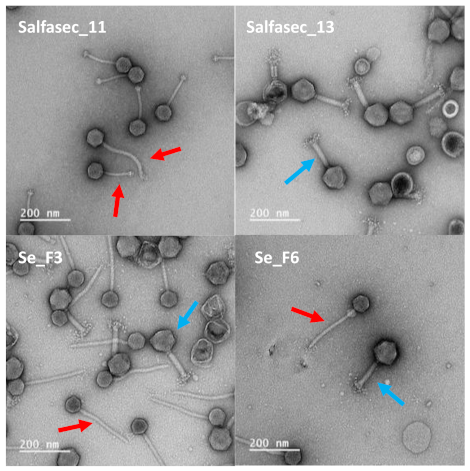
3.3. Phage Morphological Characterization by TEM
For Transmission Electronic Microscopy (TEM) exploration, the final phage pellet resuspension was performed in 2 mL of 0.22 µm-filtered TEM buffer (0.1 M ammonium acetate pH 7). Phages were pelleted once again and resuspended in 20 µL of TEM buffer. Formvar/carbon-coated copper grids were prepared at the Institut de la Méditerranée (IMM) Microscopy facility. A total of 5 µL of the phage solution was pipetted onto the grid surface and allowed to sediment for 3 min at RT. Excess liquid was then blotted and grids negatively stained according to Ackermann’s protocol with 2% uranyl acetate. Observations were made on an FEI Tecnai G2 20 TWIN (200KV), laB6, Gatan Oneview 4k × 4k CMOS transmission electron microscope. Images were visualized using FIJI [
62].
3.4. Phage DNA Purification
To remove non-encapsidated nucleic acids, 20 µL of DNAse I (6 mg/mL, EUROMEDEX, Souffelweyersheim, France), 2 µL of RNAse A (4 mg/mL, Promega), and 4 µL of DpnI restriction enzyme (20,000 U/mL, New England Biolabs, Ipswich, MA, USA) were added to 200 µL of purified phages in PB. The sample was then incubated for 1 h at 37 °C. DNAse I was inactivated by incubating the sample for 20 min at 65 °C under shaking. Phage DNA was released from the capsid by adding 20 µL of 10% SDS and 20 µL of proteinase K (50 µg/mL, EPICENTRE, LGC, Teddington, UK) and incubating the sample for 1 h at 56 °C. The sample volume was completed with PB buffer q.s. 600 µL. Phage DNA was extracted by adding 600 µL phenol/chloroform/isoamyl alcohol (25:24:1, Sigma-Aldrich, St. Louis, MO, USA) and then vortexing the sample for 30 s. The upper aqueous phase containing the nucleic acids was recovered after centrifugation (10,000× g, 10 min, 4 °C). The extraction was repeated twice. The aqueous phase-containing DNA was then ethanol-precipitated. The DNA pellet was finally resuspended in 20 µL DNAse/RNase-free water and stored at −20 °C. Phage dsDNA was quantified with a Qubit™ fluorometer in combination with the Qubit™ dsDNA HS Assay kit (Invitrogen).
Phage genomic DNA purified from Se_F1, Se_F2, Se_F3, Se_F6, and Salfasec_11 phage lysates was mechanically fragmented in 50 µL microtubes using a Covaris M220 sonifier with the following parameters: peak power 75W, duty factor 10%, Cycles/Burst 200.
Phage genomic DNA purified from Se_AO1, Se_ML1, Se_EM1, Se_EM2, Se_EM3, Se_EM4, Salfasec_9, Salfasec_10, and Salfasec_13 phage lysates was enzymatically fragmented by the tagmentase technology from the NEXTERA XT kit (Illumina®, San Diego, CA, USA) according to the manufacturer’s protocol.
DNA libraries for high throughput sequencing were prepared from the fragmented DNA at the IMM Transcriptomic and Genomic facility with the NEBNext® Ultra™ II DNA Library Prep kit for Illumina® (New England Biolabs, Ipswich, MA, USA) for Se_F1, Se_F2, Se_F3, Se_F6, Salfasec_11, and with the NEXTERA XT kit (Illumina®, San Diego, CA, USA) for Se_AO1, Se_ML1, Se_EM1, Se_EM2, Se_EM3, Se_EM4 according to the manufacturer’s protocols.
Salfasec_9, Salfasec_10, and Salfasec_13 DNA libraries preparation with the NEXTERA XT kit (Illumina®, San Diego, CA, USA) was subcontracted to AllGenetics (A Coruña, Spain).
3.5. High Throughput DNA Sequencing
Prior to sequencing, Se_AO1, Se_EM1, Se_EM2, Se_EM3, Se_EM4, Se_ML1, Se_F1, Se_F2, Se_F3, Se_F6, and Salfasec_11 DNA libraries were quantified with a Qubit™ dsDNA HS Assay kit (Invitrogen, Waltham, MA, USA) and their size distribution profiles recorded with the TapeStation 4200 System (Agilent) in combination with the D5000 DNA ScreenTape System (Agilent, Santa Clara, CA, USA). Libraries were then diluted at 4 nM in the appropriate buffer. Paired-end (2 × 150 bp) DNA sequencing was performed on the MiSeq sequencer (Illumina®, San Diego, CA, USA) hosted at the IMM Transcriptomic and Genomic facility with a MiSeq v2 (300-cycles) flow cell (Illumina®, San Diego, CA, USA) according to the manufacturer’s protocol.
Salfasec_9, Salfasec_10, and Salfasec_13 DNA libraries sequencing was subcontracted to AllGenetics (Coruña, Spain).
Raw sequencing reads (FASTQ files trimmed from their Illumina adaptors) for each BioSample were submitted to the NCBI Sequence Read Archive under the BioProject accession number PRJNA767534. Raw sequencing reads quality was then improved with Trimmomatic [
63] using the following parameters: SLIDINGWINDOW:4:25 MINLEN:75 for Se_AO1, Se_ML1, Se_EM1, Se_EM2, Se_EM3, and Se_EM4 or ILLUMINACLIP TruSe3-SE.fasta SLIDINGWINDOW:4:28 MINLEN:75 for all other phages. Only trimmed paired-reads were used for de novo genome assembly.
3.6. Genome de Novo Assembly
Genome de novo assembly was performed with SPAdes 3.14.1 with default parameters.
Table A1 (
Appendix A) summarizes the size of the 18 meaningful contig(s) obtained for the 14 BioSamples together with their mean genome coverage. By “meaningful”, we mean contigs with significant sequence coverage and length compatible with a phage genome (50 kb average size for dsDNA phages) and contigs that are not contaminants. Contaminants are usually small contigs with low sequencing coverage and whose DNA sequences match with unrelated organisms (Blastn analyses). For most contigs, we could identify identical 77 bp-DNA sequences at each contig extremity due to the SPAdes assembly algorithm. We observed this with circularly permuted genomes. We later removed the right-most copy to obtain the final contigs we considered as circular for downstream syntaxic annotation, although we are well aware that the encapsidated biological DNA molecule is linear. Indeed, we found out that some genes were spanning the contig extremities. Such was the case for Se_F2 and Se_F6b genomes.
3.7. Genomes Clustering
We used VIRIDIC (Available online:
http://rhea.icbm.uni-oldenburg.de/VIRIDIC/) (accessed on 27 July 2022) to cluster the 18 contigs with 95% and 70% for the species and genus threshold, respectively, and the following BLASTN parameters: ‘-word_size 7-reward 2-penalty-3-gapopen 5-gapextend 2’. Genomes from the
Kuttervirus,
Chivirus,
Jerseyvirus, and
Lederbergvirus sanctioned by the ICTV (Master Species List 2021_v2) were downloaded from NCBI.
3.8. Prediction of DNA Packaging
We used PhageTerm [
64] to predict putative phage genome termini as well as the DNA packaging mode. PhageTerm is based on the statistical analysis of the sequence coverage after mapping the short sequencing reads onto the assembled contig. For a statistically sound result, PhageTerm requires a minimum sequence coverage of 50. Obviously, PhageTerm requires that the genome extremities are sequenced, and this depends on the technology used to construct the DNA libraries for sequencing. Genome extremities can be recovered when the phage DNA has been mechanically fragmented, as described above. This was the case for Se_F1, Se_F2, Se_F6, and Salfasec_11. When the tagmentation has been used to construct the libraries (Nextera XT kit from Illumina
®, San Diego, CA, USA), one cannot recover the extremities of a linear dsDNA, which precludes termini identification by PhageTerm when DNA packaging is initiated by the terminase at fixed positions (
cos or
pac sites) and ends at a fixed position (
cos site). Tagmentation was used for Se_AO1, Se_ML1, Se_EM1, Se_EM2, Se_EM3, Se_EM4, Salfasec_9, Salfasec_10 and Salfasec_13.
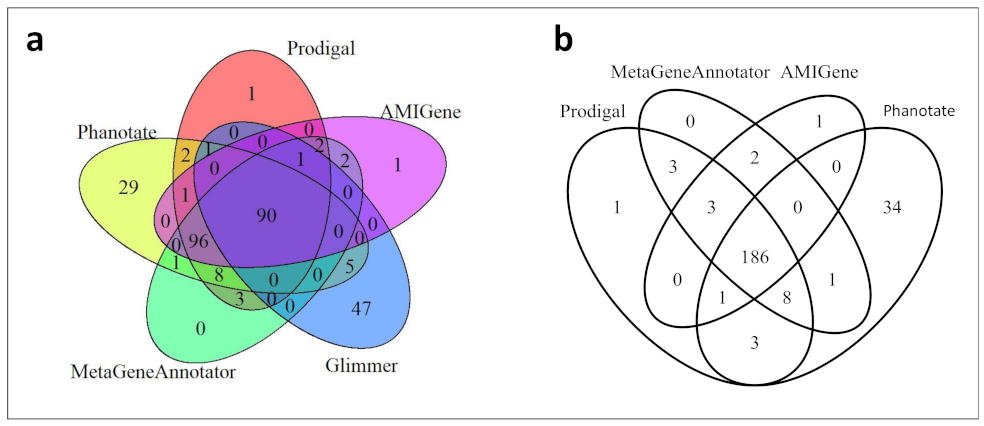
3.9. Syntaxic Annotation
For tRNA prediction, we used ARAGORN [
65]. For Open Reading Frames (ORF) prediction, we tested five different gene callers. Indeed, many gene callers are freely available to predict ORFs from nucleic acid sequences, but they rely on different algorithms, leading to variable predictions. We selected four of them optimized for bacteria (AMIGene, Glimmer, MetaGeneAnnotator, and Prodigal) and one optimized for phage genomes (Phanotate) [
66,
67,
68,
69,
70].
Locus tag names were built with the same convention for each phage: an alphanumeric string defining the phage name coupled with an alphanumeric string defining the gene product numbering. For instance, Se_AO1_GP_001 refers to the first ORF predicted for Salmonella phage SeAO1. When present, tRNAs were labeled following the same convention: Se_AO1_tRNA1 refers to the first tRNA predicted for the Salmonella phage SeAO1 genome. To ease further genomic comparisons, we decided to start the ORFs numbering with the terminase small subunit-encoding ORF as gp_001. All DNA sequences were flipped and/or rotated accordingly.
3.10. Taxonomic Classification
For taxonomic classification, we used vContact2 0.9.19 (with the ProkaryoticViralRefSeq201 database accessed on 27 September 2021) and VICTOR (Virus Classification and Tree Building Online Resource with the amino acid option selected, Available online:
https://ggdc.dsmz.de/victor.php) (accessed on 15 November 2021) Network visualization was done with Cytoscape [
71].
3.11. Detection of Protein Sequence Remote Homologies
We generated Hidden Markov Model (HMM) profiles for each predicted protein sequence of each phage with HHblits from the UniRef30_2020_06 database in order to detect sequence remote homologies [
72,
73].
3.12. PHROG-Based Functional Annotation
For functional annotation, we used the newly published PHROG database (accessed on 27 September 2021) dedicated to viral proteins. In this database, viral proteins are distributed among families called “phrog”; each phrog represents a cluster of viral proteins orthologs built using remote homology detection by HMM profile–profile comparisons. At the date of its publication, the PHROG database contains 17,473 (pro)viruses of prokaryotes or archaea for a total of 938,864 proteins. A total of 38,880 clusters were defined, and manual inspection led to the annotation of 5108 phrog clusters, representing 50.6% of the total protein dataset. Following the procedure available on the PHROG website (
https://phrogs.lmge.uca.fr/READMORE.php), we used HH-suite [
74] to compare each predicted gene product from the phages studied here with the PHROG database and ascribe a phrog and its annotation whenever possible. When several hits were found, we kept the best one for the considered protein. The corresponding phrog number and its functional annotation were then transferred to our newly predicted protein. When no hit with the PHROG database was found, or the affiliation to an existing phrog was too distant (probability < 80%, Evalue > 1 × 10
−4), the ORF was annotated as a singleton of unknown function according to the PHROG guidelines. The entire list of the best hit for each ORF for each phage before manual curation is supplied in
Table S5.
3.13. PDB-Based Functional Annotation
In order to improve the PHROG-based functional annotation for protein still annotated “unknown function”, we performed with HHblits a comparison of each predicted protein sequence HMM profile with the Protein Data Bank pdb70 database (accessed on 28 September 2021) to detect homologies with known protein structures. We kept the first hit for each protein sequence for further consideration when the probability was greater than 80% and the Evalue less than 1 × 10
−3. When these criteria were met, we also manually checked the prediction coverage to avoid transferring a PDB annotation based on a predicted structural similarity between a small portion (a domain or a sub-domain) of the PDB hit and the phage protein sequences. After all these considerations, we manually checked for phage ORF ascribed to a phrog of unknown function if a reliable structural prediction from the pdb70 could be used instead. If so, a new annotation inferred from the structural prediction was transferred to the corresponding phrog of unknown function (see
Table 4). A new annotation for this phrog cluster will be proposed to the PHROG database through a dedicated form on its website (
https://phrogs.lmge.uca.fr/suggestions.php).
3.14. ORFs Manual Curation
Phage genes sometimes overlap or may even be entirely included in one another. Nevertheless, in some cases, these overlaps can reveal misprediction from the syntaxic annotation. Thus, in an attempt to reduce false positive predictions, we manually scanned each genome to detect highly overlapping ORFs. The most common situation we encountered was two highly overlapping ORFs, with one ORF coded on one strand with a strong affiliation to a phrog family while the ORF encoded on the opposite strand had a weak affiliation to a phrog family (probability < 70%, Evalue > 1 × 10
−4). If the latter did not show any strong homology with a known structure from the PDB (probability > 80%, Evalue < 1 × 10
−3), we decided to discard the ORF. As an example of manual curation, for Salmonella phage SeAO1,
gp_014 and
gp_016 almost entirely overlap with
gp_015, all 3 ORFs predicted on the same strand. According to the phrog affiliation metrics generated by HHblits (see
Table S5), Gp_015 is strongly affiliated to phrog_519 (Prob. 99.5%, Evalue 1.4 × 10
−18, 261 proteins sequences in this phrog family), whereas Gp_014 is weakly affiliated to phrog_32723 (Prob. 55%, Evalue 1.1, 2 proteins sequences in this phrog family) and Gp_016 is weakly affiliated to phrog_8445 (Prob. 23.5%, Evalue 7.4, 13 proteins sequences in this phrog family). In this example, we discarded
gp_014 and
gp_016. In other examples, such as
gp_212,
gp_213, and
gp_214, it was not possible to decide which ORF was significant based on the phrog affiliation metrics; all three ORFs, although overlapping, code for proteins strongly affiliated to a phrog family. In this case, we chose to keep all three ORFs. The last example of manual curation is
gp_222. Based on phrog affiliation metrics, we classified Gp_222 as a “singleton” of unknown function. However,
gp_222 completely overlaps with the reliably predicted tRNA6-Val on the same strand; we, thus, decided to discard
gp_222.
Table 5 summarizes the final gene content predicted for each phage after manual curation. This procedure was carried out for every genome included in this study.
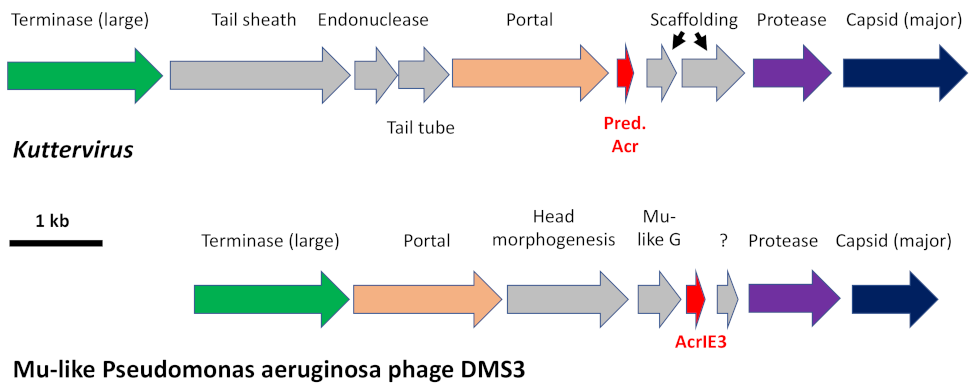
3.15. Prediction of Anti-CRISPR Proteins (Acr)
Until very recently, searching for Acr was tedious, essentially relying on “guilt by association” with anti-CRISPR-associated (Aca) proteins containing “helix-turn-helix” motives. However, not all Acr are associated with an Aca. Thanks to the new approaches based on machine learning, it is now possible to predict Acr found in undescribed genomic environments. AcrHub is a platform that incorporates state-of-the-art Acr predictors and three analytical modules (similarity analysis, phylogenetic analysis, and homology network analysis) [
27]. The AcrHub database contains 339 experimentally validated, and 71,728 predicted Acr proteins. Validated Acr are mostly short proteins (89% between 60 and 160 aa). We ran our phage proteomes in AcrHub on two different Acr prediction algorithms (PaCRISPR [
75] and AcRanker [
76]). Meaningful score thresholds are over 0.5 for PaCRISPR and over −5 for AcRanker. We then used the AcrHub “Similarity analysis” module to relate our predicted Acr with experimentally validated Acr proteins in the AcrHub database. We excluded overlong proteins (>200 aa) and those whose query cover was below 40%. When pertinent, we compared the genomic surrounding of our predicted
acr gene with an experimentally validated
acr gene product.
4. Conclusions
In this study, we present a genomic description of 10 phage strains representing 8 new species isolated in wastewater and freshwater ponds in the Sevilla area in Spain and infecting
S. enterica serovar Typhimurium strain ATCC 14028S. Based on genome-wide analyses independent from prior knowledge on functional annotation, we could ascribe taxonomic classification down to the genus level. These phages all belong to the
Caudoviricetes class (dsDNA-tailed bacteriophages with an HK97 major capsid fold) and to four different genera (
Figure 3 and
Figure 4).
Kuttervirus phages are overrepresented, with six strains identified, five of them being new species. Only Salmonella phage Salfasec13b, belonging to the
Lederbergvirus genus, was predicted as a temperate phage.
We wish to highlight in our study two important methodological points. The first one is that by combining the results of four different gene callers, including Phanotate especially designed for phage genomes (but that nevertheless tends to over-predict ORFs), we believe we could predict as many ORFs as is reasonably possible. However, we also showed that manual inspection and curation are still needed to get rid of obvious false positive predictions. The second point concerns the improvement of functional annotation with PHROG, a database dedicated to viral proteins at large (bacterial and archaeal viruses and their prophages). We believe this is a promising approach for functional annotation as it relies on viral protein orthologs clustering based on remote homology detection. PHROG capitalizes on decades worth of past work of many research teams that have led to the experimental validations of many functions, and PHROG also benefits from the manual annotation of various experts in the field (we could propose in this study a functional annotation to 24 phrog clusters previously annotated with unknown function).
We mined our 10 annotated genomes for relevant functions in the context of future biocontrol application of
Salmonella spp. We could, thus, identify phage-encoded proteins targeting bacterial anti-phage defenses such as Abortive infection, Restriction-Modification, and CRISPR-Cas systems. Our study also illustrates that in otherwise very similar genomes such as our six
Kuttervirus (ANI values between 0.94 and 0.98), we could identify subtle variations in gene equipment that can single out one phage from its closest relatives. Indeed, we predicted that the Salmonella phage SeF3a genome alone codes for a new anti-Abi system based on a phage-encoded Phage Shock Protein A ortholog, potentially conferring Salmonella phage SeF3a a selective advantage over the other five viruses of the same
Kuttervirus genus. This example emphasizes the need to extensively mine bacteriophage genomes in order to tailor therapeutic cocktails matching the targeted pathogen. Conversely, one has to obtain as much genomic information as possible about the pathogen itself in order to identify its anti-phage defenses and select appropriate phages to formulate a therapeutic cocktail that can overcome these cellular defenses. The recent publication by Tesson et al. is helpful in that respect with the description of DefenseFinder, a bioinformatic tool specially designed to systematically predict anti-phage systems in prokaryotic genomes [
77].
The coevolution of bacteria and their viruses for ages beyond count is often described as an arms race that gifted both protagonists with highly diverse molecular weapons and shields. To go beyond empiric approaches and rationally design efficient phage-based biocontrol of bacteria, we need to adopt a holistic approach to the bacteriophage/host binomen. In that respect, genomic and functional analyses of both partners are crucial to meeting the existing challenges in tackling bacterial infections.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}