
Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases

 ,
,
Abstract
1. Introduction
1.1. Neuronal NMDAR Structure, Distribution, and Functions
1.1.1. NMDARs on Neurons
1.1.2. NMDARs in Brain Development
1.1.3. NMDARs in LTP and LTD
1.1.4. NMDARs in Neuronal Death and Survival
1.2. NMDARs on Other Cell Types: Structure, Distributions, and Functions
1.2.1. NMDARs on Endothelial Cells and Tight Junctions of the BBB
1.2.2. NMDARs on Glial Cells: Astrocytes and Oligodendrocytes
1.2.3. NMDARs on Immune Cells: Microglia, Macrophages, and Immunological Synapse
2. CNS Diseases and NMDAR Dysfunctions
2.1. Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease
2.1.2. Parkinson’s Disease
2.1.3. Huntington’s Disease
2.1.4. Amyotrophic Lateral Sclerosis
2.2. Neurovascular and Traumatic Disorders
2.2.1. Stroke
2.2.2. Traumatic Brain Injury
2.3. Autoimmune Diseases
2.3.1. Multiple Sclerosis
2.3.2. Anti-NMDAR Encephalitis
2.4. Mental Diseases
2.4.1. Depression
2.4.2. Schizophrenia
2.5. Neurodevelopmental Diseases
2.5.1. Autism
2.5.2. Epilepsy
3. Modulators of NMDARs for the Treatment of Neurological Disorders: Preclinical and Clinical Data
3.1. Channel NMDAR Blockers and Modulators
3.2. Glutamate and Glycine Site Antagonists
3.3. Positive and Negative Allosteric Modulators
3.4. Compounds Acting Downstream of NMDARs
3.5. Antibody Targeting tPA-NMDAR Interaction
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Paoletti, P. Molecular Basis of NMDA Receptor Functional Diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.; Paoletti, P.; Mony, L. Positive Allosteric Modulation of NMDA Receptors: Mechanisms, Physiological Impact and Therapeutic Potential. J. Physiol. 2022, 600, 233–259. [Google Scholar] [CrossRef]
- Vance, K.M.; Hansen, K.B.; Traynelis, S.F. GluN1 Splice Variant Control of GluN1/GluN2D NMDA Receptors. J. Physiol. 2012, 590, 3857–3875. [Google Scholar] [CrossRef]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef]
- Garcia, E.; Ismail, S. Spatiotemporal Regulation of Signaling: Focus on T Cell Activation and the Immunological Synapse. Int. J. Mol. Sci. 2020, 21, 3283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sheng, M. NMDA Receptors in Nervous System Diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef]
- Vyklicky, V.; Krausova, B.; Cerny, J.; Balik, A.; Zapotocky, M.; Novotny, M.; Lichnerova, K.; Smejkalova, T.; Kaniakova, M.; Korinek, M.; et al. Block of NMDA Receptor Channels by Endogenous Neurosteroids: Implications for the Agonist Induced Conformational States of the Channel Vestibule. Sci. Rep. 2015, 5, 10935. [Google Scholar] [CrossRef] [PubMed]
- Huettner, J.E.; Bean, B.P. Block of N-Methyl-D-Aspartate-Activated Current by the Anticonvulsant MK-801: Selective Binding to Open Channels. Proc. Natl. Acad. Sci. USA 1988, 85, 1307–1311. [Google Scholar] [CrossRef]
- Albers, G.W.; Goldstein, L.B.; Hall, D.; Lesko, L.M.; Albers, G.W.; Goldstein, L.B.; Hall, D.; Lesko, L.M.; Albers, G.W.; Goldstein, L.B.; et al. Aptiganel Hydrochloride in Acute Ischemic Stroke: A Randomized Controlled Trial. JAMA 2001, 286, 2673–2682. [Google Scholar] [CrossRef]
- Steinberg, G.K.; Yoon, E.J.; Kunis, D.M.; Sun, G.H.; Maier, C.M.; Grant, G.A. Neuroprotection by N-Methyl-D-Aspartate Antagonists in Focal Cerebral Ischemia Is Dependent on Continued Maintenance Dosing. Neuroscience 1995, 64, 99–107. [Google Scholar] [CrossRef]
- Ory-Magne, F.; Corvol, J.C.; Azulay, J.P.; Bonnet, A.M.; Brefel-Courbon, C.; Damier, P.; Dellapina, E.; Destée, A.; Durif, F.; Galitzky, M.; et al. Withdrawing Amantadine in Dyskinetic Patients with Parkinson Disease: The AMANDYSK Trial. Neurology 2014, 82, 300–307. [Google Scholar] [CrossRef]
- Singh, J.B.; Fedgchin, M.; Daly, E.; Xi, L.; Melman, C.; de Bruecker, G.; Tadic, A.; Sienaert, P.; Wiegand, F.; Manji, H.; et al. Intravenous Esketamine in Adult Treatment-Resistant Depression: A Double-Blind, Double-Randomization, Placebo-Controlled Study. Biol. Psychiatry 2016, 80, 424–431. [Google Scholar] [CrossRef]
- Bernstein, G.; Davis, K.; Mills, C.; Wang, L.; McDonnell, M.; Oldenhof, J.; Inturrisi, C.; Manfredi, P.L.; Vitolo, O.V. Characterization of the Safety and Pharmacokinetic Profile of D-Methadone, a Novel N-Methyl-D-Aspartate Receptor Antagonist in Healthy, Opioid-Naive Subjects: Results of Two Phase 1 Studies. J. Clin. Psychopharmacol. 2019, 39, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K.; Salter, M.W. NMDA Receptors in Clinical Neurology: Excitatory Times Ahead. Lancet Neurol. 2008, 7, 742–755. [Google Scholar] [CrossRef]
- Kleckner, N.W.; Dingledine, R. Requirement for Glycine in Activation of NMDA-Receptors Expressed in Xenopus Oocytes. Science 1988, 241, 835–837. [Google Scholar] [CrossRef]
- Furukawa, H.; Gouaux, E. Mechanisms of Activation, Inhibition and Specificity: Crystal Structures of the NMDA Receptor NR1 Ligand-Binding Core. EMBO J. 2003, 22, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Sebih, F.; Rousset, M.; Bellahouel, S.; Rolland, M.; de Jesus Ferreira, M.C.; Guiramand, J.; Cohen-Solal, C.; Barbanel, G.; Cens, T.; Abouazza, M.; et al. Characterization of l -Theanine Excitatory Actions on Hippocampal Neurons: Toward the Generation of Novel N-Methyl- d -Aspartate Receptor Modulators Based on Its Backbone. ACS Chem. Neurosci. 2017, 8, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Mony, L.; Zhu, S.; Carvalho, S.; Paoletti, P. Molecular Basis of Positive Allosteric Modulation of GluN2B NMDA Receptors by Polyamines. EMBO J. 2011, 30, 3134–3146. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Lehmann, J.; Hutchison, A.J.; McPherson, S.E.; Mondadori, C.; Schmutz, M.; Sinton, C.M.; Tsai, C.; Murphy, D.E.; Steel, D.J.; Williams, M.; et al. CGS 19755, a Selective and Competitive N-Methyl-D-Aspartate-Type Excitatory Amino Acid Receptor Antagonist. J. Pharmacol. Exp. Ther. 1988, 246, 65–75. [Google Scholar]
- Moskal, J.; Burgdorf, J.; Stanton, P.; Kroes, R.; Disterhoft, J.; Burch, R.; Khan, M. The Development of Rapastinel (Formerly GLYX-13); A Rapid Acting and Long Lasting Antidepressant. Curr. Neuropharmacol. 2017, 15, 47–56. [Google Scholar] [CrossRef]
- Bowers, M.S.; Cacheaux, L.P.; Sahu, S.U.; Schmidt, M.E.; Sennello, J.A.; Leaderbrand, K.; Khan, M.A.; Kroes, R.A.; Moskal, J.R. NYX-2925 Induces Metabotropic N-Methyl-d-Aspartate Receptor (NMDAR) Signaling That Enhances Synaptic NMDAR and α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor. J. Neurochem. 2020, 152, 523–541. [Google Scholar] [CrossRef]
- Henter, I.D.; Park, L.T.; Zarate, C.A. Novel Glutamatergic Modulators for the Treatment of Mood Disorders: Current Status. CNS Drugs 2021, 35, 527–543. [Google Scholar] [CrossRef]
- Bordi, F.; Pietra, C.; Ziviani, L.; Reggiani, A. The Glycine Antagonist GV150526 Protects Somatosensory Evoked Potentials and Reduces the Infarct Area in the MCAo Model of Focal Ischemia in the Rat. Exp. Neurol. 1997, 145, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Piantadosi, S.C.; Wu, H.Q.; Pribut, H.J.; Dell, M.J.; Can, A.; Snodgrass, H.R.; Zarate, C.A.; Schwarcz, R.; Gould, T.D. The Prodrug 4-Chlorokynurenine Causes Ketamine-Like Antidepressant Effects, but Not Side Effects, by NMDA/GlycineB-Site Inhibition. J. Pharmacol. Exp. Ther. 2015, 355, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Hagino, Y.; Kasai, S.; Han, W.; Yamamoto, H.; Nabeshima, T.; Mishina, M.; Ikeda, K. Essential Role of NMDA Receptor Channel Ε4 Subunit (GluN2D) in the Effects of Hencyclidine, but Not Methamphetamine. PLoS ONE 2010, 5, e13722. [Google Scholar] [CrossRef] [PubMed]
- Korinek, M.; Vyklicky, V.; Borovska, J.; Lichnerova, K.; Kaniakova, M.; Krausova, B.; Krusek, J.; Balik, A.; Smejkalova, T.; Horak, M.; et al. Cholesterol Modulates Open Probability and Desensitization of NMDA Receptors. J. Physiol. 2015, 593, 2279–2293. [Google Scholar] [CrossRef] [PubMed]
- Dorota Majewska, M.; Schwartz, R.D. Pregnenolone-Sulfate: An Endogenous Antagonist of the y-Aminobutyric Acid Receptor Complex in Brain? Brain Res. 1987, 404, 355–360. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.N.; Pankratov, Y. ATP from Synaptic Terminals and Astrocytes Regulates NMDA Receptors and Synaptic Plasticity through PSD-95 Multi-Protein Complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef]
- Koenig, A.; Murck, H.; Luo, Y.; Webster, I.; Quirk, M.; Kanes, S.; Doherty, J. ACNP 58 Th Annual Meeting: Poster Session I. Neuropsychopharmacology 2019, 44, 78–229. [Google Scholar] [CrossRef]
- Cho, S.I.; Park, U.J.; Chung, J.M.; Gwag, B.J. Neu2000, an NR2B-Selective, Moderate NMDA Receptor Antagonist and Potent Spin Trapping Molecule for Stroke. Drug News Perspect. 2010, 23, 549–556. [Google Scholar] [CrossRef]
- Gwag, B.J.; Lee, Y.A.; Ko, S.Y.; Lee, M.J.; Im, D.S.; Yun, B.S.; Lim, H.R.; Park, S.M.; Byun, H.Y.; Son, S.J.; et al. Marked Prevention of Ischemic Brain Injury by Neu2000, an NMDA Antagonist and Antioxidant Derived from Aspirin and Sulfasalazine. J. Cereb. Blood Flow Metab. 2007, 27, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Martí-Fábregas, J.; Gállego, J.; Krupinski, J.; Gomis, M.; Cánovas, D.; et al. Safety and Efficacy of Uric Acid in Patients with Acute Stroke (URICO-ICTUS): A Randomised, Double-Blind Phase 2b/3 Trial. Lancet Neurol. 2014, 13, 453–460. [Google Scholar] [CrossRef]
- Ganesh, A.; Goyal, M.; Wilson, A.T.; Ospel, J.M.; Demchuk, A.M.; Mikulis, D.; Poublanc, J.; Krings, T.; Anderson, R.; Tymianski, M.; et al. Association of Iatrogenic Infarcts with Clinical and Cognitive Outcomes in the Evaluating Neuroprotection in Aneurysm Coiling Therapy Trial. Neurology 2022, 98, E1446–E1458. [Google Scholar] [CrossRef] [PubMed]
- Ugalde-Triviño, L.; Díaz-Guerra, M. PSD-95: An Effective Target for Stroke Therapy Using Neuroprotective Peptides. Int. J. Mol. Sci. 2021, 22, 12585. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, Y.T. Postsynaptic Signaling at Glutamatergic Synapses as Therapeutic Targets. Curr. Opin. Neurobiol. 2022, 75, 102585. [Google Scholar] [CrossRef]
- Bach, A.; Pedersen, S.W.; Dorr, L.A.; Vallon, G.; Ripoche, I.; Ducki, S.; Lian, L.Y. Biochemical Investigations of the Mechanism of Action of Small Molecules ZL006 and IC87201 as Potential Inhibitors of the NNOS-PDZ/PSD-95-PDZ Interactions. Sci. Rep. 2015, 5, 12157. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Scheltens, P.; Sadowsky, C.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Wang, P.; et al. Clinical Effects of Tramiprosate in APOE4/4 Homozygous Patients with Mild Alzheimer’s Disease Suggest Disease Modification Potential. J. Prev. Alzheimers Dis. 2017, 4, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Okuda, Y.; Hasegawa, K.; Hirai, K.; Kanai-Ochiai, R.; Morimoto, M.; Sugimoto, T. Effects of Edaravone on N-Methyl-d-Aspartate (NMDA)-Mediated Cytochrome c Release and Apoptosis in Neonatal Rat Cerebrocortical Slices. Int. J. Dev. Neurosci. 2006, 24, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Matsuba, Y.; Kamano, N.; Mihira, N.; Sahara, N.; Takano, J.; Muramatsu, S.I.; Saido, T.C.; Saito, T. Tau Binding Protein CAPON Induces Tau Aggregation and Neurodegeneration. Nat. Commun. 2019, 10, 2394. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and Interplay of Parkin, PINK1, and DJ1 in Neurodegenerative and Neuroinflammatory Disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Taghibiglou, C.; Girling, K.; Dong, Z.; Lin, S.Z.; Lee, W.; Shyu, W.C.; Wang, Y.T. Critical Role of Increased PTEN Nuclear Translocation in Excitotoxic and Ischemic Neuronal Injuries. J. Neurosci. 2013, 33, 7997–8008. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Docagne, F.; Ali, C.; Margaill, I.; Carmeliet, P.; MacKenzie, E.T.; Vivien, D.; Buisson, A. The Proteolytic Activity of Tissue-Plasminogen Activator Enhances NMDA Receptor-Mediated Signaling. Nat. Med. 2001, 7, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Lesept, F.; Chevilley, A.; Jezequel, J.; Ladépêche, L.; Macrez, R.; Aimable, M.; Lenoir, S.; Bertrand, T.; Rubrecht, L.; Galea, P.; et al. Tissue-Type Plasminogen Activator Controls Neuronal Death by Raising Surface Dynamics of Extrasynaptic NMDA Receptors. Cell Death Dis. 2016, 7, e2466. [Google Scholar] [CrossRef]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA Receptors: From Structure to Synaptic Physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kartvelishvily, E.; Shleper, M.; Balan, L.; Dumin, E.; Wolosker, H. Neuron-Derived D-Serine Release Provides a Novel Means to Activate N-Methyl-D-Aspartate Receptors. J. Biol. Chem. 2006, 281, 14151–14162. [Google Scholar] [CrossRef]
- Panatier, A.; Theodosis, D.T.; Mothet, J.P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H.R. Glia-Derived D-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Oliet, S.H.R.; Mothet, J.P. Regulation of N-Methyl-D-Aspartate Receptors by Astrocytic D-Serine. Neuroscience 2009, 158, 275–283. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Dore, K.; Stein, I.S.; Brock, J.A.; Castillo, P.E.; Zito, K.; Sjöström, P.J. Unconventional NMDA Receptor Signaling. J. Neurosci. 2017, 37, 10800–10807. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA Receptor Function Is Required for NMDA Receptor-Dependent Long-Term Depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and Regional Expression in the Rat Brain and Functional Properties of Four NMDA Receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Standaert, D.G.; Landwehrmeyer, G.B.; Kerner, J.A.; Penney, J.B.; Young, A.B. Expression of NMDAR2D Glutamate Receptor Subunit MRNA in Neurochemically Identified Interneurons in the Rat Neostriatum, Neocortex and Hippocampus. Brain Res. Mol. Brain Res. 1996, 42, 89–102. [Google Scholar] [CrossRef]
- Corlew, R.; Brasier, D.J.; Feldman, D.E.; Philpot, B.D. Presynaptic NMDA Receptors: Newly Appreciated Roles in Cortical Synaptic Function and Plasticity. Neuroscientist 2008, 14, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Engelman, H.S.; MacDermott, A.B. Presynaptic Ionotropic Receptors and Control of Transmitter Release. Nat. Rev. Neurosci. 2004, 5, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Duguid, I.; Sjöström, P.J. Novel Presynaptic Mechanisms for Coincidence Detection in Synaptic Plasticity. Curr. Opin. Neurobiol. 2006, 16, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.S.; Mulle, C. Presynaptic Glutamate Receptors: Physiological Functions and Mechanisms of Action. Nat. Rev. Neurosci. 2008, 9, 423–436. [Google Scholar] [CrossRef]
- le Meur, K.; Galante, M.; Angulo, M.C.; Audinat, E. Tonic Activation of NMDA Receptors by Ambient Glutamate of Non-Synaptic Origin in the Rat Hippocampus. J. Physiol. 2007, 580, 373–383. [Google Scholar] [CrossRef]
- Groc, L.; Heine, M.; Cousins, S.L.; Stephenson, F.A.; Lounis, B.; Cognet, L.; Choquet, D. NMDA Receptor Surface Mobility Depends on NR2A-2B Subunits. Proc. Natl. Acad. Sci. USA 2006, 103, 18769–18774. [Google Scholar] [CrossRef]
- Tovar, K.R.; Westbrook, G.L. The Incorporation of NMDA Receptors with a Distinct Subunit Composition at Nascent Hippocampal Synapses in Vitro. J. Neurosci. 1999, 19, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Bard, L.; Sainlos, M.; Bouchet, D.; Cousins, S.; Mikasova, L.; Breillat, C.; Stephenson, F.A.; Imperiali, B.; Choquet, D.; Groc, L. Dynamic and Specific Interaction between Synaptic NR2-NMDA Receptor and PDZ Proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 19561–19566. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.G.; Zukin, R.S. NMDA Receptor Trafficking in Synaptic Plasticity and Neuropsychiatric Disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Groc, L.; Lafourcade, M.; Heine, M.; Renner, M.; Racine, V.; Sibarita, J.-B.; Lounis, B.; Choquet, D.; Cognet, L. Surface Trafficking of Neurotransmitter Receptor: Comparison between Single-Molecule/Quantum Dot Strategies. J. Neurosci. 2007, 27, 12433–12437. [Google Scholar] [CrossRef]
- Michaluk, P.; Mikasova, L.; Groc, L.; Frischknecht, R.; Choquet, D.; Kaczmarek, L. Matrix Metalloproteinase-9 Controls NMDA Receptor Surface Diffusion through Integrin Beta1 Signaling. J. Neurosci. 2009, 29, 6007–6012. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H.R. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Booker, S.A.; Wyllie, D.J.A. NMDA Receptor Function in Inhibitory Neurons. Neuropharmacology 2021, 196, 108609. [Google Scholar] [CrossRef] [PubMed]
- Cornford, J.; Mercier, M.S.; Leite, M.; Magloire, V.; Häusser, M.; Kullmann, D.M. Dendritic NMDA Receptors in Parvalbumin Neurons Enable Strong and Stable Neuronal Assemblies. Elife 2019, 8, e49872. [Google Scholar] [CrossRef]
- Epping, L.; Schroeter, C.B.; Nelke, C.; Bock, S.; Gola, L.; Ritter, N.; Herrmann, A.M.; Räuber, S.; Henes, A.; Wasser, B.; et al. Activation of Non-Classical NMDA Receptors by Glycine Impairs Barrier Function of Brain Endothelial Cells. Cell Mol. Life Sci. 2022, 79, 479. [Google Scholar] [CrossRef]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA Receptors Are Expressed in Oligodendrocytes and Activated in Ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef]
- Skowrońska, K.; Obara-Michlewska, M.; Zielińska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of Microglial N-Methyl-D-Aspartate Receptors Triggers Inflammation and Neuronal Cell Death in the Developing and Mature Brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef]
- Affaticati, P.; Mignen, O.; Jambou, F.; Potier, M.C.; Klingel-Schmitt, I.; Degrouard, J.; Peineau, S.; Gouadon, E.; Collingridge, G.L.; Liblau, R.; et al. Sustained Calcium Signalling and Caspase-3 Activation Involve NMDA Receptors in Thymocytes in Contact with Dendritic Cells. Cell Death Differ. 2011, 18, 99–108. [Google Scholar] [CrossRef]
- Mantuano, E.; Azmoon, P.; Brifault, C.; Banki, M.A.; Gilder, A.S.; Campana, W.M.; Gonias, S.L. Tissue-Type Plasminogen Activator Regulates Macrophage Activation and Innate Immunity. Blood 2017, 130, 1364–1374. [Google Scholar] [CrossRef]
- Del Arroyo, A.G.; Hadjihambi, A.; Sanchez, J.; Turovsky, E.; Kasymov, V.; Cain, D.; Nightingale, T.D.; Lambden, S.; Grant, S.G.; Gourine, A.V.; et al. NMDA Receptor Modulation of Glutamate Release in Activated Neutrophils. EBioMedicine 2019, 47, 457–469. [Google Scholar] [CrossRef]
- Pasquet, N.; Douceau, S.; Naveau, M.; Lesept, F.; Louessard, M.; Lebouvier, L.; Hommet, Y.; Vivien, D.; Bardou, I. Tissue-Type Plasminogen Activator Controlled Corticogenesis Through a Mechanism Dependent of NMDA Receptors Expressed on Radial Glial Cells. Cereb. Cortex 2019, 29, 2482–2498. [Google Scholar] [CrossRef]
- Hall, B.J.; Ripley, B.; Ghosh, A. NR2B Signaling Regulates the Development of Synaptic AMPA Receptor Current. J. Neurosci. 2007, 27, 13446–13456. [Google Scholar] [CrossRef] [PubMed]
- Kutsuwada, T.; Sakimura, K.; Manabe, T.; Takayama, C.; Katakura, N.; Kushiya, E.; Natsume, R.; Watanabe, M.; Inoue, Y.; Yagi, T.; et al. Impairment of Suckling Response, Trigeminal Neuronal Pattern Formation, and Hippocampal LTD in NMDA Receptor Epsilon 2 Subunit Mutant Mice. Neuron 1996, 16, 333–344. [Google Scholar] [CrossRef]
- Gu, X.; Lu, W. Genetic Deletion of NMDA Receptors Suppresses GABAergic Synaptic Transmission in Two Distinct Types of Central Neurons. Neurosci. Lett. 2018, 668, 147–153. [Google Scholar] [CrossRef]
- Hou, G.; Zhang, Z.W. NMDA Receptors Regulate the Development of Neuronal Intrinsic Excitability through Cell-Autonomous Mechanisms. Front. Cell Neurosci. 2017, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Frangeul, L.; Kehayas, V.; Sanchez-Mut, J.V.; Fièvre, S.; Krishna-K, K.; Pouchelon, G.; Telley, L.; Bellone, C.; Holtmaat, A.; Gräff, J.; et al. Input-Dependent Regulation of Excitability Controls Dendritic Maturation in Somatosensory Thalamocortical Neurons. Nat. Commun. 2017, 8, 2015. [Google Scholar] [CrossRef]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of Disorganization in the Neocortex of Children with Autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Packer, A. Neocortical Neurogenesis and the Etiology of Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2016, 64, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Groc, L.; Choquet, D. Measurement and Characteristics of Neurotransmitter Receptor Surface Trafficking (Review). Mol. Membr. Biol. 2009, 25, 344–352. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Javitt, D.C. Thinking Glutamatergically: Changing Concepts of Schizophrenia Based upon Changing Neurochemical Models. Clin. Schizophr. Relat. Psychoses 2010, 4, 189–200. [Google Scholar] [CrossRef]
- Nabavi, S.; Fox, R.; Proulx, C.D.; Lin, J.Y.; Tsien, R.Y.; Malinow, R. Engineering a Memory with LTD and LTP. Nature 2014, 511, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Son, G.H.; Winters, B.L.; Kim, M.J.; Whitcomb, D.J.; Dickinson, B.A.; Lee, Y.B.; Futai, K.; Amici, M.; Sheng, M.; et al. Muscarinic Receptors Induce LTD of NMDAR EPSCs via a Mechanism Involving Hippocalcin, AP2 and PSD-95. Nat. Neurosci. 2010, 13, 1216–1224. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory Amino Acids in Synaptic Transmission in the Schaffer Collateral-Commissural Pathway of the Rat Hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D. Hippocampal LTP and Contextual Learning Require Surface Diffusion of AMPA Receptors. Nat. Publ. Group 2017, 549, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Peineau, S.; Howland, J.G.; Wang, Y.T. Long-Term Depression in the CNS. Nat. Rev. Neurosci. 2010, 11, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Bading, H. Synaptic Activity-Mediated Suppression of P53 and Induction of Nuclear Calcium-Regulated Neuroprotective Genes Promote Survival through Inhibition of Mitochondrial Permeability Transition. J. Neurosci. 2009, 29, 4420–4429. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic Cell Death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate Neurotoxicity and Diseases of the Nervous System. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular Mechanisms of Glutamate-Dependent Neurodegeneration in Ischemia and Traumatic Brain Injury. Cell Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the Pathogenesis of Neurological and Psychiatric Disorders: Therapeutic Implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Mony, L.; Kew, J.N.C.; Gunthorpe, M.J.; Paoletti, P. Allosteric Modulators of NR2B-Containing NMDA Receptors: Molecular Mechanisms and Therapeutic Potential. Br. J. Pharmacol. 2009, 157, 1301–1317. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA Receptor-Mediated Excitotoxicity Depends on the Coactivation of Synaptic and Extrasynaptic Receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [PubMed]
- Macrez, R.; Obiang, P.; Gauberti, M.; Roussel, B.; Baron, A.; Parcq, J.; Cassé, F.; Hommet, Y.; Orset, C.; Agin, V.; et al. Antibodies Preventing the Interaction of Tissue-Type Plasminogen Activator with N-Methyl-D-Aspartate Receptors Reduce Stroke Damages and Extend the Therapeutic Window of Thrombolysis. Stroke J. Cereb. Circ. 2011, 42, 2315–2322. [Google Scholar] [CrossRef]
- Baron, A.; Montagne, A.; Cassé, F.; Launay, S.; Maubert, E.; Ali, C.; Vivien, D. NR2D-Containing NMDA Receptors Mediate Tissue Plasminogen Activator-Promoted Neuronal Excitotoxicity. Cell Death Differ. 2010, 17, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Jullienne, A.; Montagne, A.; Orset, C.; Lesept, F.; Jane, D.E.; Monaghan, D.T.; Maubert, E.; Vivien, D.; Ali, C. Selective Inhibition of GluN2D-Containing N-Methyl-D-Aspartate Receptors Prevents Tissue Plasminogen Activator-Promoted Neurotoxicity Both In Vitro and In Vivo. Mol. Neurodegener. 2011, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Benchenane, K.; Castel, H.; Boulouard, M.; Bluthé, R.; Fernández-Monreal, M.; Roussel, B.D.; Lopez-Atalaya, J.P.; Butt-Gueulle, S.; Agin, V.; Maubert, E.; et al. Anti-NR1 N-Terminal-Domain Vaccination Unmasks the Crucial Action of TPA on NMDA-Receptor-Mediated Toxicity and Spatial Memory. J. Cell Sci. 2007, 120, 578–585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, J.T.; Chen, T.G.; Chang, Y.C.; Chen, C.Y.; Chen, R.M. Roles of NMDARs in Maintenance of the Mouse Cerebrovascular Endothelial Cell-Constructed Tight Junction Barrier. Toxicology 2016, 339, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes Drive Cortical Vasodilatory Signaling by Activating Endothelial NMDA Receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef]
- Macrez, R.; Ortega, M.C.; Bardou, I.; Mehra, A.; Fournier, A.; van der Pol, S.M.A.; Haelewyn, B.; Maubert, E.; Lesept, F.; Chevilley, A.; et al. Neuroendothelial NMDA Receptors as Therapeutic Targets in Experimental Autoimmune Encephalomyelitis. Brain 2016, 139, 2406–2419. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson IV, T.H.; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate Causes a Loss in Human Cerebral Endothelial Barrier Integrity through Activation of NMDA Receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef] [PubMed]
- Anfray, A.; Drieu, A.; Hingot, V.; Hommet, Y.; Yetim, M.; Rubio, M.; Deffieux, T.; Tanter, M.; Orset, C.; Vivien, D. Circulating TPA Contributes to Neurovascular Coupling by a Mechanism Involving the Endothelial NMDA Receptors. J. Cereb. Blood Flow Metab. 2020, 40, 2038–2054. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.D.; Fowler, M.; Jackson, T.H.; Houghton, J.; Warren, A.; Nanda, A.; Chandler, I.; Cappell, B.; Long, A.; Minagar, A.; et al. Human Neuroepithelial Cells Express NMDA Receptors. BMC Neurosci. 2003, 4, 28. [Google Scholar] [CrossRef]
- Mehra, A.; Guérit, S.; Macrez, R.; Gosselet, F.; Sevin, E.; Lebas, H.; Maubert, E.; de Vries, H.E.; Bardou, I.; Vivien, D.; et al. Nonionotropic Action of Endothelial NMDA Receptors on Blood-Brain Barrier Permeability via Rho/ROCK-Mediated Phosphorylation of Myosin. J. Neurosci. 2020, 40, 1778–1787. [Google Scholar] [CrossRef]
- Seillier, C.; Hélie, P.; Petit, G.; Vivien, D.; Clemente, D.; le Mauff, B.; Docagne, F.; Toutirais, O. Roles of the Tissue-Type Plasminogen Activator in Immune Response. Cell Immunol. 2022, 371, 104451. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Lu, P.; Anderson, C.M. Endothelial NMDA Receptors Mediate Activitydependent Brain Hemodynamic Responses in Mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10229–10231. [Google Scholar] [CrossRef] [PubMed]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of Glutamate Toxicity in Multiple Sclerosis: Biomarker and Therapeutic Opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Jeanneret, V.; Yepes, M. Tissue-Type Plasminogen Activator Is a Homeostatic Regulator of Synaptic Function in the Central Nervous System. Neural Regen. Res. 2017, 12, 362–365. [Google Scholar] [CrossRef]
- Armstead, W.M.; Hekierski, H.; Pastor, P.; Yarovoi, S.; Higazi, A.A.R.; Cines, D.B. Release of IL-6 After Stroke Contributes to Impaired Cerebral Autoregulation and Hippocampal Neuronal Necrosis Through NMDA Receptor Activation and Upregulation of ET-1 and JNK. Transl. Stroke Res. 2019, 10, 104–111. [Google Scholar] [CrossRef]
- Reijerkerk, A.; Kooij, G.; van der Pol, S.M.A.; Leyen, T.; Lakeman, K.; van het Hof, B.; Vivien, D.; de Vries, H.E. The NR1 Subunit of NMDA Receptor Regulates Monocyte Transmigration through the Brain Endothelial Cell Barrier. J. Neurochem. 2010, 113, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, Y.; Wei, J.; Huang, F.; Mao, F.; Nong, W.; Cao, X.; Huang, W. NMDA Mediates Disruption of Blood-Brain Barrier Permeability via Rho/ROCK Signaling Pathway. Neurochem. Int. 2022, 154, 105278. [Google Scholar] [CrossRef]
- O’Kane, E.M.; Stone, T.W.; Morris, B.J. Activation of Rho GTPases by Synaptic Transmission in the Hippocampus. J. Neurochem. 2003, 87, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. NMDA Receptors Are Expressed in Developing Oligodendrocyte Processes and Mediate Injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Jiang, Q.; Coderre, E.; Ridsdale, A.; Zhang, L.; Woulfe, J.; Yin, X.; Trapp, B.D.; McRory, J.E.; Rehak, R.; et al. NMDA Receptors Mediate Calcium Accumulation in Myelin during Chemical Ischaemia. Nature 2006, 439, 988–992. [Google Scholar] [CrossRef]
- Pérez-Otaño, I.; Larsen, R.S.; Wesseling, J.F. Emerging Roles of GluN3-Containing NMDA Receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Piña-Crespo, J.C.; Talantova, M.; Micu, I.; States, B.; Chen, H.S.V.; Tu, S.; Nakanishi, N.; Tong, G.; Zhang, D.; Heinemann, S.F.; et al. Excitatory Glycine Responses of CNS Myelin Mediated by NR1/NR3 “NMDA” Receptor Subunits. J. Neurosci. 2010, 30, 11501–11505. [Google Scholar] [CrossRef]
- Dravid, S.M.; Prakash, A.; Traynelis, S.F. Activation of Recombinant NR1/NR2C NMDA Receptors. J. Physiol. 2008, 586, 4425–4439. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E. Pro-Survival Signalling from the NMDA Receptor. Biochem. Soc. Trans. 2006, 34, 936–938. [Google Scholar] [CrossRef]
- Paoletti, P.; Neyton, J. NMDA Receptor Subunits: Function and Pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Besancon, E.; Guo, S.; Lok, J.; Tymianski, M.; Lo, E.H. Beyond NMDA and AMPA Glutamate Receptors: Emerging Mechanisms for Ionic Imbalance and Cell Death in Stroke. Trends Pharmacol. Sci. 2008, 29, 268–275. [Google Scholar] [CrossRef]
- Tong, G.; Takahashi, H.; Tu, S.; Shin, Y.; Talantova, M.; Zago, W.; Xia, P.; Nie, Z.; Goetz, T.; Zhang, D.; et al. Modulation of NMDA Receptor Properties and Synaptic Transmission by the NR3A Subunit in Mouse Hippocampal and Cerebrocortical Neurons. J. Neurophysiol. 2008, 99, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Doretto, S.; Malerba, M.; Ramos, M.; Ikrar, T.; Kinoshita, C.; de Mei, C.; Tirotta, E.; Xu, X.; Borrelli, E. Oligodendrocytes as Regulators of Neuronal Networks during Early Postnatal Development. PLoS ONE 2011, 6, e19849. [Google Scholar] [CrossRef] [PubMed]
- Matute, C. Oligodendrocyte NMDA Receptors: A Novel Therapeutic Target. Trends Mol. Med. 2006, 12, 289–292. [Google Scholar] [CrossRef]
- Lee, M.C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef]
- de Balderas, P.M.O.; Aguilera, P. A Metabotropic-Like Flux-Independent NMDA Receptor Regulates Ca2+ Exit from Endoplasmic Reticulum and Mitochondrial Membrane Potential in Cultured Astrocytes. PLoS ONE 2015, 10, e0126314. [Google Scholar] [CrossRef]
- Palygin, O.; Lalo, U.; Pankratov, Y. Distinct Pharmacological and Functional Properties of NMDA Receptors in Mouse Cortical Astrocytes. Br. J. Pharmacol. 2011, 163, 1755–1766. [Google Scholar] [CrossRef]
- Jimenez-Blasco, D.; Santofimia-Castanõ, P.; Gonzalez, A.; Almeida, A.; Bolanõs, J.P. Astrocyte NMDA Receptors’ Activity Sustains Neuronal Survival through a Cdk5-Nrf2 Pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Tilleux, S.; Hermans, E. Down-Regulation of Astrocytic GLAST by Microglia-Related Inflammation Is Abrogated in Dibutyryl CAMP-Differentiated Cultures. J. Neurochem. 2008, 105, 2224–2236. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 1201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chu, J.M.T.; Wong, G.T.C. Cerebral Glutamate Regulation and Receptor Changes in Perioperative Neuroinflammation and Cognitive Dysfunction. Biomolecules 2022, 12, 597. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte Loss Influences Alzheimer-like Neurodegeneration in Mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood-Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.V.; Grond, A.; Zuniga, E.; Barnes, S.R.; et al. APOE4 Accelerates Advanced-Stage Vascular and Neurodegenerative Disorder in Old Alzheimer’s Mice via Cyclophilin A Independently of Amyloid-β. Nat. Aging 2021, 1, 506–520. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes Mediate Neurovascular Signaling to Capillary Pericytes but Not to Arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B. v From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Eulefi, E.; Nery, A.A.; Rodrigues, L.C.; Sánchez, R.; Romero, F.; Ulrich, H. Neural Differentiation of Rat Aorta Pericyte Cells. Cytom. Part A 2012, 81, 65–71. [Google Scholar] [CrossRef]
- Domoki, F.; Kis, B.; Gáspár, T.; Bari, F.; Busija, D.W. Cerebromicrovascular endothelial cells are resistant to l-glutamate. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2008, 295, R1099–R1108. [Google Scholar] [CrossRef] [PubMed]
- Kahlfuß, S.; Simma, N.; Mankiewicz, J.; Bose, T.; Lowinus, T.; Klein-Hessling, S.; Sprengel, R.; Schraven, B.; Heine, M.; Bommhardt, U. Immunosuppression by N-Methyl-d-Aspartate Receptor Antagonists Is Mediated through Inhibition of Kv1.3 and KCa3.1 Channels in T Cells. Mol. Cell Biol. 2014, 34, 820–831. [Google Scholar] [CrossRef]
- Zalfa, C.; Azmoon, P.; Mantuano, E.; Gonias, S.L. Tissue-Type Plasminogen Activator Neutralizes LPS but Not Protease-Activated Receptor-Mediated Inflammatory Responses to Plasmin. J. Leukoc. Biol. 2019, 105, 729–740. [Google Scholar] [CrossRef]
- Gottlieb, M.; Matute, C. Expression of Ionotropic Glutamate Receptor Subunits in Glial Cells of the Hippocampal CA1 Area Following Transient Forebrain Ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Tikka, T.M.; Koistinaho, J.E. Minocycline Provides Neuroprotection against N-Methyl-D-Aspartate Neurotoxicity by Inhibiting Microglia. J. Immunol. 2001, 166, 7527–7533. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA Receptors Drive Pro-Inflammatory Responses via PARP-1/TRMP2 Signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef]
- Eyo, U.B.; Peng, J.; Swiatkowski, P.; Mukherjee, A.; Bispo, A.; Wu, L.J. Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. J. Neurosci. 2014, 34, 10528–10540. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; de Felice, F.G. Soluble Amyloid-β Oligomers as Synaptotoxins Leading to Cognitive Impairment in Alzheimer’s Disease. Front. Cell Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M. The Plasminogen Activating System in the Pathogenesis of Alzheimer’s Disease. Neural Regen. Res. 2021, 16, 1973–1977. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041. [Google Scholar] [CrossRef]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-Regulation of Vesicular Glutamate Transporters Precedes Cell Loss and Pathology in Alzheimer’s Disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.A.; Gebhardt, F.M.; Mitrovic, A.D.; Vandenberg, R.J.; Dodd, P.R. Glutamate Transporter Variants Reduce Glutamate Uptake in Alzheimer’s Disease. Neurobiol. Aging 2011, 32, 553-e1. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.; Macdonald, N.; Kirkpatrick, P. Memantine Hydrochloride. Nat. Rev. Drug Discov. 2004, 3, 109–110. [Google Scholar] [CrossRef]
- Montagne, A.; Hébert, M.; Jullienne, A.; Lesept, F.; le Béhot, A.; Louessard, M.; Gauberti, M.; Orset, C.; Ali, C.; Agin, V.; et al. Memantine Improves Safety of Thrombolysis for Stroke. Stroke 2012, 43, 2774–2781. [Google Scholar] [CrossRef]
- Xia, P.; Chen, H.S.V.; Zhang, D.; Lipton, S.A. Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Federoff, H.J. Immune Responses in Parkinson’s Disease: Interplay between Central and Peripheral Immune Systems. Biomed. Res. Int. 2014, 2014, 275178. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef]
- Reuland, C.J.; Church, F.C. Synergy between Plasminogen Activator Inhibitor-1, α-Synuclein, and Neuroinflammation in Parkinson’s Disease. Med. Hypotheses 2020, 138, 109602. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Pignataro, A.; Marino, G.; Campanelli, F.; Calabrese, V.; Cardinale, A.; Pelucchi, S.; Marcello, E.; Gardoni, F.; Viscomi, M.T.; et al. Transcranial Magnetic Stimulation Exerts “Rejuvenation” Effects on Corticostriatal Synapses after Partial Dopamine Depletion. Mov. Disord. 2021, 36, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Robelet, S.; Melon, C.; Guillet, B.; Salin, P.; Kerkerian-Le Goff, L. Chronic L-DOPA Treatment Increases Extracellular Glutamate Levels and GLT1 Expression in the Basal Ganglia in a Rat Model of Parkinson’s Disease. Eur. J. Neurosci. 2004, 20, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; di Paolo, T. Levodopa Response Motor Complications—GABA Receptors and Preproenkephalin Expression in Human Brain. Parkinsonism Relat. Disord. 2002, 8, 449–454. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.K.S.; Yung, K.K.L. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [PubMed]
- Mellone, M.; Pelucchi, S.; Alberti, L.; Genazzani, A.A.; di Luca, M.; Gardoni, F. Zinc Transporter-1: A Novel NMDA Receptor-Binding Protein at the Postsynaptic Density. J. Neurochem. 2015, 132, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, A.; de Iure, A.; Bagetta, V.; Tantucci, M.; Durante, V.; Quiroga-Varela, A.; Costa, C.; di Filippo, M.; Ghiglieri, V.; Latagliata, E.C.; et al. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting with GluN2D N-Methyl-D-Aspartate Receptor Subunit. Biol. Psychiatry 2016, 79, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Durante, V.; de Iure, A.; Loffredo, V.; Vaikath, N.; de Risi, M.; Paciotti, S.; Quiroga-Varela, A.; Chiasserini, D.; Mellone, M.; Mazzocchetti, P.; et al. Alpha-Synuclein Targets GluN2A NMDA Receptor Subunit Causing Striatal Synaptic Dysfunction and Visuospatial Memory Alteration. Brain 2019, 142, 1365–1385. [Google Scholar] [CrossRef]
- Pan, H.; Zhao, Y.; Zhai, Z.; Zheng, J.; Zhou, Y.; Zhai, Q.; Cao, X.; Tian, J.; Zhao, L. Role of Plasminogen Activator Inhibitor-1 in the Diagnosis and Prognosis of Patients with Parkinson’s Disease. Exp. Ther. Med. 2018, 15, 5517–5522. [Google Scholar] [CrossRef]
- Stevenson, T.K.; Lawrence, D.A. Characterization of Tissue Plasminogen Activator Expression and Trafficking in the Adult Murine Brain. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Sashindranath, M.; Samson, A.L.; Downes, C.E.; Crack, P.J.; Lawrence, A.J.; Li, Q.X.; Ng, A.Q.P.; Jones, N.C.; Farrugia, J.J.; Abdella, E.; et al. Compartment- and Context-Specific Changes in Tissue-Type Plasminogen Activator (TPA) Activity Following Brain Injury and Pharmacological Stimulation. Lab. Investig. 2011, 91, 1079–1091. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Misra, C.; Brickley, S.G.; Wyllie, D.J.A.; Cull-Candy, S.G. Slow Deactivation Kinetics of NMDA Receptors Containing NR1 and NR2D Subunits in Rat Cerebellar Purkinje Cells. J. Physiol. 2000, 525, 299. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between Synaptic versus Extrasynaptic NMDA Receptor Activity Influences Inclusions and Neurotoxicity of Mutant Huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef]
- Jocoy, E.L.; André, V.M.; Cummings, D.M.; Rao, S.P.; Wu, N.; Ramsey, A.J.; Caron, M.G.; Cepeda, C.; Levine, M.S. Dissecting the Contribution of Individual Receptor Subunits to the Enhancement of N-Methyl-d-Aspartate Currents by Dopamine D1 Receptor Activation in Striatum. Front. Syst. Neurosci. 2011, 5, 28. [Google Scholar] [CrossRef]
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In Vivo Evidence for NMDA Receptor-Mediated Excitotoxicity in a Murine Genetic Model of Huntington Disease. J. Neurosci. 2009, 29, 3200–3205. [Google Scholar] [CrossRef]
- Boillée, S.; vande Velde, C.; Cleveland, D.W.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene Discovery in Amyotrophic Lateral Sclerosis: Implications for Clinical Management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef]
- Johnston, C.A.; Stanton, B.R.; Turner, M.R.; Gray, R.; Blunt, A.H.M.; Butt, D.; Ampong, M.A.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis in an Urban Setting: A Population Based Study of Inner City London. J. Neurol. 2006, 253, 1642–1643. [Google Scholar] [CrossRef]
- Marin, B.; Boumédiene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in Worldwide Incidence of Amyotrophic Lateral Sclerosis: A Meta-Analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic Lateral Sclerosis: Mechanisms and Therapeutics in the Epigenomic Era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef]
- Mitchell, J.; Paul, P.; Chen, H.J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial Amyotrophic Lateral Sclerosis Is Associated with a Mutation in D-Amino Acid Oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef]
- Kondori, N.R.; Paul, P.; Robbins, J.P.; Liu, K.; Hildyard, J.C.W.; Wells, D.J.; de Belleroche, J.S. Focus on the Role of D-Serine and D-Amino Acid Oxidase in Amyotrophic Lateral Sclerosis/Motor Neuron Disease (ALS). Front Mol. Biosci. 2018, 5, 8. [Google Scholar] [CrossRef]
- Paul, P.; de Belleroche, J. Experimental Approaches for Elucidating Co-Agonist Regulation of NMDA Receptor in Motor Neurons: Therapeutic Implications for Amyotrophic Lateral Sclerosis (ALS). J. Pharm. Biomed. Anal. 2015, 116, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; de Belleroche, J. The Role of D-Serine and Glycine as Co-Agonists of NMDA Receptors in Motor Neuron Degeneration and Amyotrophic Lateral Sclerosis (ALS). Front. Synaptic Neurosci. 2014, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Arachchige, B.J.; Henderson, R.; Pow, D.; Reed, S.; Aylward, J.; McCombe, P.A. Elevated Plasma Levels of D-Serine in Some Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Ono, S.I.; Ishige, K.; Naganuma, A.; Ito, Y.; Suzuki, T. Metallothionein Proteins Expression, Copper and Zinc Concentrations, and Lipid Peroxidation Level in a Rodent Model for Amyotrophic Lateral Sclerosis. Toxicology 2007, 229, 33–41. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted Neuronal Trafficking in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef] [PubMed]
- Debono, M.W.; le Guern, J.; Canton, T.; Doble, A.; Pradier, L. Inhibition by Riluzole of Electrophysiological Responses Mediated by Rat Kainate and NMDA Receptors Expressed in Xenopus Oocytes. Eur. J. Pharmacol. 1993, 235, 283–289. [Google Scholar] [CrossRef]
- Spalloni, A.; Nutini, M.; Longone, P. Role of the N-Methyl-d-Aspartate Receptors Complex in Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 312–322. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Huber, R.J. Calmodulin Binding Proteins and Neuroinflammation in Multiple Neurodegenerative Diseases. BMC Neurosci. 2022, 23, 10. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Olney, J.W. Excitotoxicity and the NMDA Receptor--Still Lethal after Eight Years. Trends Neurosci. 1995, 18, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Shyu, W.-C.; Wang, Y.T. Stroke Intervention Pathways: NMDA Receptors and Beyond. Trends Mol. Med. 2011, 17, 266–275. [Google Scholar] [CrossRef]
- Simon, R.P.; Swan, J.H.; Griffiths, T.; Meldrum, B.S. Blockade of N-Methyl-D-Aspartate Receptors May Protect against Ischemic Damage in the Brain. Science 1984, 226, 850–852. [Google Scholar] [CrossRef]
- Chen, M.; Lu, T.-J.; Chen, X.-J.; Zhou, Y.; Chen, Q.; Feng, X.-Y.; Xu, L.; Duan, W.-H.; Xiong, Z.-Q. Differential Roles of NMDA Receptor Subtypes in Ischemic Neuronal Cell Death and Ischemic Tolerance. Stroke J. Cereb. Circ. 2008, 39, 3042–3048. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tak, P.W.; Aarts, M.; Rooyakkers, A.; Liu, L.; Ted, W.L.; Dong, C.W.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA Receptor Subunits Have Differential Roles in Mediating Excitotoxic Neuronal Death Both In Vitro and In Vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, T.; Lesept, F.; Chevilley, A.; Lenoir, S.; Aimable, M.; Briens, A.; Hommet, Y.; Bardou, I.; Parcq, J.; Vivien, D. Conformations of Tissue Plasminogen Activator (TPA) Orchestrate Neuronal Survival by a Crosstalk between EGFR and NMDAR. Cell Death Dis. 2015, 6, e1924. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion--from Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Zhang, C.; An, J.; Haile, W.B.; Echeverry, R.; Strickland, D.K.; Yepes, M. Microglial Low-Density Lipoprotein Receptor-Related Protein 1 Mediates the Effect of Tissue-Type Plasminogen Activator on Matrix Metalloproteinase-9 Activity in the Ischemic Brain. J. Cereb. Blood Flow Metab. 2009, 29, 1946–1954. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Ziai, W.C.; Cordonnier, C.; Dowlatshahi, D.; Francis, B.; Goldstein, J.N.; Hemphill, J.C.; Johnson, R.; Keigher, K.M.; Mack, W.J.; et al. 2022 Guideline for the Management of Patients with Spontaneous Intracerebral Hemorrhage: A Guideline from the American Heart Association/American Stroke Association. Stroke 2022, 53, e282–e361. [Google Scholar] [CrossRef]
- Adeoye, O.; Albright, K.C.; Carr, B.G.; Wolff, C.; Mullen, M.T.; Abruzzo, T.; Ringer, A.; Khatri, P.; Branas, C.; Kleindorfer, D. Geographic Access to Acute Stroke Care in the United States. Stroke 2014, 45, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M. Tissue-Type Plasminogen Activator Is a Neuroprotectant in the Central Nervous System. Front. Cell Neurosci. 2015, 9, 304. [Google Scholar] [CrossRef]
- Wu, F.; Echeverry, R.; Wu, J.; An, J.; Haile, W.B.; Cooper, D.S.; Catano, M.; Yepes, M. Tissue-Type Plasminogen Activator Protects Neurons from Excitotoxin-Induced Cell Death via Activation of the ERK1/2-CREB-ATF3 Signaling Pathway. Mol. Cell Neurosci. 2013, 52, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. Stroke Research at a Crossroad: Asking the Brain for Directions. Nat. Neurosci. 2011, 14, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Arba, F.; Piccardi, B.; Palumbo, V.; Biagini, S.; Galmozzi, F.; Iovene, V.; Giannini, A.; Testa, G.D.; Sodero, A.; Nesi, M.; et al. Blood-Brain Barrier Leakage and Hemorrhagic Transformation: The Reperfusion Injury in Ischemic StroKe (RISK) Study. Eur. J. Neurol. 2021, 28, 3147–3154. [Google Scholar] [CrossRef]
- Bruns, J.; Hauser, W.A. The Epidemiology of Traumatic Brain Injury: A Review. Epilepsia 2003, 44, 2–10. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; David Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Zaloshnja, E.; Miller, T.; Langlois, J.A.; Selassie, A.W. Prevalence of Long-Term Disability from Traumatic Brain Injury in the Civilian Population of the United States, 2005. J. Head Trauma Rehabil. 2008, 23, 394–400. [Google Scholar] [CrossRef]
- Bazarian, J.J.; Cernak, I.; Noble-Haeusslein, L.; Potolicchio, S.; Temkin, N. Long-Term Neurologic Outcomes after Traumatic Brain Injury. J. Head Trauma Rehabil. 2009, 24, 439–451. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Gauberti, M.; Parcq, J.; Macrez, R.; Hommet, Y.; Obiang, P.; Hernangómez, M.; Montagne, A.; Liot, G.; Guaza, C.; et al. Tissue Plasminogen Activator Prevents White Matter Damage Following Stroke. J. Exp. Med. 2011, 208, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Pu, H.; Leak, R.K.; Shi, Y.; Mu, H.; Hu, X.; Lu, Z.; Foley, L.M.; Hitchens, T.K.; Dixon, C.E.; et al. Tissue Plasminogen Activator Promotes White Matter Integrity and Functional Recovery in a Murine Model of Traumatic Brain Injury. Proc. Natl. Acad. Sci. USA 2018, 115, E9230–E9238. [Google Scholar] [CrossRef]
- Gaberel, T.; MacRez, R.; Gauberti, M.; Montagne, A.; Hebert, M.; Petersen, K.U.; Touze, E.; Agin, V.; Emery, E.; Ali, C.; et al. Immunotherapy Blocking the Tissue Plasminogen Activator-Dependent Activation of N-Methyl-D-Aspartate Glutamate Receptors Improves Hemorrhagic Stroke Outcome. Neuropharmacology 2013, 67, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.; Kim, E.H.; Kim, S.J.; Sinn, D.I.; Ko, S.Y.; Kim, M.; Roh, J.K. Memantine Reduces Hematoma Expansion in Experimental Intracerebral Hemorrhage, Resulting in Functional Improvement. J. Cereb. Blood Flow Metab. 2006, 26, 536–544. [Google Scholar] [CrossRef]
- Lotocki, G.; de Rivero Vaccari, J.P.; Perez, E.R.; Sanchez-Molano, J.; Furones-Alonso, O.; Bramlett, H.M.; Dietrich, W.D. Alterations in Blood-Brain Barrier Permeability to Large and Small Molecules and Leukocyte Accumulation after Traumatic Brain Injury: Effects of Post-Traumatic Hypothermia. J. Neurotrauma 2009, 26, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Dixon, C.E.; Banik, N.L. Molecular Mechanisms in the Pathogenesis of Traumatic Brain Injury. Histol. Histopathol. 2002, 17, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, M.M.; Parks, E.A.; Dedman, J.R.; Kaetzel, M.A.; Lyeth, B.G.; Berman, R.F. Phosphorylation of Calcium Calmodulin-Dependent Protein Kinase II Following Lateral Fluid Percussion Brain Injury in Rats. J. Neurotrauma 2007, 24, 638–650. [Google Scholar] [CrossRef]
- Atkins, C.M.; Oliva, A.A.; Alonso, O.F.; Pearse, D.D.; Bramlett, H.M.; Dietrich, W.D. Modulation of the CAMP Signaling Pathway after Traumatic Brain Injury. Exp. Neurol. 2007, 208, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Geddes-Klein, D.M.; Serbest, G.; Mesfin, M.N.; Cohen, A.S.; Meaney, D.F. Pharmacologically Induced Calcium Oscillations Protect Neurons from Increases in Cytosolic Calcium after Trauma. J. Neurochem. 2006, 97, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.T.; Cheng, N.C.; Wu, C.Y.; Yang, Y.L. NKCC1-Mediated Traumatic Brain Injury-Induced Brain Edema and Neuron Death via Raf/MEK/MAPK Cascade. Crit. Care Med. 2008, 36, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Doshi, S.; Spaethling, J.M.; Hockenberry, A.J.; Patel, T.P.; Geddes-Klein, D.M.; Lynch, D.R.; Meaney, D.F. N-Methyl-D-Aspartate Receptor Mechanosensitivity Is Governed by C Terminus of NR2B Subunit. J. Biol. Chem. 2012, 287, 4348–4359. [Google Scholar] [CrossRef]
- Maneshi, M.M.; Maki, B.; Gnanasambandam, R.; Belin, S.; Popescu, G.K.; Sachs, F.; Hua, S.Z. Mechanical Stress Activates NMDA Receptors in the Absence of Agonists. Sci. Rep. 2017, 7, 39610. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Giovannoni, G. Multiple Sclerosis Should Be Treated Using a Step-down Strategy Rather than a Step-up Strategy-YES. Mult. Scler. J. 2016, 22, 1397–1400. [Google Scholar] [CrossRef]
- Venkatesan, A.; Adatia, K. Anti-NMDA-Receptor Encephalitis: From Bench to Clinic. ACS Chem. Neurosci. 2017, 8, 2586–2595. [Google Scholar] [CrossRef]
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An Update on Anti-NMDA Receptor Encephalitis for Neurologists and Psychiatrists: Mechanisms and Models. Lancet Neurol. 2019, 18, 1045–1057. [Google Scholar] [CrossRef]
- Dalmau, J.; Tüzün, E.; Wu, H.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic Anti-N-Methyl-D-Aspartate Receptor Encephalitis Associated with Ovarian Teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Xiong, H. Anti-NMDA Receptor Encephalitis: A Review of Mechanistic Studies. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 1–11. Available online: https://pubmed.ncbi.nlm.nih.gov/33815666/ (accessed on 4 July 2022).
- Nosadini, M.; Mohammad, S.S.; Corazza, F.; Ruga, E.M.; Kothur, K.; Perilongo, G.; Frigo, A.C.; Toldo, I.; Dale, R.C.; Sartori, S. Herpes Simplex Virus-Induced Anti-N-Methyl-d-Aspartate Receptor Encephalitis: A Systematic Literature Review with Analysis of 43 Cases. Dev. Med. Child Neurol. 2017, 59, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Planagumà, J.; Leypoldt, F.; Mannara, F.; Gutiérrez-Cuesta, J.; Martín-García, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-Methyl D-Aspartate Receptor Antibodies Alter Memory and Behaviour in Mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, E.; Horvath, J.; Shiloh-Malawsky, Y.; Sangha, N.; Martinez-Lage, M.; Dalmau, J. Analysis of Complement and Plasma Cells in the Brain of Patients with Anti-NMDAR Encephalitis. Neurology 2011, 77, 589–593. [Google Scholar] [CrossRef]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical Experience and Laboratory Investigations in Patients with Anti-NMDAR Encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef]
- Dalmau, J. Limbic Encephalitis and Variants Related to Neuronal Cell Membrane Autoantigens. Rinsho Shinkeigaku 2008, 48, 871–874. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wagnon, I.; Hélie, P.; Bardou, I.; Regnauld, C.; Lesec, L.; Leprince, J.; Naveau, M.; Delaunay, B.; Toutirais, O.; Lemauff, B.; et al. Autoimmune Encephalitis Mediated by B-Cell Response against N-Methyl-d-Aspartate Receptor. Brain 2020, 143, 2957–2972. [Google Scholar] [CrossRef]
- Ladépêche, L.; Planagumà, J.; Thakur, S.; Suárez, I.; Hara, M.; Borbely, J.S.; Sandoval, A.; Laparra-Cuervo, L.; Dalmau, J.; Lakadamyali, M. NMDA Receptor Autoantibodies in Autoimmune Encephalitis Cause a Subunit-Specific Nanoscale Redistribution of NMDA Receptors. Cell Rep. 2018, 23, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Tovar, K.R.; Goehring, A.; Jalali-Yazdi, F.; Okada, N.J.; Gouaux, E.; Westbrook, G.L. Autoimmune Receptor Encephalitis in Mice Induced by Active Immunization with Conformationally Stabilized Holoreceptors. Sci. Transl. Med. 2019, 11, eaaw0044. [Google Scholar] [CrossRef]
- Warikoo, N.; Brunwasser, S.J.; Benz, A.; Shu, H.J.; Paul, S.M.; Lewis, M.; Doherty, J.; Quirk, M.; Piccio, L.; Zorumski, C.F.; et al. Positive Allosteric Modulation as a Potential Therapeutic Strategy in Anti-NMDA Receptor Encephalitis. J. Neurosci. 2018, 38, 3218–3229. [Google Scholar] [CrossRef]
- Mannara, F.; Radosevic, M.; Planagumà, J.; Soto, D.; Aguilar, E.; García-Serra, A.; Maudes, E.; Pedreño, M.; Paul, S.; Doherty, J.; et al. Allosteric Modulation of NMDA Receptors Prevents the Antibody Effects of Patients with Anti-NMDAR Encephalitis. Brain 2020, 143, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Q.; Wang, Z.Z.; Chen, N.H. The Receptor Hypothesis and the Pathogenesis of Depression: Genetic Bases and Biological Correlates. Pharmacol. Res. 2021, 167, 105542. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J. Role of Tissue-Type Plasminogen Activator and Plasminogen Activator Inhibitor-1 in Psychological Stress and Depression. Oncotarget 2017, 8, 113258–113268. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, A.M.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. Reduced Levels of NR2A and NR2B Subunits of NMDA Receptor and PSD-95 in the Prefrontal Cortex in Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Amidfar, M.; Woelfer, M.; Réus, G.Z.; Quevedo, J.; Walter, M.; Kim, Y.K. The Role of NMDA Receptor in Neurobiology and Treatment of Major Depressive Disorder: Evidence from Translational Research. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 94, 109668. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Quiroz, J.A.; de Jesus, G.; Denicoff, K.K.; Luckenbaugh, D.A.; Manji, H.K.; Charney, D.S. A Double-Blind, Placebo-Controlled Study of Memantine in the Treatment of Major Depression. Am. J. Psychiatry 2006, 163, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.T.; Ballard, E.D.; Bloch, M.H.; Mathew, S.J.; Murrough, J.W.; Feder, A.; Sos, P.; Wang, G.; Zarate, C.A.; Sanacora, G. The Effect of a Single Dose of Intravenous Ketamine on Suicidal Ideation: A Systematic Review and Individual Participant Data Meta-Analysis. Am. J. Psychiatry 2018, 175, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Castaldelli-Maia, J.M.; Bhugra, D. Analysis of Global Prevalence of Mental and Substance Use Disorders within Countries: Focus on Sociodemographic Characteristics and Income Levels. Int. Rev. Psychiatry 2022, 34, 6–15. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping Genomic Loci Implicates Genes and Synaptic Biology in Schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Tsutsui, K.; Kanbayashi, T.; Tanaka, K.; Boku, S.; Ito, W.; Tokunaga, J.; Mori, A.; Hishikawa, Y.; Shimizu, T.; Nishino, S. Anti-NMDA-Receptor Antibody Detected in Encephalitis, Schizophrenia, and Narcolepsy with Psychotic Features. BMC Psychiatry 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Walter, M.; Glanz, W.; Sarnyai, Z.; Bernstein, H.G.; Vielhaber, S.; Kästner, A.; Skalej, M.; Jordan, W.; Schiltz, K.; et al. Increased Prevalence of Diverse N-Methyl-D-Aspartate Glutamate Receptor Antibodies in Patients with an Initial Diagnosis of Schizophrenia: Specific Relevance of IgG NR1a Antibodies for Distinction from N-Methyl-D-Aspartate Glutamate Receptor Encephalitis. JAMA Psychiatry 2013, 70, 271–278. [Google Scholar] [CrossRef]
- Pollak, T.A.; McCormack, R.; Peakman, M.; Nicholson, T.R.; David, A.S. Prevalence of Anti-N-Methyl-D-Aspartate (NMDA) Receptor [Corrected] Antibodies in Patients with Schizophrenia and Related Psychoses: A Systematic Review and Meta-Analysis. Psychol. Med. 2014, 44, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- le Guen, E.; Doukhan, R.; Hamdani, N.; Tamouza, R.; Groc, L.; Honnorat, J.; Leboyer, M. Anti-NR1 Antibodies in Anti-N-Methyl-D-Aspartate Receptor Encephalitis and Schizophrenia. Med. Sci. 2015, 31, 60–67. [Google Scholar] [CrossRef]
- Masdeu, J.C.; GonzáLez-Pinto, A.; Matute, C.; de Azúa, S.R.; Palomino, A.; de Leon, J.; Berman, K.F.; Dalmau, J. Serum IgG Antibodies against the NR1 Subunit of the NMDA Receptor Not Detected in Schizophrenia. Am. J. Psychiatry 2012, 169, 1120–1121. [Google Scholar] [CrossRef] [PubMed]
- Elmi, S.; Sahu, G.; Malavade, K.; Jacob, T. Role of Tissue Plasminogen Activator and Plasminogen Activator Inhibitor as Potential Biomarkers in Psychosis. Asian J. Psychiatr. 2019, 43, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Jézéquel, J.; Johansson, E.M.; Dupuis, J.P.; Rogemond, V.; Gréa, H.; Kellermayer, B.; Hamdani, N.; le Guen, E.; Rabu, C.; Lepleux, M.; et al. Dynamic Disorganization of Synaptic NMDA Receptors Triggered by Autoantibodies from Psychotic Patients. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Lyall, K.; Croen, L.; Daniels, J.; Fallin, M.D.; Ladd-Acosta, C.; Lee, B.K.; Park, B.Y.; Snyder, N.W.; Schendel, D.; Volk, H.; et al. The Changing Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2017, 38, 81–102. [Google Scholar] [CrossRef]
- Stone, W.S.; Iguchi, L. Do Apparent Overlaps between Schizophrenia and Autistic Spectrum Disorders Reflect Superficial Similarities or Etiological Commonalities? N. Am. J. Med. Sci. 2011, 4, 124. [Google Scholar] [CrossRef] [PubMed]
- Tzang, R.F.; Chang, C.H.; Chang, Y.C.; Lane, H.Y. Autism Associated with Anti-NMDAR Encephalitis: Glutamate-Related Therapy. Front. Psychiatry 2019, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef]
- Gogolla, N.; LeBlanc, J.J.; Quast, K.B.; Südhof, T.C.; Fagiolini, M.; Hensch, T.K. Common Circuit Defect of Excitatory-Inhibitory Balance in Mouse Models of Autism. J. Neurodev. Disord. 2009, 1, 172–181. [Google Scholar] [CrossRef]
- Yuan, H.; Low, C.M.; Moody, O.A.; Jenkins, A.; Traynelis, S.F. Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Mol. Pharmacol. 2015, 88, 203–217. [Google Scholar] [CrossRef]
- Myers, S.J.; Yuan, H.; Kang, J.Q.; Tan, F.C.K.; Traynelis, S.F.; Low, C.M. Distinct Roles of GRIN2A and GRIN2B Variants in Neurological Conditions. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Pothakos, K.; Robinson, J.K.; Gravanis, I.; Marsteller, D.A.; Dewey, S.L.; Tsirka, S.E. Decreased Serotonin Levels Associated with Behavioral Disinhibition in Tissue Plasminogen Activator Deficient (TPA−/−) Mice. Brain Res. 2010, 1326, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Maiya, R.; Norris, E.H.; Kreek, M.J.; Strickland, S. Involvement of Tissue Plasminogen Activator in Stress Responsivity during Acute Cocaine Withdrawal in Mice. Stress 2010, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Şimşek, Ş.; Çetin, İ.; Çim, A.; Kaya, S. Elevated Levels of Tissue Plasminogen Activator and E-Selectin in Male Children with Autism Spectrum Disorder. Autism. Res. 2016, 9, 1241–1247. [Google Scholar] [CrossRef]
- Hu, Y.; Shan, Y.; Du, Q.; Ding, Y.; Shen, C.; Wang, S.; Ding, M.; Xu, Y. Gender and Socioeconomic Disparities in Global Burden of Epilepsy: An Analysis of Time Trends From 1990 to 2017. Front. Neurol. 2021, 12, 473. [Google Scholar] [CrossRef]
- Borsato, G.S.; Siegel, J.L.; Rose, M.Q.; Ojard, M.; Feyissa, A.M.; Quinones-Hinojosa, A.; Jackson, D.A.; Rogers, E.R.; Freeman, W.D. Ketamine in Seizure Management and Future Pharmacogenomic Considerations. Pharm. J. 2020, 20, 351–354. [Google Scholar] [CrossRef]
- Mendes, N.F.; Pansani, A.P.; Carmanhães, E.R.F.; Tange, P.; Meireles, J.V.; Ochikubo, M.; Chagas, J.R.; da Silva, A.V.; Monteiro de Castro, G.; le Sueur-Maluf, L. The Blood-Brain Barrier Breakdown During Acute Phase of the Pilocarpine Model of Epilepsy Is Dynamic and Time-Dependent. Front. Neurol. 2019, 10, 382. [Google Scholar] [CrossRef]
- Lei, S.; He, Y.; Zhu, Z.; Liu, Z.; Lin, Y.; He, Y.; Du, S.; Chen, X.; Xu, P.; Zhu, X. Inhibition of NMDA Receptors Downregulates Astrocytic AQP4 to Suppress Seizures. Cell Mol. Neurobiol. 2020, 40, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, X. Ketamine for the Treatment of Refractory Status Epilepticus. Seizure 2015, 30, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and Stroke: Identifying Novel Targets for Neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Markandaiah, S.S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Excess Glutamate Secreted from Astrocytes Drives Upregulation of P-Glycoprotein in Endothelial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2019, 316, 27–38. [Google Scholar] [CrossRef]
- Ayala, G.X.; Tapia, R. Late N-Methyl-D-Aspartate Receptor Blockade Rescues Hippocampal Neurons from Excitotoxic Stress and Death after 4-Aminopyridine-Induced Epilepsy. Eur. J. Neurosci. 2005, 22, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Kocaeli, H.; Korfali, E.; Öztürk, H.; Kahveci, N.; Yilmazlar, S. MK-801 Improves Neurological and Histological Outcomes after Spinal Cord Ischemia Induced by Transient Aortic Cross-Clipping in Rats. Surg. Neurol. 2005, 64 (Suppl. S2), S22–S26. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, A.G.; Ivanova, S.A.; Knoblach, S.M.; Faden, A.I. New In Vitro Model of Traumatic Neuronal Injury: Evaluation of Secondary Injury and Glutamate Receptor-Mediated Neurotoxicity. J. Neurotrauma 2009, 14, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, A.G.; Ivanova, S.A.; Faden, A.I. MGluR Modulation of Post-Traumatic Neuronal Death: Role of NMDA Receptors. Neuroreport 1997, 8, 2561–2566. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Ikonomidou, C.; Mosinger, J.L.; Frierdich, G. MK-801 Prevents Hypobaric-Ischemic Neuronal Degeneration in Infant Rat Brain. J. Neurosci. 1989, 9, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.K.; Ridley, R.M. The Effect of Dizocilpine (MK-801) on Conditional Discrimination Learning in the Rat. Behav. Pharmacol. 1997, 8, 383–388. [Google Scholar] [CrossRef]
- Harder, J.A.; Aboobaker, A.A.; Hodgetts, T.C.; Ridley, R.M. Learning Impairments Induced by Glutamate Blockade Using Dizocilpine (MK-801) in Monkeys. Br. J. Pharmacol. 1998, 125, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Aronowski, J.; Ostrow, P.; Samways, E.; Strong, R.; Zivin, J.A.; Grotta, J.G. Graded Bioassay for Demonstration of Brain Rescue from Experimental Acute Ischemia in Rats. Stroke 1994, 25, 2235–2240. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A.; Hasegawa, Y.; Fisher, M. Effects of a Novel NMDA Receptor Antagonist on Experimental Stroke Quantitatively Assessed by Spectral EEG and Infarct Volume. Neurol. Res. 1994, 16, 443–448. [Google Scholar] [CrossRef]
- Muir, K.W.; Grosset, D.G.; Lees, K.R. Effects of Prolonged Infusions of the NMDA Antagonist Aptiganel Hydrochloride (CNS 1102) in Normal Volunteers. Clin. Neuropharmacol. 1997, 20, 311–321. [Google Scholar] [CrossRef]
- Hoyte, L.; Barber, P.; Buchan, A.; Hill, M. The Rise and Fall of NMDA Antagonists for Ischemic Stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef]
- Mousavi, S.A.; Saadatnia, M.; Khorvash, F.; Hoseini, T.; Sariaslani, P. Evaluation of the Neuroprotective Effect of Dextromethorphan in the Acute Phase of Ischaemic Stroke. Arch. Med. Sci. 2011, 7, 465–469. [Google Scholar] [CrossRef] [PubMed]
- O’Suilleabhain, P.; Dewey, R.B. A Randomized Trial of Amantadine in Huntington Disease. Arch. Neurol. 2003, 60, 996–998. [Google Scholar] [CrossRef]
- Wang, C.C.; Wu, T.L.; Lin, F.J.; Tai, C.H.; Lin, C.H.; Wu, R.M. Amantadine Treatment and Delayed Onset of Levodopa-Induced Dyskinesia in Patients with Early Parkinson’s Disease. Eur. J. Neurol. 2022, 29, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Moryl, E.; Danysz, W.; Quack, G. Potential Antidepressive Properties of Amantadine, Memantine and Bifemelane. Pharmacol. Toxicol. 1993, 72, 394–397. [Google Scholar] [CrossRef]
- Vanle, B.; Olcott, W.; Jimenez, J.; Bashmi, L.; Danovitch, I.; Ishak, W.W. NMDA Antagonists for Treating the Non-Motor Symptoms in Parkinson’s Disease. Transl. Psychiatry 2018, 8, 117. [Google Scholar] [CrossRef]
- Czarnecka, K.; Chuchmacz, J.; Wójtowicz, P.; Szymański, P. Memantine in Neurological Disorders—Schizophrenia and Depression. J. Mol. Med. 2021, 99, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Aboujaoude, E.; Barry, J.J.; Gamel, N. Memantine Augmentation in Treatment-Resistant Obsessive-Compulsive Disorder: An Open-Label Trial. J. Clin. Psychopharmacol. 2009, 29, 51–55. [Google Scholar] [CrossRef]
- Herrmann, N.; Cappell, J.; Eryavec, G.M.; Lancôtt, K.L. Changes in Nursing Burden Following Memantine for Agitation and Aggression in Long-Term Care Residents with Moderate to Severe Alzheimer’s Disease: An Open-Label Pilot Study. CNS Drugs 2011, 25, 425–433. [Google Scholar] [CrossRef]
- Feusner, J.D.; Kerwin, L.; Saxena, S.; Bystritsky, A. Differential Efficacy of Memantine for Obsessive-Compulsive Disorder vs. Generalized Anxiety Disorder: An Open-Label Trial. Psychopharm. Bull 2009, 42, 81–93. Available online: https://pubmed.ncbi.nlm.nih.gov/19204653/ (accessed on 30 June 2022).
- Beladi Moghadam, N.; Pourheidar, E.; Ahmadpour, F.; Kafi, H.; Salamzadeh, J.; Nasiri, S.; Sistanizad, M. The Effects of Memantine on the Serum Concentrations of Matrix Metalloproteinases and Neurologic Function of Patients with Ischemic Stroke. J. Clin. Neurosci. 2021, 90, 268–272. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and Its Implication for Treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Sapkota, K. The Origin of NMDA Receptor Hypofunction in Schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef]
- Li, L.; Vlisides, P.E. Ketamine: 50 Years of Modulating the Mind. Front. Hum. Neurosci. 2016, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Fogaça, M.V.; Fukumoto, K.; Franklin, T.; Liu, R.J.; Duman, C.H.; Vitolo, O.V.; Duman, R.S. N-Methyl-D-Aspartate Receptor Antagonist d-Methadone Produces Rapid, MTORC1-Dependent Antidepressant Effects. Neuropsychopharmacology 2019, 44, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Hung, D. Dimebon: A Phase 3 Investigational Agent for Alzheimer’s Disease with a Novel Mitochondrial Mechanism of Action [Abstract S4-04-05]. In Proceedings of the 12th International Conference on Alzheimer’s Disease and Related Disorders, Vienna, Austria, 11–16 June 2008. [Google Scholar]
- Sweetlove, M. Phase III CONCERT Trial of Latrepirdine. Pharm. Med. 2012, 26, 113–115. [Google Scholar] [CrossRef]
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A Randomized, Placebo-Controlled Trial of Latrepirdine in Huntington Disease. Arch. Neurol. 2010, 67, 154–160. [Google Scholar] [CrossRef]
- Dorsey, E.R. A Randomized, Double-Blind, Placebo-Controlled Study of Latrepirdine in Patients with Mild to Moderate Huntington Disease. JAMA Neurol. 2013, 70, 25–33. [Google Scholar] [CrossRef]
- Boast, C.A.; Gerhardt, S.C.; Pastor, G.; Lehmann, J.; Etienne, P.E.; Liebman, J.M. The N-Methyl-D-Aspartate Antagonists CGS 19755 and CPP Reduce Ischemic Brain Damage in Gerbils. Brain Res. 1988, 442, 345–348. [Google Scholar] [CrossRef]
- Grotta, J.; Clark, W.; Coull, B.; Pettigrew, L.C.; Mackay, B.; Goldstein, L.B.; Meissner, I.; Murphy, D.; Larue, L. Safety and Tolerability of the Glutamate Antagonist CGS 19755 (Selfotel) in Patients with Acute Ischemic Stroke. Results of a Phase IIa Randomized Trial. Stroke 1995, 26, 602–605. [Google Scholar] [CrossRef]
- Davis, S.M.; Lees, K.R.; Albers, G.W.; Diener, H.C.; Markabi, S.; Karlsson, G.; Norris, J. Selfotel in Acute Ischemic Stroke: Possible Neurotoxic Effects of an NMDA Antagonist. Stroke 2000, 31, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Preskorn, S.; Macaluso, M.; Mehra, V.; Zammit, G.; Moskal, J.R.; Burch, R.M. Randomized Proof of Concept Trial of GLYX-13, an N-Methyl-D-Aspartate Receptor Glycine Site Partial Agonist, in Major Depressive Disorder Nonresponsive to a Previous Antidepressant Agent. J. Psychiatr. Pract. 2015, 21, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Gajda, J.M.; Asiedu, M.; Morrison, G.; Dunning, J.A.; Ghoreishi-Haack, N.; Barth, A.L. NYX-2925, a novel, non-opioid, small-molecule modulator of the n-methyl-d-aspartate receptor (nmdar), demonstrates potential to treat chronic, supraspinal centralized pain conditions. Med. Drug. Discov. 2021, 9, 100067. [Google Scholar] [CrossRef]
- Ghoreishi-Haack, N.; Priebe, J.M.; Aguado, J.D.; Colechio, E.M.; Burgdorf, J.S.; Bowers, M.S.; Cearley, C.N.; Khan, M.A.; Moskal, J.R. NYX-2925 Is a Novel N-Methyl-d-Aspartate Receptor Modulator That Induces Rapid and Long-Lasting Analgesia in Rat Models of Neuropathic Pain. J. Pharmacol. Exp. Ther. 2018, 366, 485–497. [Google Scholar] [CrossRef]
- Barth, A.L.; Schneider, J.S.; Johnston, T.H.; Hill, M.P.; Brotchie, J.M.; Moskal, J.R.; Cearley, C.N. NYX-458 Improves Cognitive Performance in a Primate Parkinson’s Disease Model. Mov. Disord. 2020, 35, 640–649. [Google Scholar] [CrossRef]
- Fasipe, O.J. The Emergence of New Antidepressants for Clinical Use: Agomelatine Paradox versus Other Novel Agents. IBRO Rep. 2019, 6, 95–110. [Google Scholar] [CrossRef]
- Muir, K.W.; Lees, K.R. Excitatory Amino Acid Antagonists for Acute Stroke. Cochrane Database Syst. Rev. 2003, 2003, CD001244. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.J.; Ruppa, K.P.; Wilson, L.J.; Tahirovic, Y.A.; Lyuboslavsky, P.; Menaldino, D.S.; Dentmon, Z.W.; Koszalka, G.W.; Zaczek, R.; Dingledine, R.J.; et al. A Glutamate N-Methyl-d-Aspartate (NMDA) Receptor Subunit 2B–Selective Inhibitor of NMDA Receptor Function with Enhanced Potency at Acidic PH and Oral Bioavailability for Clinical Use. J. Pharmacol. Exp. Ther. 2021, 379, 41–52. [Google Scholar] [CrossRef]
- Yuan, H.; Myers, S.J.; Wells, G.; Nicholson, K.L.; Swanger, S.A.; Lyuboslavsky, P.; Tahirovic, Y.A.; Menaldino, D.S.; Ganesh, T.; Wilson, L.J.; et al. Context-Dependent GluN2B-Selective Inhibitors of NMDA Receptor Function Are Neuroprotective with Minimal Side Effects. Neuron 2015, 85, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.M.; Choi, M.H.; Sohn, S.I.; Hwang, Y.H.; Ahn, S.H.; Lee, Y.B.; Shin, D.I.; Chamorro, Á.; Choi, D.W. Safety and Optimal Neuroprotection of Neu2000 in Acute Ischemic Stroke with ReCanalization: Study Protocol for a Randomized, Double-Blinded, Placebo-Controlled, Phase-II Trial. Trials 2018, 19, 375. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, M.J.; Horrocks, M.H.; Zhu, F.; Muresan, L.; Benavides-Piccione, R.; DeFelipe, J.; Fricker, D.; Kopanitsa, M.V.; Duncan, R.R.; Klenerman, D.; et al. PSD95 Nanoclusters Are Postsynaptic Building Blocks in Hippocampus Circuits. Sci. Rep. 2016, 6, 24626. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor- PSD-95 Protein Interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific Coupling of NMDA Receptor Activation to Nitric Oxide Neurotoxicity by PSD-95 Protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.X.; Lin, Y.H.; Qian, X.D.; Tang, Y.; Zhou, H.H.; Jin, X.; Ni, H.Y.; Zhang, F.Y.; Qin, C.; Li, F.; et al. Interaction of NNOS with PSD-95 Negatively Controls Regenerative Repair after Stroke. J. Neurosci. 2014, 34, 13535–13548. [Google Scholar] [CrossRef] [PubMed]
- Kurrikoff, K.; Langel, Ü. Recent CPP-Based Applications in Medicine. Expert Opin. Drug Deliv. 2019, 16, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Nunez, D.M.; Ji, Z.; Sun, X.; Teves, L.; Garman, J.D.; Tymianski, M. Plasmin-Resistant PSD-95 Inhibitors Resolve Effect-Modifying Drug-Drug Interactions between Alteplase and Nerinetide in Acute Stroke. Sci. Transl. Med. 2021, 13, eabb1498. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; terBrugge, K.G.; Milot, G.; Clark, W.M.; MacDonald, R.L.; Kelly, M.E.; et al. Safety and Efficacy of NA-1 in Patients with Iatrogenic Stroke after Endovascular Aneurysm Repair (ENACT): A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef]
- Bach, A.; Clausen, B.H.; Møller, M.; Vestergaard, B.; Chi, C.N.; Round, A.; Sørensen, P.L.; Nissen, K.B.; Kastrup, J.S.; Gajhede, M.; et al. A High-Affinity, Dimeric Inhibitor of PSD-95 Bivalently Interacts with PDZ1-2 and Protects against Ischemic Brain Damage. Proc. Natl. Acad. Sci. USA 2012, 109, 3317–3322. [Google Scholar] [CrossRef]
- Meloni, B.P.; Brookes, L.M.; Clark, V.W.; Cross, J.L.; Edwards, A.B.; Anderton, R.S.; Hopkins, R.M.; Hoffmann, K.; Knuckey, N.W. Poly-Arginine and Arginine-Rich Peptides Are Neuroprotective in Stroke Models. J. Cereb. Blood Flow Metab. 2015, 35, 993. [Google Scholar] [CrossRef]
- Milani, D.; Cross, J.L.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W.; Meloni, B.P. Neuroprotective Efficacy of Poly-Arginine R18 and NA-1 (TAT-NR2B9c) Peptides Following Transient Middle Cerebral Artery Occlusion in the Rat. Neurosci. Res. 2017, 114, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Tian, H.; Tao, L.; Wang, Y.; Xiang, J.; Wang, S.; Ding, H. Tramiprosate Protects Neurons against Ischemic Stroke by Disrupting the Interaction between PSD95 and NNOS. Neuropharmacology 2014, 83, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, A.; Meng, X.; Yalkun, G.; Xu, A.; Gao, Z.; Chen, H.; Ji, Y.; Xu, J.; Geng, D.; et al. Edaravone Dexborneol Versus Edaravone Alone for the Treatment of Acute Ischemic Stroke A Phase III, Randomized, Double-Blind, Comparative Trial. Stroke 2021, 52, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Li, L.L.; Ginet, V.; Liu, X.; Vergun, O.; Tuittila, M.; Mathieu, M.; Bonny, C.; Puyal, J.; Truttmann, A.C.; Courtney, M.J. The NNOS-P38MAPK Pathway Is Mediated by NOS1AP during Neuronal Death. J. Neurosci. 2013, 33, 8185–8201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Li, T.Y.; Luo, C.X.; Jiang, N.; Chang, L.; Lin, Y.H.; Zhou, H.H.; Chen, C.; Zhang, Y.; Lu, W.; et al. CAPON-NNOS Coupling Can Serve as a Target for Developing New Anxiolytics. Nat. Med. 2014, 20, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.Y.; Song, Y.X.; Lin, Y.H.; Cao, B.; Wang, D.L.; Zhang, Y.; Dong, J.; Liang, H.Y.; Xu, K.; Li, T.Y.; et al. Dissociating NNOS (Neuronal NO Synthase)-CAPON (Carboxy-Terminal Postsynaptic Density-95/Discs Large/Zona Occludens-1 Ligand of NNOS) Interaction Promotes Functional Recovery After Stroke via Enhanced Structural Neuroplasticity. Stroke 2019, 50, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Pei, L.; Liao, M.; Liu, B.; Zhang, Y.; Jiang, W.; Mielke, J.G.; Li, L.; Chen, Y.; El-Hayek, Y.H.; et al. Dual Neuroprotective Signaling Mediated by Downregulating Two Distinct Phosphatase Activities of PTEN. J. Neurosci. 2004, 24, 4052–4060. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Benoist, M.; Lario, A.; Knafo, S.; Petrok, C.N.; Esteban, J.A. PTEN Is Recruited to the Postsynaptic Terminal for NMDA Receptor-Dependent Long-Term Depression. EMBO J. 2010, 29, 2827–2840. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jia, J.; Zhou, X.; Xiao, Y.; Wang, Y.; Mao, X.; Zhen, X.; Guan, Y.; Alkayed, N.J.; Cheng, J. Delayed Administration of a PTEN Inhibitor BPV Improves Functional Recovery after Experimental Stroke. Neuroscience 2013, 231, 272–281. [Google Scholar] [CrossRef]
- Fernández-Monreal, M.; López-Atalaya, J.P.; Benchenane, K.; Cacquevel, M.; Dulin, F.; le Caer, J.-P.; Rossier, J.; Jarrige, A.-C.; Mackenzie, E.T.; Colloc’h, N.; et al. Arginine 260 of the Amino-Terminal Domain of NR1 Subunit Is Critical for Tissue-Type Plasminogen Activator-Mediated Enhancement of N-Methyl-D-Aspartate Receptor Signaling. J. Biol. Chem. 2004, 279, 50850–50856. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Nagai, N.; Urano, T.; Napiorkowska-Pawlak, D.; Ihara, H.; Takada, Y.; Collen, D.; Takada, A. Rapid, Specific and Active Site-Catalyzed Effect of Tissue-Plasminogen Activator on Hippocampus-Dependent Learning in Mice. Neuroscience 2002, 113, 995–1001. [Google Scholar] [CrossRef]
- Samson, A.L.; Nevin, S.T.; Croucher, D.; Niego, B.; Daniel, P.B.; Weiss, T.W.; Moreno, E.; Monard, D.; Lawrence, D.A.; Medcalf, R.L. Tissue-Type Plasminogen Activator Requires a Co-Receptor to Enhance NMDA Receptor Function. J. Neurochem. 2008, 107, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Stroebel, D.; Yao, C.A.; Taly, A.; Paoletti, P. Allosteric Signaling and Dynamics of the Clamshell-like NMDA Receptor GluN1 N-Terminal Domain. Nat. Struct. Mol. Biol. 2013, 20, 477–485. [Google Scholar] [CrossRef]
- Chevilley, A.; Lesept, F.; Lenoir, S.; Ali, C.; Parcq, J.; Vivien, D. Impacts of Tissue-Type Plasminogen Activator (TPA) on Neuronal Survival. Front. Cell Neurosci. 2015, 9, 415. [Google Scholar] [CrossRef]
- Cassé, F.; Bardou, I.; Danglot, L.; Briens, A.; Montagne, A.; Parcq, J.; Alahari, A.; Galli, T.; Vivien, D.; Docagne, F. Glutamate Controls TPA Recycling by Astrocytes, Which in Turn Influences Glutamatergic Signals. J. Neurosci. 2012, 32, 5186–5199. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Atalaya, J.P.; Roussel, B.D.; Levrat, D.; Parcq, J.; Nicole, O.; Hommet, Y.; Benchenane, K.; Castel, H.; Leprince, J.; To Van, D.; et al. Toward Safer Thrombolytic Agents in Stroke: Molecular Requirements for NMDA Receptor-Mediated Neurotoxicity. J. Cereb. Blood Flow Metab. 2008, 28, 1212–1221. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Yepes, M.; Jiang, Q.; Li, Q.; Arniego, P.; Coleman, T.A.; Lawrence, D.A.; Chopp, M. Adjuvant Treatment with Neuroserpin Increases the Therapeutic Window for Tissue-Type Plasminogen Activator Administration in a Rat Model of Embolic Stroke. Circulation 2002, 106, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Buisson, A.; Nicole, O.; Docagne, F.; Sartelet, H.; Mackenzie, E.T.; Vivien, D. Up-Regulation of a Serine Protease Inhibitor in Astrocytes Mediates the Neuroprotective Activity of Transforming Growth Factor Beta1. FASEB J. 1998, 12, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Chung, C. NMDA Receptor as a Newly Identified Member of the Metabotropic Glutamate Receptor Family: Clinical Implications for Neurodegenerative Diseases. Mol. Cells 2013, 36, 99–104. [Google Scholar] [CrossRef]
- Torrente, D.; Su, E.J.; Schielke, G.P.; Warnock, M.; Stevenson, T.; Mann, K.; Vivien, D.; Lawrence, D.A. Tissue Plasminogen Activator Interaction with NMDAR1 Promotes Dopaminergic Neuron Degeneration in a Model of A-Synuclein-Mediated Neurotoxicity. SSRN Electron. J. 2022. [Google Scholar] [CrossRef]



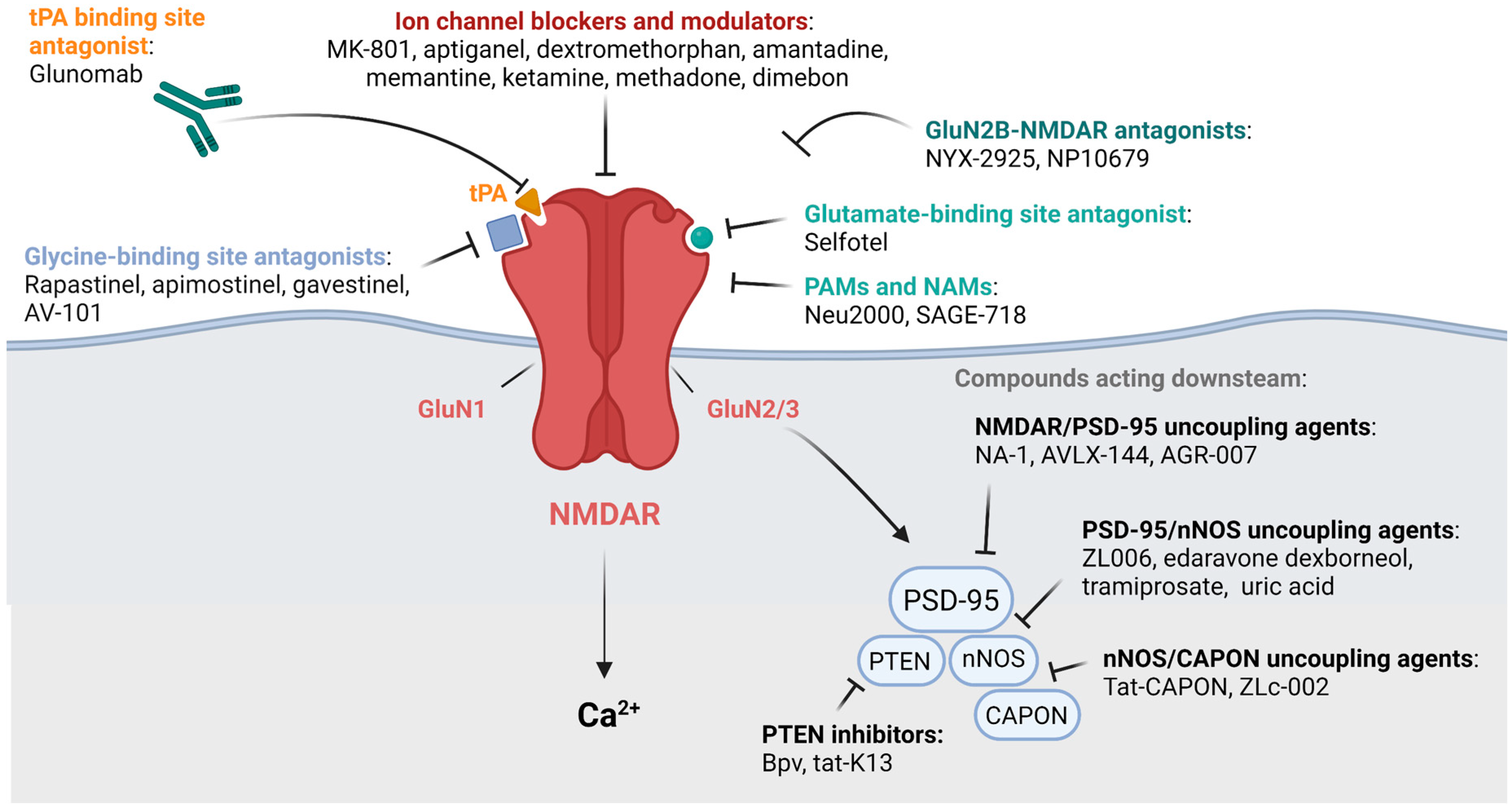{kind=link}
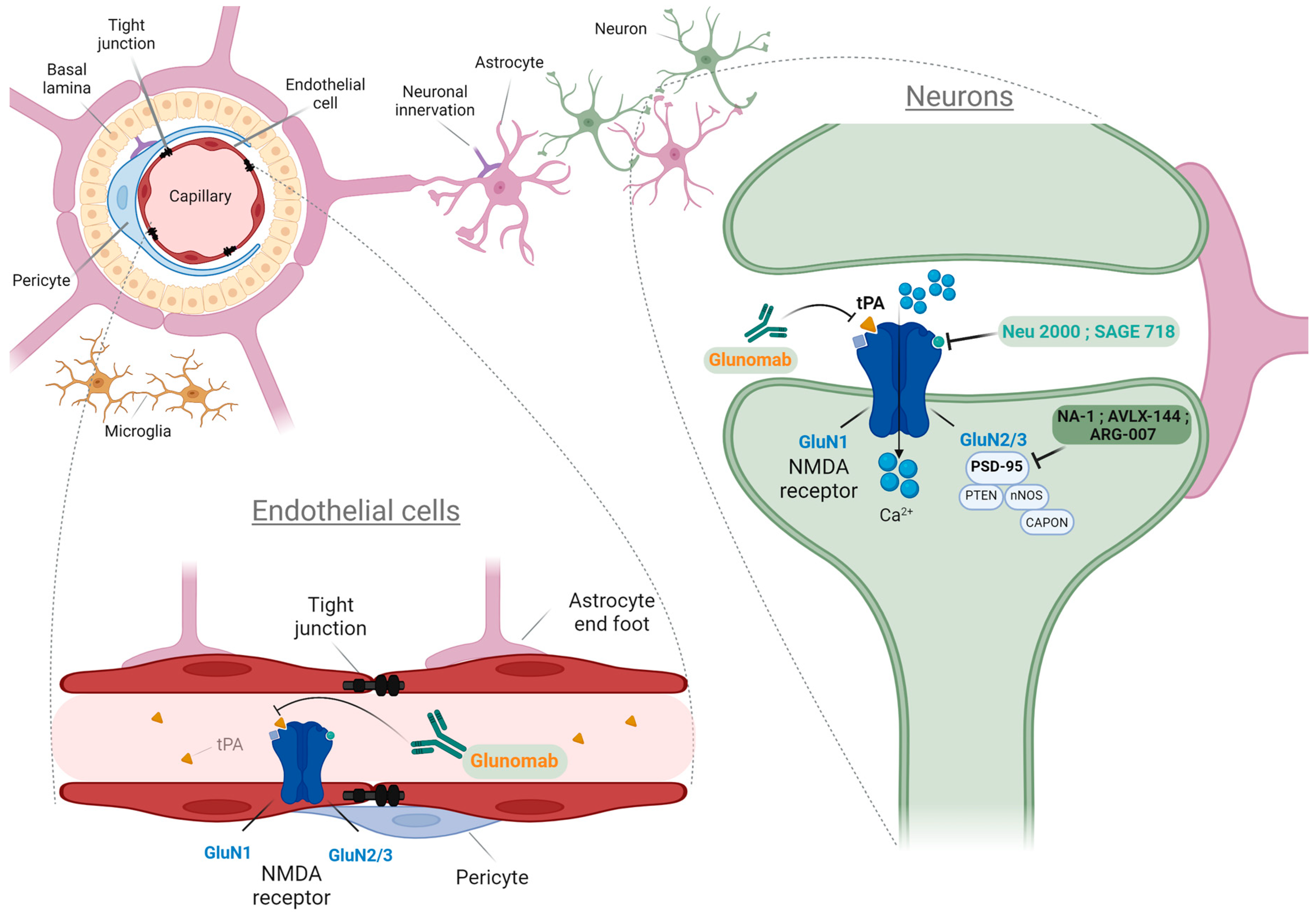{kind=link}
| Endogenous Molecules | Pharmacological Molecules |
|---|---|
| Channel blockers and modulators: Mg2+ [1] Neurosteroids [8] | Channel blockers and modulators: MK-801 [9] Aptiganel [10] Dextromethorphan [11] Amantadine [12] Memantine [13] Ketamine [13] Methadone [14] Dimebon [15] |
| Agonists: Glutamate [16] Glycine/D-serine [17] L-Theanine [18] | |
| Glycine/Glutamate site antagonists: Polyamines [19] Zn2+; Cu2+ [20] | Glycine/Glutamate site antagonists: Selfotel [21] Rapastinel [22] NYX-2925 [23] Apimostinel [24] Gavestinel [25] AV-101 [26] Phencyclidine [27] |
| PAMs and NAMs: Spermines and Spermidines [19] Cholesterol [28] Pregnelonone sulfate [29] ATP [30] | PAMs and NAMs: SAGE-718 [31] NP10679 [19] Neu2000 [32,33] |
| Downstream antagonist: Uric acid [34] | Downstream antagonists: Nerinetide [35] AVLX-144 [36] ARG-007 [37] ZL006 [38] Tramiprosate [39] Edaravone Dexborneol [40] Tat-CAPON and ZLc-002 [41] Bpv and Tat-K13 [42,43] |
| Modulators: amyloid-β (Aβ) [44] tPA [45] | Antibody: Glunomab [46] |
| Cell Types | Subunit Types | |
|---|---|---|
| NVU | Neurons [20] | GluN1/GluN2(A,B,C,D)/GluN3 (A,B) |
| Endothelial cells [70] | GluN1/GluN2A/2C/GluN3(A,B) | |
| Oligodendrocytes [71] | GluN1/GluN2C/GluN3(A,B) | |
| Astrocytes [72] | GluN1/GluN2(A,B,C,D)/GluN3(A,B) | |
| Microglia [73] | GluN1/GluN2(A,B,C,D)/GluN3A | |
| T cells [74] | GluN1/GluN2(A,B) | |
| Macrophages [75] | GluN1/GluN2(A,D) | |
| Neutrophils [76] | GluN1/GluN2B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seillier, C.; Lesept, F.; Toutirais, O.; Potzeha, F.; Blanc, M.; Vivien, D. Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases. Int. J. Mol. Sci. 2022, 23, 10336. https://doi.org/10.3390/ijms231810336
Seillier C, Lesept F, Toutirais O, Potzeha F, Blanc M, Vivien D. Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases. International Journal of Molecular Sciences. 2022; 23(18):10336. https://doi.org/10.3390/ijms231810336
Chicago/Turabian StyleSeillier, Célia, Flavie Lesept, Olivier Toutirais, Fanny Potzeha, Manuel Blanc, and Denis Vivien. 2022. "Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases" International Journal of Molecular Sciences 23, no. 18: 10336. https://doi.org/10.3390/ijms231810336
APA StyleSeillier, C., Lesept, F., Toutirais, O., Potzeha, F., Blanc, M., & Vivien, D. (2022). Targeting NMDA Receptors at the Neurovascular Unit: Past and Future Treatments for Central Nervous System Diseases. International Journal of Molecular Sciences, 23(18), 10336. https://doi.org/10.3390/ijms231810336