
NX210c Peptide Promotes Glutamatergic Receptor-Mediated Synaptic Transmission and Signaling in the Mouse Central Nervous System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
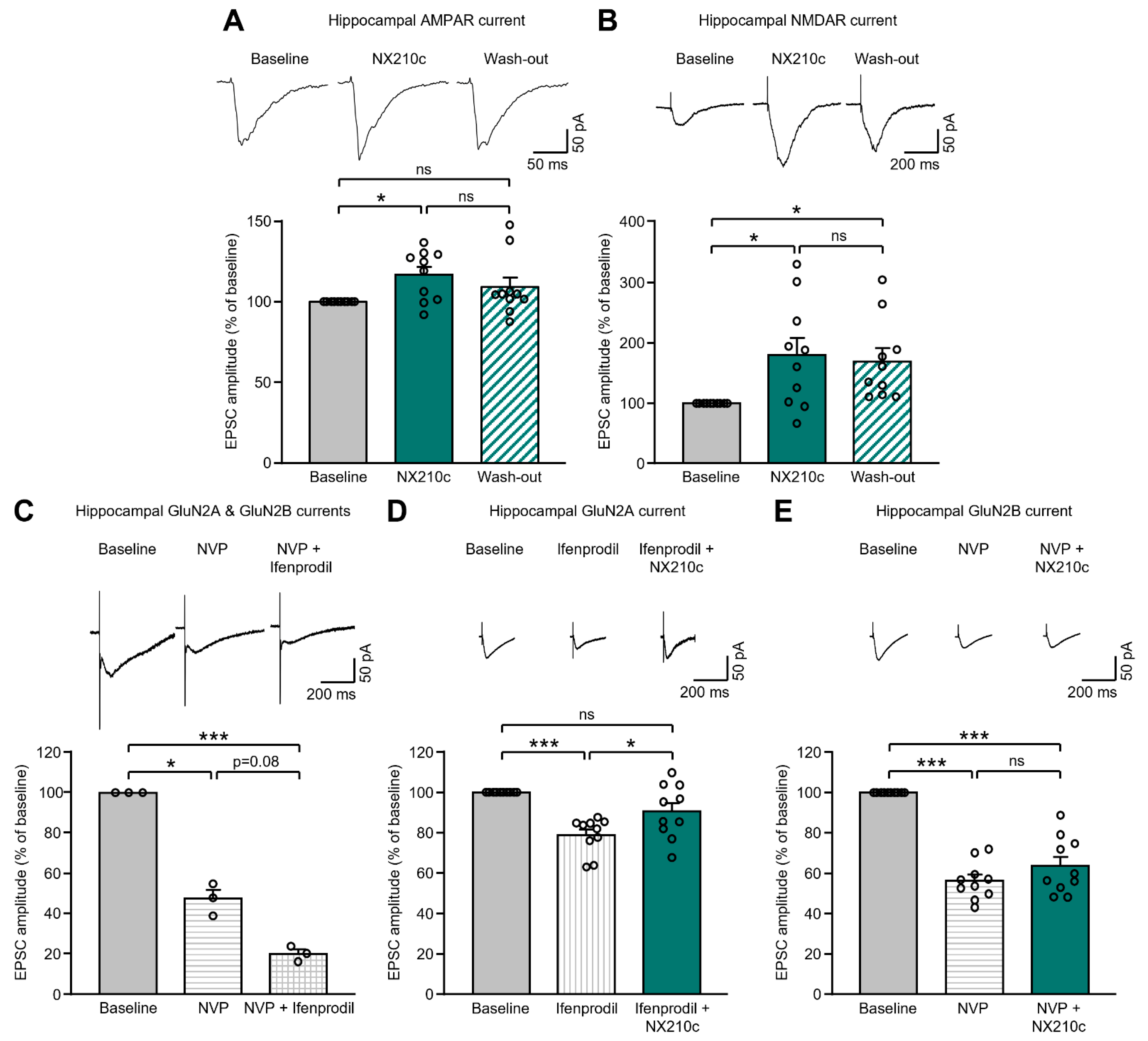
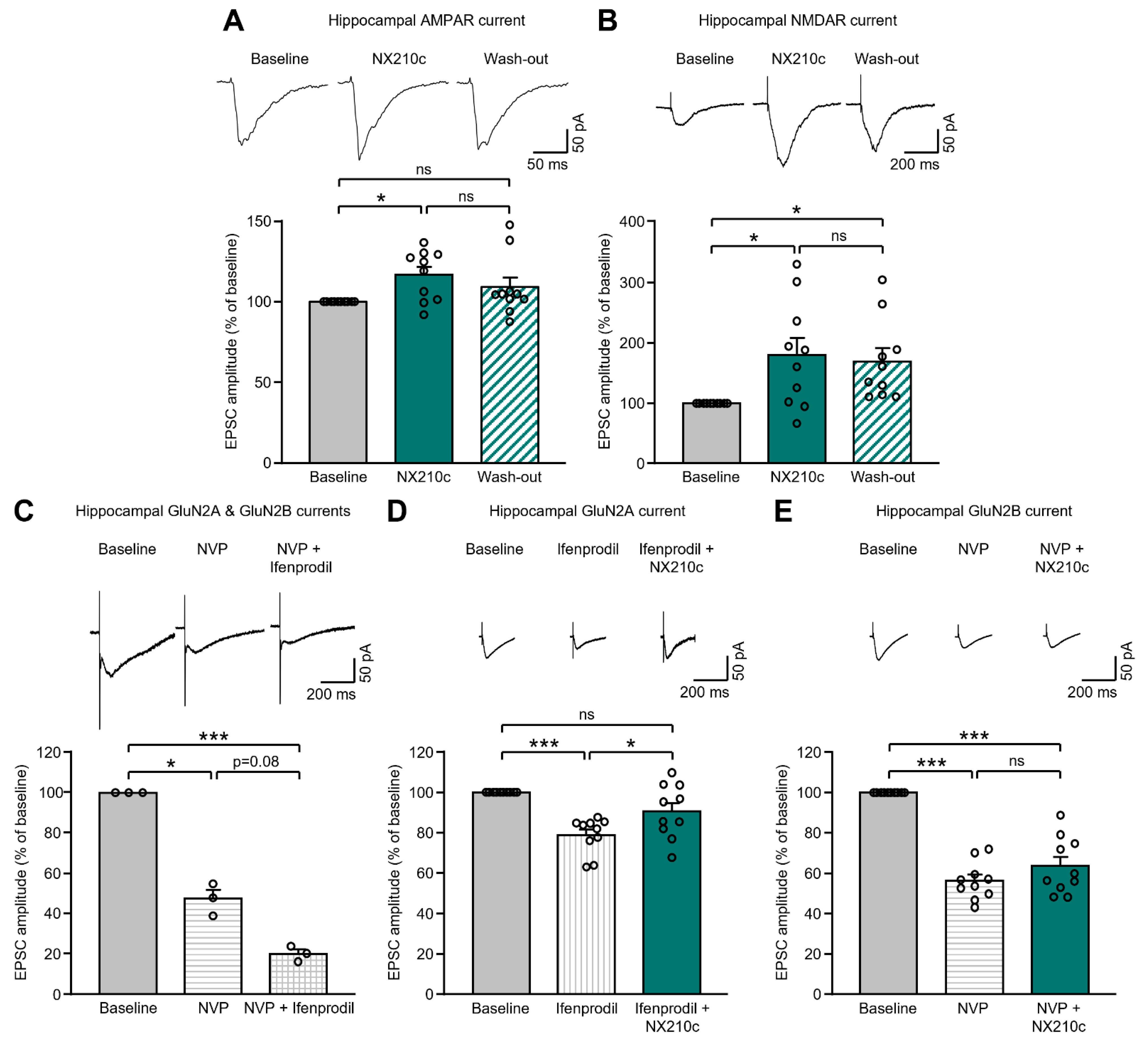
2.1. NX210c Potentiates AMPAR- and NMDAR-Mediated Postsynaptic Currents in the Mouse Hippocampus
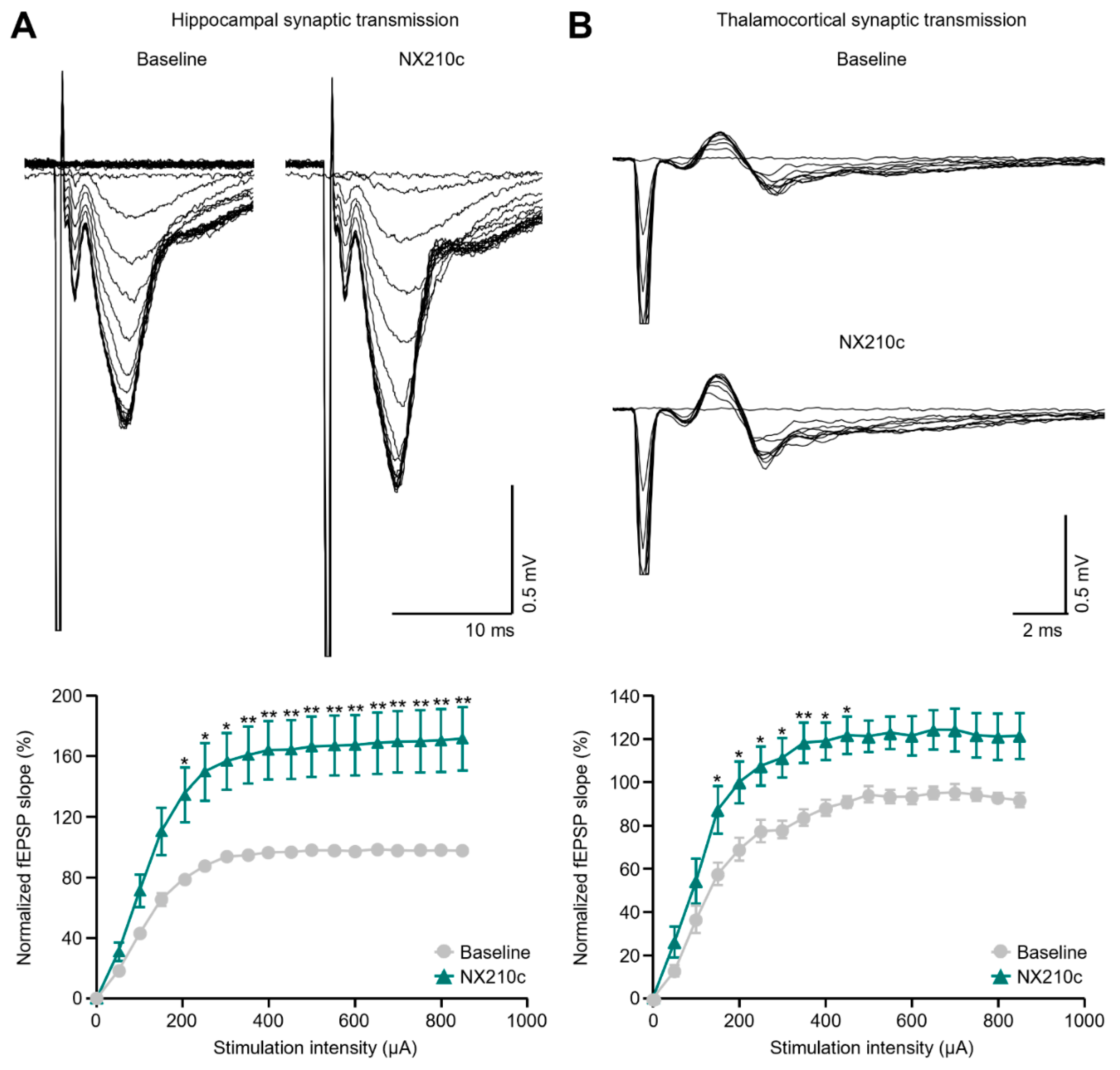
2.2. NX210c Increases Hippocampal and Thalamocortical Basal Synaptic Transmission in Mice
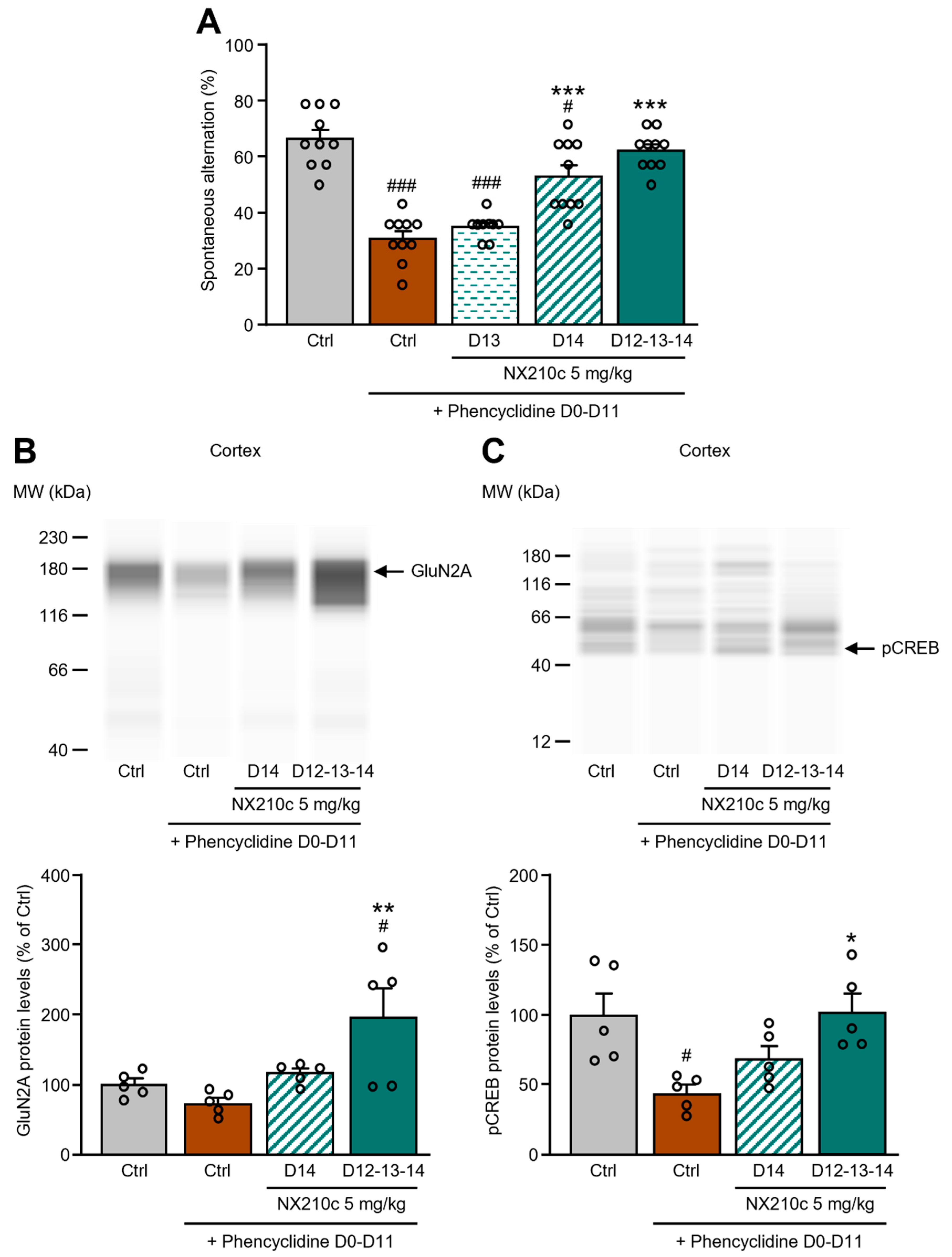
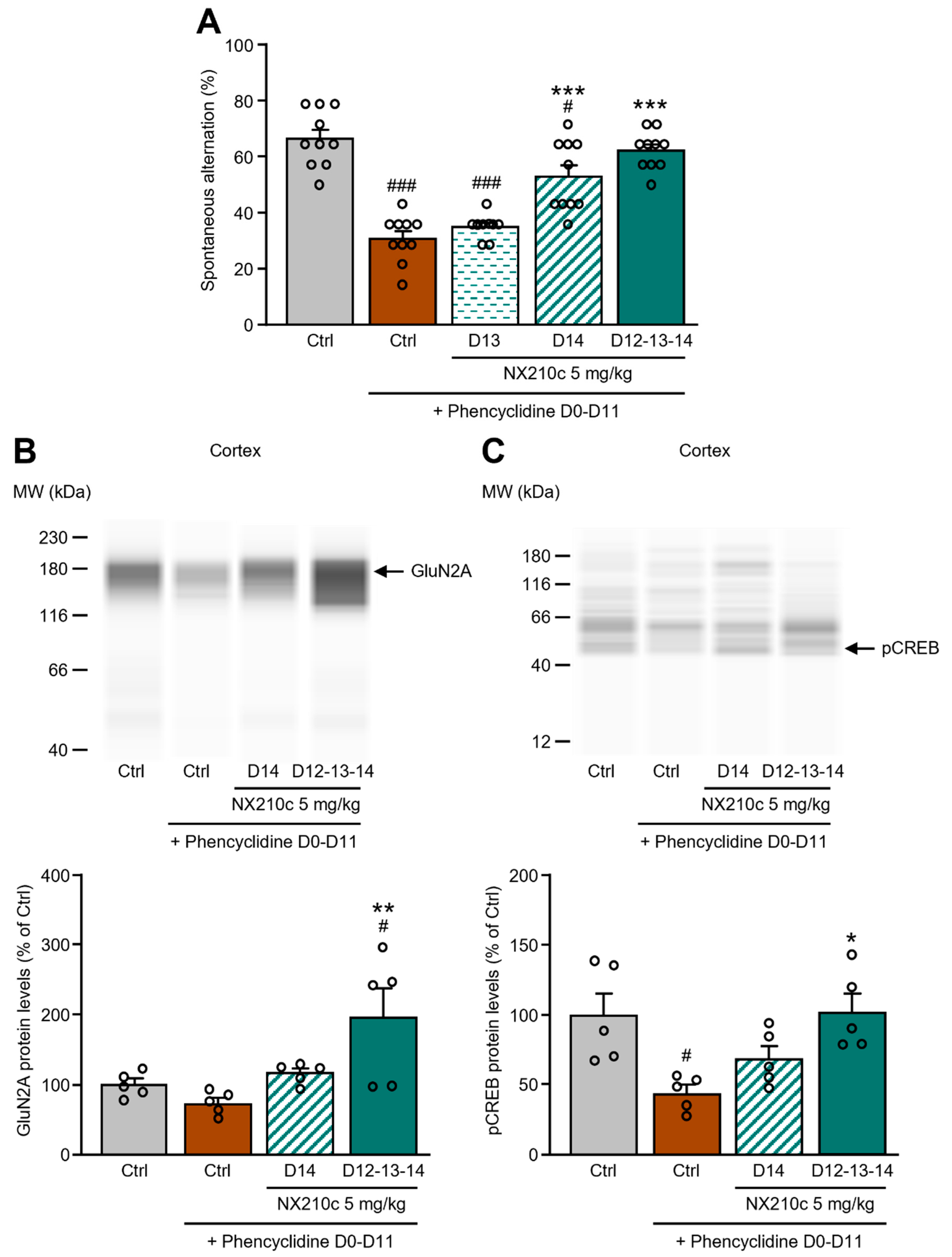
2.3. Acute or Repeated Administrations of NX210c Restored Memory Deficits in a Mouse Pharmacological Model of Synaptic Dysfunction Induced by Chronic Administrations of the NMDAR Antagonist Phencyclidine
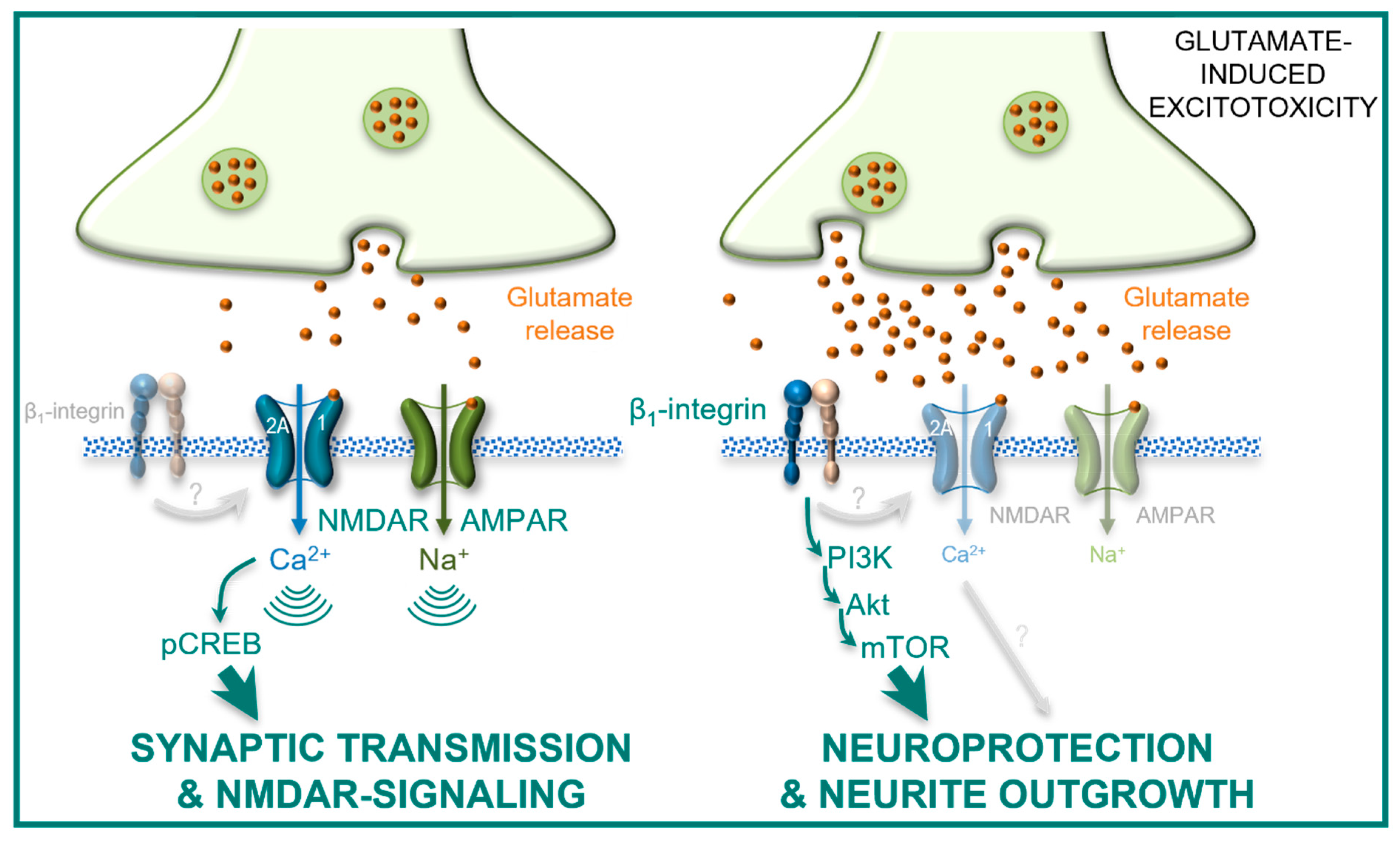
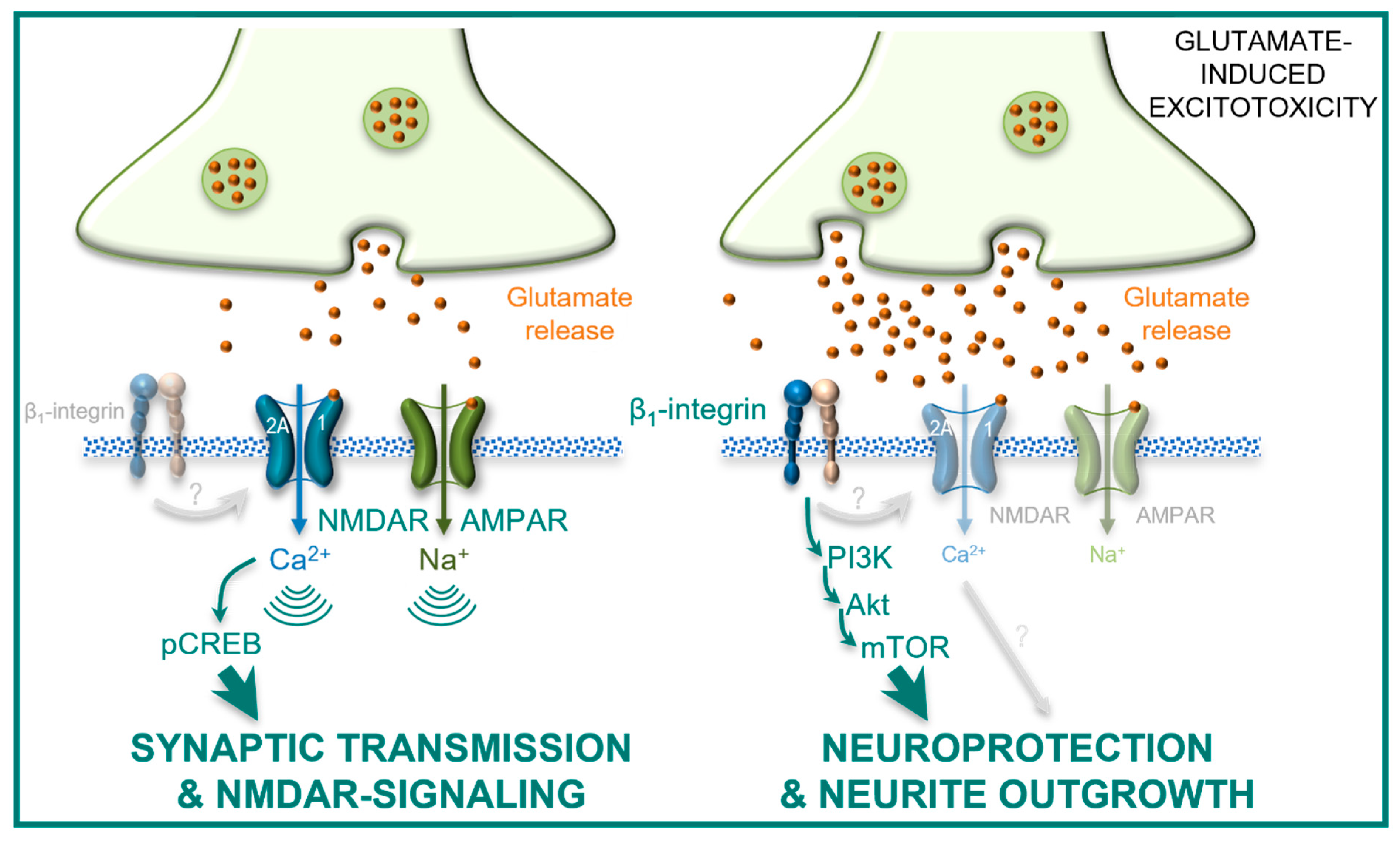
3. Discussion
4. Materials and Methods
4.1. SCO-Spondin-Derived Peptide NX210c
4.2. Animals
4.3. Mouse Pharmacological Model of Synaptic Dysfunction
4.3.1. Treatment Groups
4.3.2. T-Maze Behavioral Test
4.3.3. Western Blot
4.4. Electrophysiology on Mouse Brain Slices
4.4.1. Basal Synaptic Transmission in the Hippocampus Ex Vivo
- -
- Signals that showed a fiber volley amplitude bigger than fEPSP amplitude (low fEPSP amplitude-to-fiber volley ratio (<3));
- -
- Slices that showed a decreasing slope reaching a close to null value during vehicle perfusion;
- -
- Slices that failed to exhibit a linear increasing I/O curve, because increasing stimulation intensities should result in a linear increase in the fEPSP slope until a maximal plateau during vehicle perfusion;
- -
- Slices that failed to show stable fEPSP slopes (>10% change) during the stabilization period preceding I/O assessment;
- -
- Slices that failed to reach acceptable amplitude ranges during vehicle perfusion (0.5 mV minimum).
4.4.2. NMDAR and AMPAR Postsynaptic Currents Ex Vivo
4.4.3. NMDAR Subunit Postsynaptic Currents Ex Vivo
4.4.4. Basal Synaptic Transmission in Thalamocortical Projections Ex Vivo
- -
- Signals that showed a fiber volley amplitude bigger than fEPSP amplitude (low fEPSP amplitude-to-fiber volley ratio (<3));
- -
- Slices that showed a decreasing slope reaching a close to null value during vehicle perfusion;
- -
- Slices that failed to exhibit a linear increasing I/O curve, because increasing stimulation intensities should result in a linear increase in the fEPSP slope until a maximal plateau during vehicle perfusion;
- -
- Slices that failed to show stable fEPSP slopes (>10% change) during the stabilization period preceding I/O assessment;
- -
- Slices that failed to reach acceptable amplitude ranges during vehicle perfusion (0.2 mV minimum).
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sepúlveda, V.; Maurelia, F.; González, M.; Aguayo, J.; Caprile, T. SCO-spondin, a giant matricellular protein that regulates cerebrospinal fluid activity. Fluids Barriers CNS 2021, 18, 45. [Google Scholar]
- Gobron, S.; Monnerie, H.; Meiniel, R.; Creveaux, I.; Lehmann, W.; Lamalle, D.; Dastugue, B.; Meiniel, A. SCO-spondin: A new member of the thrombospondin family secreted by the subcommissural organ is a candidate in the modulation of neuronal aggregation. J. Cell Sci. 1996, 109, 1053–1061. [Google Scholar]
- Oksche, A. The subcommissural organ. Neurohormones Neurohumors 1969, 31, 111–139. [Google Scholar]
- Gonçalves-Mendes, N.; Simon-Chazottes, D.; Creveaux, I.; Meiniel, A.; Guénet, J.L.; Meiniel, R. Mouse SCO-spondin, a gene of the thrombospondin type 1 repeat (TSR) superfamily expressed in the brain. Gene 2003, 312, 263–270. [Google Scholar]
- Tsintou, M.; Dalamagkas, K.; Makris, N. Taking central nervous system regenerative therapies to the clinic: Curing rodents versus nonhuman primates versus humans. Neural Regen. Res. 2020, 15, 425–437. [Google Scholar]
- Monnerie, H.; Dastugue, B.; Meiniel, A. Effect of synthetic peptides derived from SCO-spondin conserved domains on chick cortical and spinal-cord neurons in cell cultures. Cell Tissue Res. 1998, 293, 407–418. [Google Scholar]
- Sakka, L.; Delétage, N.; Lalloué, F.; Duval, A.; Chazal, J.; Lemaire, J.J.; Meiniel, A.; Monnerie, H.; Gobron, S. SCO-spondin derived peptide NX210 induces neuroprotection in vitro and promotes fiber regrowth and functional recovery after spinal cord injury. PLoS ONE 2014, 9, e93179. [Google Scholar]
- Le Douce, J.; Delétage, N.; Bourdès, V.; Lemarchant, S.; Godfrin, Y. Subcommissural Organ-Spondin-Derived Peptide Restores Memory in a Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 651094. [Google Scholar]
- Delétage, N.; Le Douce, J.; Callizot, N.; Godfrin, Y.; Lemarchant, S. SCO-spondin-derived Peptide Protects Neurons from Glutamate-induced Excitotoxicity. Neuroscience 2021, 463, 317–336. [Google Scholar]
- Bourdès, V.; Dogterom, P.; Aleman, A.; Parmantier, P.; Colas, D.; Lemarchant, S.; Marie, S.; Chou, T.; Abd-Elaziz, K.; Godfrin, Y. Safety, Tolerability, Pharmacokinetics and Initial Pharmacodynamics of a Subcommissural Organ-Spondin-Derived Peptide: A Randomized, Placebo-Controlled, Double-Blind, Single Ascending Dose First-in-Human Study. Neurol. Ther. 2022, 11, 1353–1374. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. Methods Mol. Biol. 2017, 1677, 1–80. [Google Scholar]
- Hashimoto, K. Targeting of NMDA receptors in new treatments for schizophrenia. Expert Opin. Ther. Targets 2014, 18, 1049–1063. [Google Scholar]
- Tang, W.; Liu, D.; Traynelis, S.F.; Yuan, H. Positive allosteric modulators that target NMDA receptors rectify loss-of-function GRIN variants associated with neurological and neuropsychiatric disorders. Neuropharmacology 2020, 177, 108247. [Google Scholar]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar]
- Balu, D.T. The NMDA Receptor and Schizophrenia: From Pathophysiology to Treatment. Adv. Pharmacol. 2016, 76, 351–382. [Google Scholar]
- Luo, Y.; Yu, Y.; Zhang, M.; He, H.; Fan, N. Chronic administration of ketamine induces cognitive deterioration by restraining synaptic signaling. Mol. Psychiatry 2021, 26, 4702–4718. [Google Scholar]
- Benitez, D.P.; Jiang, S.; Wood, J.; Wang, R.; Hall, C.M.; Peerboom, C.; Wong, N.; Stringer, K.M.; Vitanova, K.S.; Smith, V.C.; et al. Knock-in models related to Alzheimer’s disease: Synaptic transmission, plaques and the role of microglia. Mol. Neurodegener. 2021, 16, 47. [Google Scholar]
- Conway, M.E. Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 2020, 21, 257–274. [Google Scholar]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar]
- Kumar, S.; Veldhuis, A.; Malhotra, T. Neuropsychiatric and Cognitive Sequelae of COVID-19. Front. Psychol. 2021, 12, 577529. [Google Scholar]
- Finke, C.; Kopp, U.A.; Prüss, H.; Dalmau, J.; Wandinger, K.P.; Ploner, C.J. Cognitive deficits following anti-NMDA receptor encephalitis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 195–198. [Google Scholar]
- Bagnato, S.; Boccagni, C.; Sant’angelo, A.; Fingelkurts, A.A.; Galardi, G. Emerging from an unresponsive wakefulness syndrome: Brain plasticity has to cross a threshold level. Neurosci. Biobehav. Rev. 2013, 37, 2721–2736. [Google Scholar]
- Kumar, A.; Thinschmidt, J.S.; Foster, T.C. Subunit contribution to NMDA receptor hypofunction and redox sensitivity of hippocampal synaptic transmission during aging. Aging (Albany NY) 2019, 11, 5140–5157. [Google Scholar]
- Shi, L.; Adams, M.; Brunso-Bechtold, J.K. Subtle alterations in glutamatergic synapses underlie the aging-related decline in hippocampal function. In Brain Aging: Models, Methods, and Mechanisms; Riddle, D.R., Ed.; CRC Press: Boca Raton, FL, USA, 2007; Chapter 8; 24p. [Google Scholar]
- Ge, Y.; Chen, W.; Axerio-Cilies, P.; Wang, Y.T. NMDARs in Cell Survival and Death: Implications in Stroke Pathogenesis and Treatment. Trends Mol. Med. 2020, 26, 533–551. [Google Scholar]
- Neurofit. T-Maze Continuous Alternation Task (T-CAT) in Mice. Available online: https://www.neurofit.com/tech-anim-tmaze.html#pcp (accessed on 26 July 2022).
- Wang, H.; Peng, R.Y. Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil. Med. Res. 2016, 3, 26. [Google Scholar]
- Royo, M.; Escolano, B.A.; Madrigal, M.P.; Jurado, S. AMPA Receptor Function in Hypothalamic Synapses. Front. Synaptic Neurosci. 2022, 14, 833449. [Google Scholar]
- Collingridge, G.L.; Volianskis, A.; Bannister, N.; France, G.; Hanna, L.; Mercier, M.; Tidball, P.; Fang, G.; Irvine, M.W.; Costa, B.M.; et al. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 2013, 64, 13–26. [Google Scholar]
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar]
- Rao, V.R.; Finkbeiner, S. NMDA and AMPA receptors: Old channels, new tricks. Trends Neurosci. 2007, 30, 284–291. [Google Scholar]
- Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. [Google Scholar] [CrossRef] [Green Version]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar]
- Liu, Y.; Ouyang, P.; Zheng, Y.; Mi, L.; Zhao, J.; Ning, Y.; Guo, W. A Selective Review of the Excitatory-Inhibitory Imbalance in Schizophrenia: Underlying Biology, Genetics, Microcircuits, and Symptoms. Front. Cell Dev. Biol. 2021, 9, 664535. [Google Scholar]
- Liguz-Lecznar, M.; Lehner, M.; Kaliszewska, A.; Zakrzewska, R.; Sobolewska, A.; Kossut, M. Altered glutamate/GABA equilibrium in aged mice cortex influences cortical plasticity. Brain Struct. Funct. 2015, 220, 1681–1693. [Google Scholar]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar]
- Bamdad, M.; Volle, D.; Dastugue, B.; Meiniel, A. Alpha1beta1-integrin is an essential signal for neurite outgrowth induced by thrombospondin type 1 repeats of SCO-spondin. Cell Tissue Res. 2004, 315, 15–25. [Google Scholar]
- Wang, W.; Kantorovich, S.; Babayan, A.H.; Hou, B.; Gall, C.M.; Lynch, G. Estrogen’s Effects on Excitatory Synaptic Transmission Entail Integrin and TrkB Transactivation and Depend Upon β1-integrin function. Neuropsychopharmacology 2016, 41, 2723–2732. [Google Scholar]
- Lin, C.Y.; Hilgenberg, L.G.; Smith, M.A.; Lynch, G.; Gall, C.M. Integrin regulation of cytoplasmic calcium in excitatory neurons depends upon glutamate receptors and release from intracellular stores. Mol. Cell. Neurosci. 2008, 37, 770–780. [Google Scholar]
- Huang, Z.; Shimazu, K.; Woo, N.H.; Zang, K.; Müller, U.; Lu, B.; Reichardt, L.F. Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J. Neurosci. 2006, 26, 11208–11219. [Google Scholar]
- Kramár, E.A.; Bernard, J.A.; Gall, C.M.; Lynch, G. Integrins modulate fast excitatory transmission at hippocampal synapses. J. Biol. Chem. 2003, 278, 10722–10730. [Google Scholar]
- Bernard-Trifilo, J.A.; Kramár, E.A.; Torp, R.; Lin, C.Y.; Pineda, E.A.; Lynch, G.; Gall, C.M. Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J. Neurochem. 2005, 93, 834–849. [Google Scholar]
- Kramár, E.A.; Lin, B.; Rex, C.S.; Gall, C.M.; Lynch, G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 5579–5584. [Google Scholar]
- Lin, B.; Arai, A.C.; Lynch, G.; Gall, C.M. Integrins regulate NMDA receptor-mediated synaptic currents. J. Neurophysiol. 2003, 89, 2874–2878. [Google Scholar]
- Zhang, B.; Xiong, F.; Ma, Y.; Li, B.; Mao, Y.; Zhou, Z.; Yu, H.; Li, J.; Li, C.; Fu, J.; et al. Chronic phencyclidine treatment impairs spatial working memory in rhesus monkeys. Psychopharmacology 2019, 236, 2223–2232. [Google Scholar]
- Knox, L.T.; Jing, Y.; Bawazier-Edgecombe, J.; Collie, N.D.; Zhang, H.; Liu, P. Effects of withdrawal from repeated phencyclidine administration on behavioural function and brain arginine metabolism in rats. Pharmacol. Biochem. Behav. 2017, 153, 45–59. [Google Scholar]
- Zhu, S.; Wang, H.; Shi, R.; Zhang, R.; Wang, J.; Kong, L.; Sun, Y.; He, J.; Kong, J.; Wang, J.F.; et al. Chronic phencyclidine induces inflammatory responses and activates GSK3β in mice. Neurochem. Res. 2014, 39, 2385–2393. [Google Scholar]
- Domino, E.F.; Luby, E.D. Phencyclidine/schizophrenia: One view toward the past, the other to the future. Schizophr. Bull. 2012, 38, 914–919. [Google Scholar]
- Jentsch, J.D.; Tran, A.; Le, D.; Youngren, K.D.; Roth, R.H. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology 1997, 17, 92–99. [Google Scholar]
- Rajdev, S.; Fix, A.S.; Sharp, F.R. Acute phencyclidine neurotoxicity in rat forebrain: Induction of haem oxygenase-1 and attenuation by the antioxidant dimethylthiourea. Eur. J. Neurosci. 1998, 10, 3840–3852. [Google Scholar]
- Gigg, J.; McEwan, F.; Smausz, R.; Neill, J.; Harte, M.K. Synaptic biomarker reduction and impaired cognition in the sub-chronic PCP mouse model for schizophrenia. J. Psychopharmacol. 2020, 34, 115–124. [Google Scholar]
- Xia, Y.; Wang, C.Z.; Liu, J.; Anastasio, N.C.; Johnson, K.M. Brain-derived neurotrophic factor prevents phencyclidine-induced apoptosis in developing brain by parallel activation of both the ERK and PI-3K/Akt pathways. Neuropharmacology 2010, 58, 330–336. [Google Scholar]
- Tanqueiro, S.R.; Mouro, F.M.; Ferreira, C.B.; Freitas, C.F.; Fonseca-Gomes, J.; Simões do Couto, F.; Sebastião, A.M.; Dawson, N.; Diógenes, M.J. Sustained NMDA receptor hypofunction impairs brain-derived neurotropic factor signalling in the PFC, but not in the hippocampus, and disturbs PFC-dependent cognition in mice. J. Psychopharmacol. 2021, 35, 730–743. [Google Scholar]
- Arime, Y.; Akiyama, K. Abnormal neural activation patterns underlying working memory impairment in chronic phencyclidine-treated mice. PLoS ONE 2017, 12, e0189287. [Google Scholar]
- Castañé, A.; Santana, N.; Artigas, F. PCP-based mice models of schizophrenia: Differential behavioral, neurochemical and cellular effects of acute and subchronic treatments. Psychopharmacology 2015, 232, 4085–4097. [Google Scholar]
- Molteni, R.; Pasini, M.; Moraschi, S.; Gennarelli, M.; Drago, F.; Racagni, G.; Riva, M.A. Reduced activation of intracellular signaling pathways in rat prefrontal cortex after chronic phencyclidine administration. Pharmacol. Res. 2008, 57, 296–302. [Google Scholar]
- Lindahl, J.S.; Keifer, J. Glutamate receptor subunits are altered in forebrain and cerebellum in rats chronically exposed to the NMDA receptor antagonist phencyclidine. Neuropsychopharmacology 2004, 29, 2065–2073. [Google Scholar]
- Katanuma, Y.; Numakawa, T.; Adachi, N.; Yamamoto, N.; Ooshima, Y.; Odaka, H.; Inoue, T.; Kunugi, H. Phencyclidine rapidly decreases neuronal mRNA of brain-derived neurotrophic factor. Synapse 2014, 68, 257–265. [Google Scholar]
- Adachi, N.; Numakawa, T.; Kumamaru, E.; Itami, C.; Chiba, S.; Iijima, Y.; Richards, M.; Katoh-Semba, R.; Kunugi, H. Phencyclidine-induced decrease of synaptic connectivity via inhibition of BDNF secretion in cultured cortical neurons. Cereb. Cortex 2013, 23, 847–858. [Google Scholar]
- Gerlai, R. A new continuous alternation task in T-maze detects hippocampal dysfunction in mice. A strain comparison and lesion study. Behav. Brain Res. 1998, 95, 91–101. [Google Scholar]
- Guyot, A.C.; Leuxe, C.; Disdier, C.; Oumata, N.; Costa, N.; Roux, G.L.; Varela, P.F.; Duchon, A.; Charbonnier, J.B.; Herault, Y.; et al. A Small Compound Targeting Prohibitin with Potential Interest for Cognitive Deficit Rescue in Aging mice and Tau Pathology Treatment. Sci. Rep. 2020, 10, 1143. [Google Scholar]
- Combes, M.; Poindron, P.; Callizot, N. Glutamate protects neuromuscular junctions from deleterious effects of β-amyloid peptide and conversely: An in vitro study in a nerve-muscle coculture. J. Neurosci. Res. 2015, 93, 633–643. [Google Scholar]
- Burke, K.J., Jr.; Keeshen, C.M.; Bender, K.J. Two Forms of Synaptic Depression Produced by Differential Neuromodulation of Presynaptic Calcium Channels. Neuron 2018, 99, 969–984.e7. [Google Scholar]
- Morabito, A.; Zerlaut, Y.; Serraz, B.; Sala, R.; Paoletti, P.; Rebola, N. Activity-dependent modulation of NMDA receptors by endogenous zinc shapes dendritic function in cortical neurons. Cell Rep. 2022, 38, 110415. [Google Scholar]
- Li, R.; Huang, F.S.; Abbas, A.K.; Wigström, H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007, 8, 55. [Google Scholar]
- Auberson, Y.P.; Allgeier, H.; Bischoff, S.; Lingenhoehl, K.; Moretti, R.; Schmutz, M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg. Med. Chem. Lett. 2002, 12, 1099–1102. [Google Scholar]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993, 44, 851–859. [Google Scholar]
- Agmon, A.; Connors, B.W. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 1991, 41, 365–379. [Google Scholar]
- Varela, C. The gating of neocortical information by modulators. J. Neurophysiol. 2013, 109, 1229–1232. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemarchant, S.; Sourioux, M.; Le Douce, J.; Henriques, A.; Callizot, N.; Hugues, S.; Farinelli, M.; Godfrin, Y. NX210c Peptide Promotes Glutamatergic Receptor-Mediated Synaptic Transmission and Signaling in the Mouse Central Nervous System. Int. J. Mol. Sci. 2022, 23, 8867. https://doi.org/10.3390/ijms23168867
Lemarchant S, Sourioux M, Le Douce J, Henriques A, Callizot N, Hugues S, Farinelli M, Godfrin Y. NX210c Peptide Promotes Glutamatergic Receptor-Mediated Synaptic Transmission and Signaling in the Mouse Central Nervous System. International Journal of Molecular Sciences. 2022; 23(16):8867. https://doi.org/10.3390/ijms23168867
Chicago/Turabian StyleLemarchant, Sighild, Mélissa Sourioux, Juliette Le Douce, Alexandre Henriques, Noëlle Callizot, Sandrine Hugues, Mélissa Farinelli, and Yann Godfrin. 2022. "NX210c Peptide Promotes Glutamatergic Receptor-Mediated Synaptic Transmission and Signaling in the Mouse Central Nervous System" International Journal of Molecular Sciences 23, no. 16: 8867. https://doi.org/10.3390/ijms23168867
APA StyleLemarchant, S., Sourioux, M., Le Douce, J., Henriques, A., Callizot, N., Hugues, S., Farinelli, M., & Godfrin, Y. (2022). NX210c Peptide Promotes Glutamatergic Receptor-Mediated Synaptic Transmission and Signaling in the Mouse Central Nervous System. International Journal of Molecular Sciences, 23(16), 8867. https://doi.org/10.3390/ijms23168867