
Inhibition of TREM-2 Markedly Suppresses Joint Inflammation and Damage in Experimental Arthritis

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
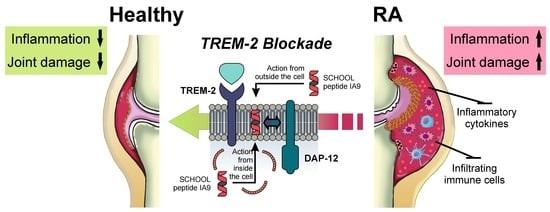
1. Introduction
2. Results
2.1. Reduction of Inflammation and Suppression of Arthritis in a Therapeutic CIA Model
2.2. Reduction of Joint Damage and Bone Erosion in CIA
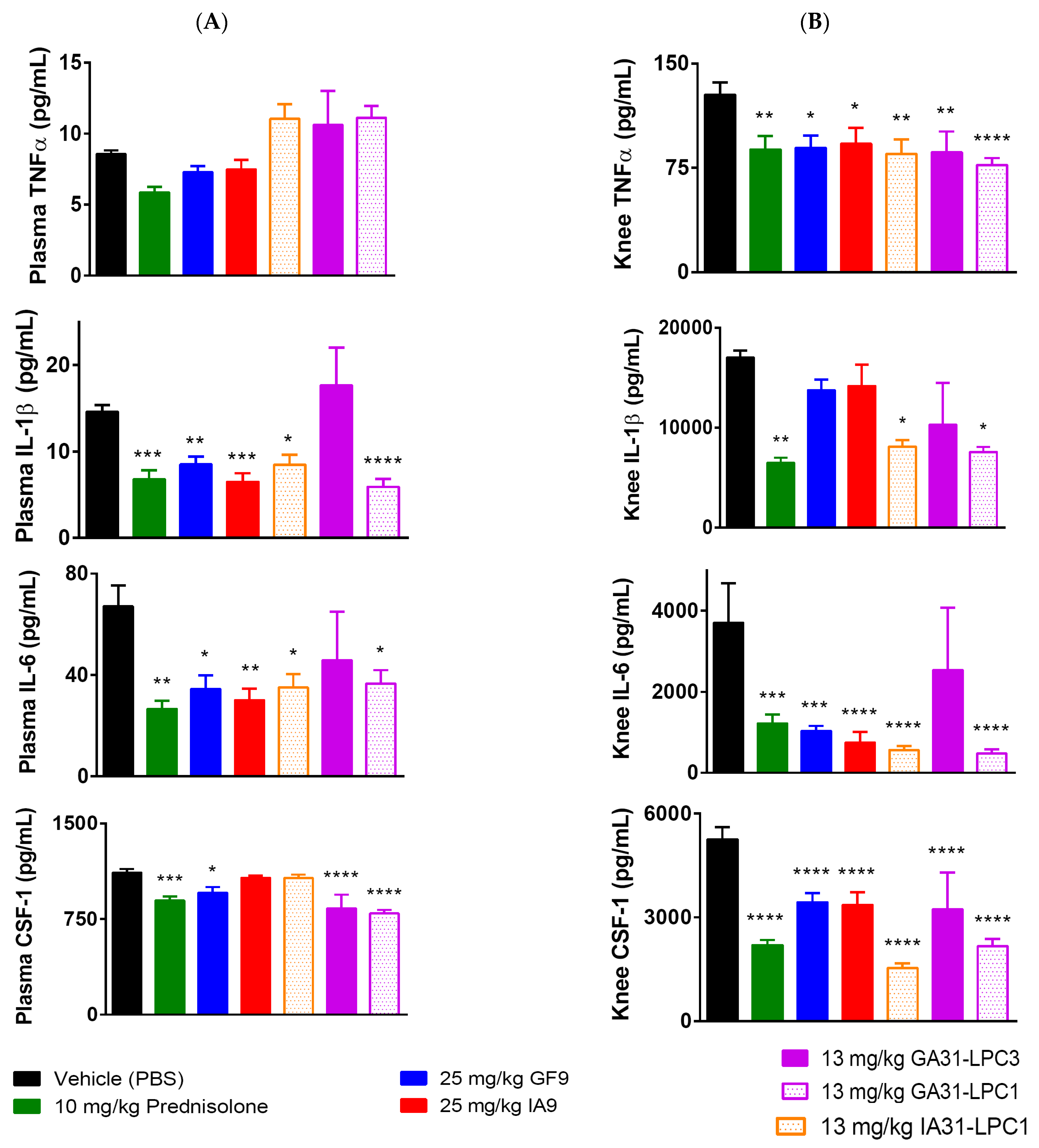
2.3. Reduction of Plasma and Joint Proinflammatory Cytokine Levels in CIA
2.4. Immunohistochemical Analysis for F4/80, CD68, Collagen IV, TREM-1 and TREM-2
3. Discussion
4. Materials and Methods
4.1. Chemicals, Lipids and Peptides
4.2. Lipopeptide Complexes
4.3. Therapeutic Collagen-Induced Arthritis (CIA) Model
4.4. Histological Assessment
4.5. Plasma and Joint Cytokine Analysis
4.6. Immunohistochemical Analysis
4.7. Statistical Analysis
4.8. Sequence Accession Numbers
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RA | Rheumatoid arthritis |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| M-CSF | Macrophage-colony stimulating factor |
| CIA | Collagen-induced arthritis |
| SEM | Standard error of the mean |
| TREM-1 | Triggering receptors expressed by myeloid cells 1 |
| TREM-2 | Triggering receptors expressed by myeloid cells 2 |
| DAP-12 | DNAX-activating protein of 12 kDa |
| SCHOOL | Signaling chain homo-oligomerization |
| MIRR | Multichain immune recognition receptor |
| LPC | Lipopeptide complex |
| HDL | High density lipoproteins |
| Apo | Apolipoprotein |
| SR-A | Type A scavenger receptor |
| IP | Intraperitoneally |
| I | Inflammation |
| P | Pannus |
| CD | Cartilage damage |
| PBF | Periosteal bone formation |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| IBD | Inflammatory bowel disease |
| mAb | Monoclonal antibody |
| ICB | Immune checkpoint blockade |
| PD-1 | Programmed cell death protein 1 |
| RP-HPLC | Reversed-phase high-performance liquid chromatography |
| DLS | Dynamic light scattering |
| PDI | Polydispersity index |
| SPF | Specific pathogen-free |
| PBS | Phosphate-buffered saline |
| NBF | Neutral buffered formalin |
| T blue | Toluidine blue |
| DAB | Diaminobenzidine |
| IHC | Immunohistochemistry |
| HRP | Horseradish peroxidase |
| ELISA | Enzyme-linked immunosorbent assay |
| FFPE | Formalin-fixed, paraffin-embedded |
References
- Prasad, P.; Verma, S.; Surbhi; Ganguly, N.K.; Chaturvedi, V.; Mittal, S.A. Rheumatoid arthritis: Advances in treatment strategies. Mol. Cell. Biochem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gibofsky, A. Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. Am. J. Manag. Care 2012, 18, S295–S302. [Google Scholar] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Pincus, T.; Callahan, L.F. Taking mortality in rheumatoid arthritis seriously--predictive markers, socioeconomic status and comorbidity. J. Rheumatol. 1986, 13, 841–845. [Google Scholar] [PubMed]
- Scott, D.L.; Symmons, D.P.; Coulton, B.L.; Popert, A.J. Long-term outcome of treating rheumatoid arthritis: Results after 20 years. Lancet 1987, 1, 1108–1111. [Google Scholar] [CrossRef]
- McInnes, I.B.; O’Dell, J.R. State-of-the-art: Rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1898–1906. [Google Scholar] [CrossRef]
- Siegel, J. Comparative effectiveness of treatments for rheumatoid arthritis. Ann. Intern. Med. 2008, 148, 162–163. [Google Scholar] [CrossRef]
- Bansard, C.; Lequerre, T.; Daveau, M.; Boyer, O.; Tron, F.; Salier, J.P.; Vittecoq, O.; Le-Loet, X. Can rheumatoid arthritis responsiveness to methotrexate and biologics be predicted? Rheumatology 2009, 48, 1021–1028. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Kinne, R.W.; Brauer, R.; Stuhlmuller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [Google Scholar] [CrossRef][Green Version]
- Alivernini, S.; Tolusso, B.; Ferraccioli, G.; Gremese, E.; Kurowska-Stolarska, M.; McInnes, I.B. Driving chronicity in rheumatoid arthritis: Perpetuating role of myeloid cells. Clin. Exp. Immunol. 2018, 193, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mulherin, D.; Fitzgerald, O.; Bresnihan, B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kinne, R.W.; Stuhlmuller, B.; Burmester, G.R. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res. Ther. 2007, 9, 224. [Google Scholar] [CrossRef]
- Bresnihan, B.; Gerlag, D.M.; Rooney, T.; Smeets, T.J.; Wijbrandts, C.A.; Boyle, D.; Fitzgerald, O.; Kirkham, B.W.; McInnes, I.B.; Smith, M.; et al. Synovial macrophages as a biomarker of response to therapeutic intervention in rheumatoid arthritis: Standardization and consistency across centers. J. Rheumatol. 2007, 34, 620–622. [Google Scholar] [PubMed]
- Li, J.; Hsu, H.C.; Mountz, J.D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 2012, 14, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Axmann, R.; Bohm, C.; Kronke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Takagi, N.; Mihara, M.; Moriya, Y.; Nishimoto, N.; Yoshizaki, K.; Kishimoto, T.; Takeda, Y.; Ohsugi, Y. Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 1998, 41, 2117–2121. [Google Scholar] [CrossRef]
- Smolen, J.S.; Avila, J.C.; Aletaha, D. Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: Disassociation of the link between inflammation and destruction. Ann. Rheum. Dis. 2012, 71, 687–693. [Google Scholar] [CrossRef]
- Benucci, M.; Saviola, G.; Baiardi, P.; Manfredi, M.; Sarzi Puttini, P.; Atzeni, F. Determinants of Risk Infection During Therapy with Anti TNF-Alpha Blocking Agents in Rheumatoid Arthritis. Open Rheumatol. J. 2012, 6, 33–37. [Google Scholar] [CrossRef][Green Version]
- Choy, E.H.; Kavanaugh, A.F.; Jones, S.A. The problem of choice: Current biologic agents and future prospects in RA. Nat. Rev. Rheumatol. 2013, 9, 154–163. [Google Scholar] [CrossRef]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Edrees, A.F.; Misra, S.N.; Abdou, N.I. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: Correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin. Exp. Rheumatol. 2005, 23, 469–474. [Google Scholar] [PubMed]
- Takeuchi, T.; Miyasaka, N.; Tatsuki, Y.; Yano, T.; Yoshinari, T.; Abe, T.; Koike, T. Baseline tumour necrosis factor alpha levels predict the necessity for dose escalation of infliximab therapy in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- American College of Rheumatology Ad Hoc Committee on Clinical Guidelines. Guidelines for monitoring drug therapy in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 723–731. [Google Scholar]
- Garrood, T.; Pitzalis, C. Targeting the inflamed synovium: The quest for specificity. Arthritis Rheum. 2006, 54, 1055–1060. [Google Scholar] [CrossRef]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef]
- Kuai, J.; Gregory, B.; Hill, A.; Pittman, D.D.; Feldman, J.L.; Brown, T.; Carito, B.; O’Toole, M.; Ramsey, R.; Adolfsson, O.; et al. TREM-1 expression is increased in the synovium of rheumatoid arthritis patients and induces the expression of pro-inflammatory cytokines. Rheumatology 2009, 48, 1352–1358. [Google Scholar] [CrossRef]
- Dower, K.; Ellis, D.K.; Saraf, K.; Jelinsky, S.A.; Lin, L.L. Innate immune responses to TREM-1 activation: Overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J. Immunol. 2008, 180, 3520–3534. [Google Scholar] [CrossRef]
- Murakami, Y.; Akahoshi, T.; Aoki, N.; Toyomoto, M.; Miyasaka, N.; Kohsaka, H. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. 2009, 60, 1615–1623. [Google Scholar] [CrossRef]
- Shen, Z.T.; Sigalov, A.B. Rationally designed ligand-independent peptide inhibitors of TREM-1 ameliorate collagen-induced arthritis. J. Cell. Mol. Med. 2017, 21, 2524–2534. [Google Scholar] [CrossRef]
- Weber, B.; Schuster, S.; Zysset, D.; Rihs, S.; Dickgreber, N.; Schurch, C.; Riether, C.; Siegrist, M.; Schneider, C.; Pawelski, H.; et al. TREM-1 Deficiency Can Attenuate Disease Severity without Affecting Pathogen Clearance. PLoS Pathog. 2014, 10, e1003900. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, A.; Derive, M.; Gibot, S.; Leemans, J.C.; Florquin, S.; Dessing, M.C. TREM-1 and its potential ligands in non-infectious diseases: From biology to clinical perspectives. Pharmacol. Ther. 2017, 177, 81–95. [Google Scholar] [CrossRef]
- Gao, X.; Dong, Y.; Liu, Z.; Niu, B. Silencing of triggering receptor expressed on myeloid cells-2 enhances the inflammatory responses of alveolar macrophages to lipopolysaccharide. Mol. Med. Rep. 2013, 7, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Feng, J.; Tang, L. Function of TREM1 and TREM2 in Liver-Related Diseases. Cells 2020, 9, 2626. [Google Scholar] [CrossRef]
- Rai, V.; Dietz, N.E.; Dilisio, M.F.; Radwan, M.M.; Agrawal, D.K. Vitamin D attenuates inflammation, fatty infiltration, and cartilage loss in the knee of hyperlipidemic microswine. Arthritis Res. Ther. 2016, 18, 203. [Google Scholar] [CrossRef]
- Wang, M.; Gao, X.; Zhao, K.; Chen, H.; Xu, M.; Wang, K. Effect of TREM2 on Release of Inflammatory Factor from LPS-stimulated Microglia and Its Possible Mechanism. Ann. Clin. Lab. Sci. 2019, 49, 249–256. [Google Scholar]
- Suchankova, M.; Bucova, M.; Tibenska, E.; Tedlova, E.; Demian, J.; Majer, I.; Novosadova, H.; Tedla, M.; Paulovicova, E.; Kantarova, D. Triggering receptor expressed on myeloid cells-1 and 2 in bronchoalveolar lavage fluid in pulmonary sarcoidosis. Respirology 2013, 18, 455–462. [Google Scholar] [CrossRef]
- Sharif, O.; Gawish, R.; Warszawska, J.M.; Martins, R.; Lakovits, K.; Hladik, A.; Doninger, B.; Brunner, J.; Korosec, A.; Schwarzenbacher, R.E.; et al. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumonia. PLoS Pathog. 2014, 10, e1004167. [Google Scholar] [CrossRef] [PubMed]
- Genua, M.; Rutella, S.; Correale, C.; Danese, S. The triggering receptor expressed on myeloid cells (TREM) in inflammatory bowel disease pathogenesis. J. Transl. Med. 2014, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Crotti, T.N.; Dharmapatni, A.A.; Alias, E.; Zannettino, A.C.; Smith, M.D.; Haynes, D.R. The immunoreceptor tyrosine-based activation motif (ITAM)-related factors are increased in synovial tissue and vasculature of rheumatoid arthritic joints. Arthritis Res. Ther. 2012, 14, R245. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Li, J.H.; Xu, J.H.; Huang, S.H.; Liu, G.W.; Zhang, W.Q.; Zheng, P.Z.; Huang, J.R. Triggering receptor expressed on myeloid cells 2 in synovial tissue of rheumatoid arthritis rats. Chin. J. Tissue Eng. Res. 2015, 19, 2807–2813. [Google Scholar] [CrossRef]
- Kober, D.L.; Brett, T.J. TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 2017, 429, 1607–1629. [Google Scholar] [CrossRef]
- Sigalov, A.B. Multichain immune recognition receptor signaling: Different players, same game? Trends Immunol. 2004, 25, 583–589. [Google Scholar] [CrossRef]
- Sigalov, A.B. Immune cell signaling: A novel mechanistic model reveals new therapeutic targets. Trends Pharmacol. Sci. 2006, 27, 518–524. [Google Scholar] [CrossRef]
- Sigalov, A.B. SCHOOL of nature: Ligand-independent immunomodulatory peptides. Drug Discov. Today 2020, 25, 1298–1306. [Google Scholar] [CrossRef]
- Greaves, D.R.; Gordon, S. Thematic review series: The immune system and atherogenesis. Recent insights into the biology of macrophage scavenger receptors. J. Lipid Res. 2005, 46, 11–20. [Google Scholar] [CrossRef]
- Lin, Y.L.; de Villiers, W.J.S.; Garvy, B.; Post, S.R.; Nagy, T.R.; Safadi, F.F.; Faugere, M.C.; Wang, G.; Malluche, H.H.; Williams, J.P. The effect of class a scavenger receptor deficiency in bone. J. Biol. Chem. 2007, 282, 4653–4660. [Google Scholar] [CrossRef]
- Takemura, K.; Sakashita, N.; Fujiwara, Y.; Komohara, Y.; Lei, X.; Ohnishi, K.; Suzuki, H.; Kodama, T.; Mizuta, H.; Takeya, M. Class A scavenger receptor promotes osteoclast differentiation via the enhanced expression of receptor activator of NF-kappaB (RANK). Biochem. Biophys. Res. Commun. 2010, 391, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B. Nature-inspired nanoformulations for contrast-enhanced in vivo MR imaging of macrophages. Contrast Media Mol. Imaging 2014, 9, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B. A novel ligand-independent peptide inhibitor of TREM-1 suppresses tumor growth in human lung cancer xenografts and prolongs survival of mice with lipopolysaccharide-induced septic shock. Int. Immunopharmacol. 2014, 21, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Corvo, M.L.; Boerman, O.C.; Oyen, W.J.; Van Bloois, L.; Cruz, M.E.; Crommelin, D.J.; Storm, G. Intravenous administration of superoxide dismutase entrapped in long circulating liposomes. II. In vivo fate in a rat model of adjuvant arthritis. Biochim. Biophys. Acta (BBA)-Biomembr. 1999, 1419, 325–334. [Google Scholar] [CrossRef][Green Version]
- Corvo, M.L.; Boerman, O.C.; Oyen, W.J.; Jorge, J.C.; Cruz, M.E.; Crommelin, D.J.; Storm, G. Subcutaneous administration of superoxide dismutase entrapped in long circulating liposomes: In vivo fate and therapeutic activity in an inflammation model. Pharm. Res. 2000, 17, 600–606. [Google Scholar] [CrossRef]
- Wang, S.; Yang, S.; Lai, X.; Song, Y.; Hu, L.; Li, C.; Shi, T.; Liu, X.; Deng, Y.; Chen, G. Sialic Acid Conjugate-Modified Liposomal Dexamethasone Palmitate Targeting Neutrophils for Rheumatoid Arthritis Therapy: Influence of Particle Size. AAPS PharmSciTech 2021, 22, 16. [Google Scholar] [CrossRef]
- Stolina, M.; Bolon, B.; Dwyer, D.; Middleton, S.; Duryea, D.; Kostenuik, P.J.; Feige, U.; Zack, D.J. The evolving systemic and local biomarker milieu at different stages of disease progression in rat collagen-induced arthritis. Biomarkers 2008, 13, 692–712. [Google Scholar] [CrossRef]
- Stolina, M.; Bolon, B.; Middleton, S.; Dwyer, D.; Brown, H.; Duryea, D.; Zhu, L.; Rohner, A.; Pretorius, J.; Kostenuik, P.; et al. The evolving systemic and local biomarker milieu at different stages of disease progression in rat adjuvant-induced arthritis. J. Clin. Immunol. 2009, 29, 158–174. [Google Scholar] [CrossRef]
- Sigalov, A.B. The SCHOOL of nature: III. From mechanistic understanding to novel therapies. Self Nonself 2010, 1, 192–224. [Google Scholar] [CrossRef][Green Version]
- Schett, G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res. Ther. 2007, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Helsen, M.M.; Saxne, T.; van De Loo, F.A.; Heinegard, D.; van Den Berg, W.B. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J. Immunol. 1999, 163, 5049–5055. [Google Scholar] [PubMed]
- Ohno, H.; Uemura, Y.; Murooka, H.; Takanashi, H.; Tokieda, T.; Ohzeki, Y.; Kubo, K.; Serizawa, I. The orally-active and selective c-Fms tyrosine kinase inhibitor Ki20227 inhibits disease progression in a collagen-induced arthritis mouse model. Eur. J. Immunol. 2008, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; He, X.; Bian, Y.; Guo, Q.; Zheng, K.; Zhao, Y.; Lu, C.; Liu, B.; Xu, X.; Zhang, G.; et al. Triptolide Modulates TREM-1 Signal Pathway to Inhibit the Inflammatory Response in Rheumatoid Arthritis. Int. J. Mol. Sci. 2016, 17, 498. [Google Scholar] [CrossRef]
- Burmester, G.R.; Stuhlmuller, B.; Keyszer, G.; Kinne, R.W. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997, 40, 5–18. [Google Scholar] [CrossRef]
- Pandupuspitasari, N.S.; Khan, F.A.; Huang, C.J.; Chen, X.; Zhang, S. Novel Attributions of TREMs in Immunity. Curr. Issues Mol. Biol. 2016, 20, 47–54. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Rommel, C.; Camps, M.; Ji, H. PI3K delta and PI3K gamma: Partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 2007, 7, 191–201. [Google Scholar] [CrossRef]
- Dinarello, C.A. Inflammation in human disease: Anticytokine therapy. Biol. Blood Marrow Transplant. 2009, 15, 134–136. [Google Scholar] [CrossRef]
- Curtis, J.R.; Singh, J.A. Use of biologics in rheumatoid arthritis: Current and emerging paradigms of care. Clin. Ther. 2011, 33, 679–707. [Google Scholar] [CrossRef]
- Mewar, D.; Wilson, A.G. Treatment of rheumatoid arthritis with tumour necrosis factor inhibitors. Br. J. Pharmacol. 2011, 162, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, F.; Sarzi-Puttini, P.; Botsios, C.; Carletto, A.; Cipriani, P.; Favalli, E.G.; Frati, E.; Foschi, V.; Gasparini, S.; Giardina, A.; et al. Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: Comparison of adalimumab, etanercept and infliximab in the GISEA registry. Autoimmun. Rev. 2012, 12, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.B.; Hyrich, K.L.; Mercer, L.K.; Dixon, W.G.; Ustianowski, A.P.; Helbert, M.; Watson, K.D.; Lunt, M.; Symmons, D.P. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2011, 70, 1810–1814. [Google Scholar] [CrossRef]
- Sica, A.; Massarotti, M. Myeloid suppressor cells in cancer and autoimmunity. J. Autoimmun. 2017, 85, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.T.; Sigalov, A.B. Novel TREM-1 Inhibitors Attenuate Tumor Growth and Prolong Survival in Experimental Pancreatic Cancer. Mol. Pharm. 2017, 14, 4572–4582. [Google Scholar] [CrossRef]
- Rojas, M.A.; Shen, Z.T.; Caldwell, R.B.; Sigalov, A.B. Blockade of TREM-1 prevents vitreoretinal neovascularization in mice with oxygen-induced retinopathy. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2761–2768. [Google Scholar] [CrossRef]
- Tornai, D.; Furi, I.; Shen, Z.T.; Sigalov, A.B.; Coban, S.; Szabo, G. Inhibition of Triggering Receptor Expressed on Myeloid Cells 1 Ameliorates Inflammation and Macrophage and Neutrophil Activation in Alcoholic Liver Disease in Mice. Hepatol. Commun. 2019, 3, 99–115. [Google Scholar] [CrossRef]
- Shen, Z.T.; Sigalov, A.B. SARS Coronavirus Fusion Peptide-Derived Sequence Suppresses Collagen-Induced Arthritis in DBA/1J Mice. Sci. Rep. 2016, 6, 28672. [Google Scholar] [CrossRef]
- Correale, C.; Genua, M.; Vetrano, S.; Mazzini, E.; Martinoli, C.; Spinelli, A.; Arena, V.; Peyrin-Biroulet, L.; Caprioli, F.; Passini, N.; et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology 2013, 144, 346–356 e343. [Google Scholar] [CrossRef]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e817. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, X.; Nie, K.; Cheng, L.; Zhang, Z.; Hu, Y.; Peng, W. Systematic Pan-Cancer Analysis Identifies TREM2 as an Immunological and Prognostic Biomarker. Front. Immunol. 2021, 12, 646523. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Pollack, J.L.; Rudolph, J.; Dash, S.; Abushawish, M.; Lee, T.; Jahchan, N.S.; Canaday, P.; Lu, E.; Norng, M.; et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021, 37, 109844. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, N.; Laverty, S.; Kraus, V.B.; Aigner, T. Basic methods in histopathology of joint tissues. Osteoarthr. Cartil. 2010, 18 (Suppl. 3), S113–S116. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigalov, A.B. Inhibition of TREM-2 Markedly Suppresses Joint Inflammation and Damage in Experimental Arthritis. Int. J. Mol. Sci. 2022, 23, 8857. https://doi.org/10.3390/ijms23168857
Sigalov AB. Inhibition of TREM-2 Markedly Suppresses Joint Inflammation and Damage in Experimental Arthritis. International Journal of Molecular Sciences. 2022; 23(16):8857. https://doi.org/10.3390/ijms23168857
Chicago/Turabian StyleSigalov, Alexander B. 2022. "Inhibition of TREM-2 Markedly Suppresses Joint Inflammation and Damage in Experimental Arthritis" International Journal of Molecular Sciences 23, no. 16: 8857. https://doi.org/10.3390/ijms23168857
APA StyleSigalov, A. B. (2022). Inhibition of TREM-2 Markedly Suppresses Joint Inflammation and Damage in Experimental Arthritis. International Journal of Molecular Sciences, 23(16), 8857. https://doi.org/10.3390/ijms23168857